

Abstract
The crystal structures of bovine pancreatic ribonuclease A (RNase A) in complex with 3′,5′-ADP, 2′,5′-ADP, 5′-ADP, U-2′-p and U-3′-p have been determined at high resolution. The structures reveal that each inhibitor binds differently in the RNase A active site by anchoring a phosphate group in subsite P1. The most potent inhibitor of all five, 5′-ADP (Ki = 1.2 μM), adopts a syn conformation (in contrast to 3′,5′-ADP and 2′,5′-ADP, which adopt an anti), and it is the β- rather than the α-phosphate group that binds to P1. 3′,5′-ADP binds with the 5′-phosphate group in P1 and the adenosine in the B2 pocket. Two different binding modes are observed in the two RNase A molecules of the asymmetric unit for 2′,5′-ADP. This inhibitor binds with either the 3′ or the 5′ phosphate groups in subsite P1, and in each case, the adenosine binds in two different positions within the B2 subsite. The two uridilyl inhibitors bind similarly with the uridine moiety in the B1 subsite but the placement of a different phosphate group in P1 (2′ versus 3′) has significant implications on their potency against RNase A. Comparative structural analysis of the RNase A, eosinophil-derived neurotoxin (EDN), eosinophil cationic protein (ECP), and human angiogenin (Ang) complexes with these and other phosphonucleotide inhibitors provides a wealth of information for structure-based design of inhibitors specific for each RNase. These inhibitors could be developed to therapeutic agents that could control the biological activities of EDN, ECP, and ANG, which play key roles in human pathologies.
Keywords: Ribonuclease A inhibitors, structure-aided drug design, angiogenin, eosinophil-derived neurotoxin, eosinophil cationic protein
Bovine pancreatic Ribonuclease A (RNase A; EC 3.1.27.5) catalyzes the cleavage of P-O5′ bonds in RNA on the 3′ side of pyrimidines to form cyclic 2′,5′-phosphates (D’Alessio and Riordan 1997; Raines 1998). In recent years, RNase A has been the molecular target for the development of small molecule inhibitors. These inhibitors are being developed with the goal to restrain the biological activities of different RNase A homologs in a variety of pathological conditions. These homologs include human Angiogenin (Ang), a potent inducer of neovascularization involved in cancer and in vascular and rheumatoid diseases (Fett et al. 1985), eosinophil-derived neurotoxin (EDN), and eosinophil cationic protein (ECP), two neurotoxins implicated in hypereosinophilic syndromes and allergy (Gleich and Adolphson 1986). EDN has been also implicated in the soluble HIV-1 inhibitory activity by lymphocytes (Rugeles et al. 2003). The ribonucleolytic activity of these homologs is closely associated with their biological function because it is impaired when assayed in the presence of ribonucleolytic inhibitors or when residues critical for their enzymic activity are chemically modified or mutated (Shapiro and Vallee 1989; Shapiro et al. 1989; Sorrentino et al. 1992; Newton et al. 1994; Domachowske et al. 1998a,b). Development of small molecule inhibitors using RNase A as a model are quite amenable because the central region of the catalytic active site of RNase A encompassing subsites P1, where the phosphate phosphodiester bond cleaveage occurs, and subsites B1 and B2, where the nucleotide bases on the 3′ and 5′ sides of the scissile bond bind, respectively, is highly conserved amongst RNase A homologs.
In the last years, a potent class of inhibitors, the adenosine 5′-pyrophosphate derivatives, has been discovered (Leonidas et al. 1997, 1999; Russo et al. 1997; Russo and Shapiro 1999; Jardine et al. 2001). Crystallographic studies on the binding of such derivatives like ppA-3′-p (Ki = 0.24 mM), ppA-2′-p (Ki = 0.52 mM; Leonidas et al. 1997; Russo et al. 1997), dUppA (Ki = 11.3 μM; Jardine et al. 2001), and pdUppA-3′-p (Ki= 0.027 μM; Leonidas et al. 1999; Russo and Shapiro 1999), with RNase A have shown that the phosphate groups constitute the anchor points for the binding of these inhibitors to RNase A. The 5′-pyrophosphate group binds with the β-phosphate at subsite P1, while the 2′ or 3′ adenylic phosphates bind at the peripheral phosphate-binding subsites Po and P2, respectively. The adenosine adopts the unusual syn conformation and binds at B2, while uridine is found at B1. These inhibitors are also potent against EDN, RNase 4, and Ang (Shapiro 1998; Russo and Shapiro 1999; Russo et al. 2001). However, recent structural studies of EDN complexes with three adenylic phosponucleotide inhibitors (Leonidas et al. 2001a), ECP in complex with 2′,5′-ADP (Mohan et al. 2002) and Ang in complex with phosphate and pyrophosphate ions (Leonidas et al. 2001b) have shown that the binding mode of phosphonucleotides to these enzymes might differ considerably from the one observed in RNase A. Thus, although the interactions of a phosphate group at P1 were still the driving force for the binding of phosphoadenosine inhibitors to EDN and ECP, the position of the nucleotide base differs considerably depending on the nucleotide (Leonidas et al. 2001a; Mohan et al. 2002). Furthermore, in Ang, the pyrophosphate ion binds with one of the phosphate groups in P1 and the other directed towards the pyrimidine binding site B1 in contrast to the binding mode of pyrophosphate derivatives to RNase A (Leonidas et al. 2001b).
In the present study we are investigating the mode of binding of three adenylic (3′,5′-ADP, 2′,5′-ADP, and 5′-ADP) and two uridylyl (U-2′-p, U-3′-p) inhibitors to RNase A at high resolution. These structures give a detailed picture of the specificity of the ribonucleolytic active site towards phosphates and nucleotide bases and show the degree of flexibility of subsites B1 and B2. The high-resolution structures also provide the basis for a comparative structural analysis of EDN, ECP, and RNase A phosphonucleotide complexes and assist the process of the development of ribonucleolytic inhibitors specific for each RNase. Furthermore, the crystallographic data of the RNase A complexed with 2′,5′-ADP and 5′-ADP at near atomic resolution (1.2 Å) revealed the positions of additional water molecules bound in the RNase A active site and indicated a complex water-mediated network of interactions between the inhibitors and the protein. This network may have important implications for the further development of new and more potent ribonucleolytic inhibitors.
Results and Discussion
Overall structures
The complexed structures of RNase A described here are similar to the free RNase A structure from monoclinic crystals reported previously (Leonidas et al. 1997), indicating that the binding of the inhibitors did not affect the overall structure of the protein. The RMSD between the structures of the free RNase A (PDB entry 1AFU) and RNase A in complex with either 3′,5′-ADP, 2′,5′-ADP, 5′-ADP, U-2′-p or U-3′-p is 0.79, 0.67, 0.76, 0.82, and 0.62 Å, respectively, for 248 equivalent Cα atoms. In all free RNase A structures reported so far the side chain of the catalytic residue His119 adopts two conformations denoted as A (χ1 = ∼160°) and B (χ1 = ∼−80°), which are related by a 100° rotation about the Cα–Cβ bond and a 180° rotation about the Cβ–Cγ bond (Borkakoti et al. 1982; Howlin et al. 1989; deMel et al. 1994). These conformations are dependent on the pH (Berisio et al. 1999) and the ionic strength of the crystallization solution (Fedorov et al. 1996). Conformation A is considered as the active conformation, which promotes catalysis, whereas conformation B is considered as the inactive conformation (Raines 1998). The side chain of His119 adopts conformation A in the 3′,5′-ADP, 5′-ADP, U-2′-p, and U-3′-p complexes while in the 2′,5′-ADP complex it is found in conformation B.
Superposition of the structures of the RNase A–3′,5′-ADP complex (molecules I and II of the asymmetric unit) on the structure of free RNase A at 1.1 Å resolution (PDB code 1KF2; Berisio et al. 2002) reveals that 3′,5′-ADP displaces two (molecule I) and three (molecule II) water molecules from the active site. Similarly, two water molecules are replaced upon binding of 2′,5′-ADP (molec I and molec II) while three (molec I) and one (molec II) water molecules are replaced upon binding of 5′-ADP. Upon binding of U-2′-p, five (molec I) and four (molec II) water molecules are replaced while upon binding of U-3′-p, five water molecules are replaced in both molecules I and II of the asymmetric unit.
The RMSD between the positions of the Cα atoms of the 3′,5′-ADP complex and those of the 2′,5′-ADP, 5′-ADP, U-2′-p, and U-3′-p complexes are 0.46, 0.42, 0.46, and 0.54 Å respectively. The corresponding values within the complexes, 2′,5′-ADP against 5′-ADP, U-2′-p, and U-3′-p are 0.24, 0.30, and 0.22 Å, respectively, whereas the RMSD between the 5′-ADP and U-2′-p, and U-3′-p complexes are 0.26 and 0.31 Å, respectively. In addition, the RMSD between the positions of the Cα atoms of the U-2′-p and U-3′-p complexes is 0.35 Å. Thus, it appears that the differences between the five complex structures are very small, mainly concentrated on the loop regions and are not related to the binding of the different inhibitors. The structures of the 3′,5′-ADP, U-2′-p, and U-3′-p complexes are defined at 1.5 Å resolution and contain 389, 299, and 415 water molecules, respectively. The structures of the 2′,5′-ADP, and 5′-ADP complexes are determined at a higher resolution (1.2 Å), and contain 558 and 540 water molecules, respectively. Numbering scheme definitions for all five inhibitors are presented in Figure 1 ▶.
Figure 1.
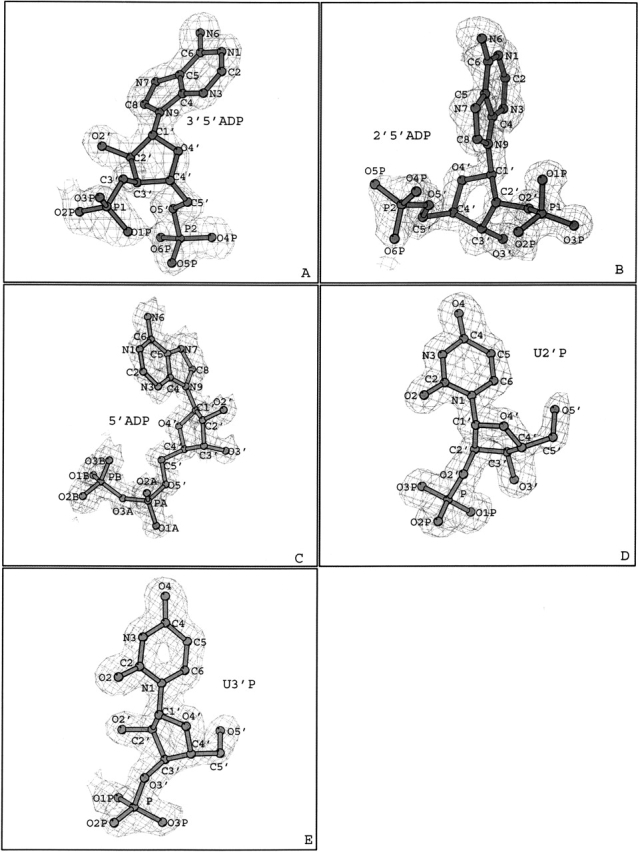
(A–E) Diagrams of the sigma 2|Fo| − |Fc| electron density maps for 3′,5′-ADP, 2′,5′-ADP, 5′-ADP, U-2′-p, and U-3′-p, respectively. Electron density maps were calculated with CNS (Brünger et al. 1998) from the RNase A model before incorporating the coordinates of each inhibitor, are contoured at 1.0 σ level, and the refined structure of the inhibitor is shown. General numbering scheme for each inhibitor is also shown. The figure was prepared with BOBSCRIPT (Esnouf 1997).
The binding of 3′,5′-ADP to RNase A
The 3′,5′-ADP molecule is well defined in the electron density map in both molecules (I and II) of RNase A in the asymmetric unit (Fig. 1A ▶). The conformation of 3′,5′-ADP when bound to RNase A is similar to that observed previously for adenosine nucleotides bound at B2 in the RNase A complexes with d(pA)4 (McPherson et al. 1986), d(ApTpApApG) (Fontecilla-Camps et al. 1994), and d(CpA) (Zegers et al. 1994), as well as to those frequently observed in the unbound and protein bound adenosines (Moodie and Thornton 1993). The glycosyl torsion angle χ adopts the anti conformation, and the rest of the backbone and phosphate torsion angles are in the preferred range for protein bound adenosines (Moodie and Thornton 1993). The γ torsion angle of 3′,5′-ADP in RNase A molecule II is in the unusual sp range, but its value (18°) is close to the favorable +sc range (30°–90°; Table 1). The ribose is found in both molecules at the C2′-endo conformation.
Table 1.
Torsion angles for 3′,5′-ADP, 2′,5′-ADP, 5′-ADP, U-2′-p, and U-3′-p when bound to RNase A
| 3′,5′-ADP | 2′,5′-ADP | 5′-ADP | U-2′-p | U-3′-p | |||
| RNase A molecule | I | II | I | II | I | I | I |
| Backbone torsion angles | |||||||
| O5′-C5′-C4′-C3′ (γ) | 32 (+sc) | 18 (sp) | 57 (+sc) | 46 (+ac) | −75 (−sc) | 60 (+sc) | 59 (+sc) |
| C5′-C4′-C3′-O3′ (δ) | 138 (+ac) | 138 (+ac) | 149 (+ac) | 134 (+ac) | 142 (+ac) | 83 (+ac) | 141 (+ac) |
| C5′-C4′-C3′-C2′ | −104 | −104 | −92 | −109 | −100 | −157 | −100 |
| C4′-C3′-C2′-O2′ | −152 | −151 | −158 | −152 | −157 | −76 | −151 |
| Glycosyl torsion angle | |||||||
| O4′-C1′-N9′-C4 (χ) | 49 (anti) | −62 (anti) | −83 (anti) | −66 (anti) | 73 (syn) | ||
| O4′-C1′-N1-C2 (χ′) | −137 (anti) | −134 (anti) | |||||
| Pseudorotation angles | |||||||
| C4′-O4′-C1′-C2′ (v0) | −29 | −28 | −15 | −31 | −32 | 6 | −23 |
| O4′-C1′-C2′-C3′ (v1) | 39 | 38 | 34 | 38 | 42 | −28 | 35 |
| C1′-C2′-C3′-C4′ (v2) | −34 | −34 | −39 | −31 | −36 | 38 | −33 |
| C2′-C3′-C4′-O4′ (v3) | 18 | 19 | 31 | 14 | 18 | −36 | 21 |
| C3′-C4′-O4′-C1′ (v4) | 6 | 5 | −10 | 11 | 8 | 19 | 1 |
| Phase | 152 | 154 | 176 | 145 | 150 | 10 | 159 |
| C2′-endo | C2′-endo | C2′-endo | C2′-endo | C2′-endo | C3′-endo | C2′-endo | |
| Phosphate torsion angles | |||||||
| O3A-PA-O5′-C5′ (α) | 59 (+sc) | ||||||
| P2-O5′-C5′-C4′ (β) | −158 (−ap) | −167 (−ap) | 178 (+ap) | 166 (+ap) | 164 (+ap) | ||
| PB-O3A-PA-O5′A (ζpp) | −94 (+ap) | ||||||
| C4′-C3′-O3′-P1 (ɛ) | −111 | −95 | 120 | ||||
| P1-O3′-C3′-C2′ | 135 | 152 | −126 | ||||
| P1-O2′-C2′-C3′ (ɛ′) | −117 (ac) | −97 (ac) | −92 (−ac) | ||||
| P1-O2′-C2′-C1′ | 130 | 150 | 157 | ||||
Definitions of the torsion angles are according to the current IUPAC-IUB nomenclature (IUPAC-IUB and [JCBN] 1983), and the phase angle of the ribose ring is calculated as described previously (Altona and Sundaralingam 1972). The α torsion angle for 3′,5′-ADP and 2′,5′-aDP cannot be defined because the position of atom O3A is ambiguous (the phosphate group is free). For atom definitions see Figure 1 ▶.
The binding mode of 3′,5′-ADP to both molecules of RNase A in the asymmetric unit is almost identical. The inhibitor binds to the P1-B2-P2 region of the catalytic site (Fig. 2A ▶) with the 5′-phosphate group in P1 involved in hydrogen bond interactions with Gln11, His12, Lys41, and Phe120, while the 3′-phosphate group is bound at the P2 subsite forming a hydrogen bond with Lys7 (Fig. 2A ▶). The adenine is bound at the B2 subsite in a manner similar to that observed previously for other adenylic nucleotides bound to RNase A (McPherson et al. 1986; Fontecilla-Camps et al. 1994; Zegers et al. 1994). The adenine ring packs with its five-member ring against the imidazole ring of His119, which adopts conformation A (χ1 = 158°). The N6 group of adenine is in hydrogen-bonding distance from the side chain atoms of residues Gln69 and Asn71 (Table 2, observed only in RNase A molecule I).
Figure 2.
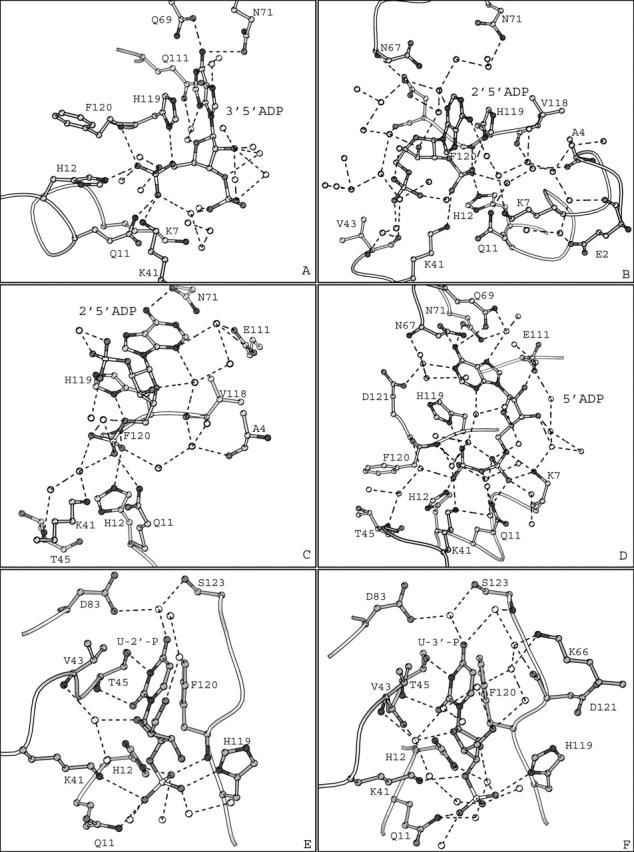
(A, B, D, E, and F) Diagrams of the interactions between RNase A molecule I of the asymmetric unit and 3′,5′-ADP, 2′,5′-ADP, 5′-ADP, 2′ UMP, and 3′ UMP, respectively. (C) Diagram of the interactions between RNase A and 2′,5′-ADP in molecule II of the asymmetric unit. Molecules I and II refer to the two RNase A molecules in the asymmetric unit. Residues are drawn as ball-and-stick models and water molecules as white spheres. Hydrogen bonds are indicated as dashed lines.
Table 2.
Potential hydrogen bonds of 3′,5′-ADP with RNase A (this study) and eDN (Leonidas et al. 2001a)
| Distance (Å) | |||||
| 3′,5′-ADP | RNase A | EDN | RNase A mol. I | RNase A mol. II | EDN |
| O1P | Lys7 Nζ | — | 2.8 | — | |
| O3P | Met0 N | — | — | 2.7 | |
| O4P | His12 Nɛ2 | 2.7 | 2.8 | 2.9 | |
| O4P | Phe120 N | Leu130 N | 3.1 | 3.0 | 3.0 |
| O5P | Gln11 Nɛ2 | 2.9 | 3.2 | 3.0 | |
| O5P | Lys41 Nζ | 3.1 | — | — | |
| O6P | His129 Nδ1 | — | — | 2.3 | |
| N1 | Asn70 Nδ2 | — | — | 2.6 | |
| N6 | Gln69 Oɛ1 | 3.3 | (3.4) | — | |
| N6 | Asn71 Oδ1 | Asn70 Oδ1 | 3.0 | (3.4) | 2.7 |
| O5′ | His119 Nδ1 | 2.7 | — | — | |
| N1 | Water | 3.0 | — | — | |
| N7 | Water | 3.0 | 2.9 | — | |
| N3 | Water | 2.9 | — | — | |
| O2′ | Water | 3.3 | — | — | |
| O2′ | Water | 3.1 | — | — | |
| O2′ | Water | 3.0 | — | — | |
| O2′ | Water | 2.7 | — | — | |
| O2′ | Water | 2.9 | — | — | |
| O3′ | Water | 3.1 | 3.0 | — | |
| O4′ | Water | 2.7 | — | — | |
| O1P | Water | 2.7 | — | 2.7 | |
| O1P | Water | 2.8 | — | — | |
| O2P | Water | 2.7 | 2.8 | — | |
| O2P | Water | 2.8 | — | — | |
| O3P | Water | — | 2.6 | — | |
| O4P | Water | 2.9 | 3.0 | 2.9 | |
| O5P | Water | 2.7 | 2.6 | 2.8 | |
| O6P | Water | 2.4 | 3.2 | — | |
| O6P | Water | 2.8 | — | — | |
Hydrogen bond interactions were calculated with the program HBPLUS (McDonald and Thornton 1994). Atom names are shown in Figure 1 ▶.
There are 10 and 11 water molecules forming hydrogen bonds with 3′,5′-ADP in RNase molecules I and II, respectively. Furthermore, six and four of them mediate polar interactions between the inhibitor and RNase A molecules I and II, of the asymmetric unit, respectively (Fig. 2A ▶). RNase A residues and 3′,5′-ADP atoms are involved in 35 and 48 van der Waals contacts in molecules I and II, respectively.
The binding of 2′,5′-ADP to RNase A
The 2′,5′-ADP molecule is well defined within the electron density map in both RNase A molecules of the crystallographic asymmetric unit (Fig. 1B ▶). The conformation of 2′,5′-ADP is similar in the two RNase A molecules of the asymmetric unit and to that of 3′,5′-ADP with the glycosyl torsion angle χ in the most commonly found anti range and the ribose at the C2′-endo conformation (Table 1).
The binding mode of 2′,5′-ADP in RNase A molecules I and II differs considerably, as different phosphate groups of the inhibitor are bound in subsite P1. In molecule I, it is the 2′-phosphate that binds at P1 (Fig. 2B ▶), while in RNase A molecule II the 5′-phosphate occupies the P1 subsite (Fig. 2C ▶). In RNase A molecule I the side chain of His119 is found in conformation B (χ1 = −55°), while in RNase A molecule II adopts conformation A (χ1 = 157°). The adenine packs with its five-member ring against the imidazole ring of His119 and it is involved in π–π stacking interactions only in molecule II (Fig. 2C ▶).
In molecule I, the adenine ring is in a different orientation but still parallel to the imidazole ring of His119 (Fig. 2B ▶). The 5′ and the 2′-phosphate groups of 2′,5′-ADP in molecules I and II, respectively, point away from the protein, occupying two distinct positions 5 Å apart. These groups are exposed to the solvent and are not involved in any hydrogen bond interactions with RNase A. However, in molecule I, the 5′ phosphate is oriented towards Lys7, the sole component of subsite P2. Although the closest distance of the phosphate oxygens to atom Nζ of Lys7 is ∼7 Å, it is sufficient for long-range coulombic interactions, which have been shown to play an important role in catalysis by RNase A (Fisher et al. 1998a,b).
The binding mode of 2′,5′-ADP in molecule I resembles that of 3′,5′-ADP, and it is similar to that observed previously for adenosine nucleotides bound at B2 in the RNase A complexes with d(pA)4 (McPherson et al. 1986), d(ApTpApApG) (Fontecilla-Camps et al. 1994), and d(CpA) (Zegers et al. 1994). The binding mode of 2′,5′-ADP in molecule II is a novel mode observed for the first time. Upon superposition of the two RNase A molecules, the 5′-phosphate group of 2′,5′-ADP in molecule I passes through the adenine ring of 2′,5′-ADP in molecule II (Fig. 3A ▶). However, apart from the side chain of His119, the rest of the active site residues are structurally conserved between molecules I and II (Fig. 3 ▶). The positions occupied by the 2′ and the 5′-phosphate groups at P1 in the two binding modes are only 0.5 Å apart. In both binding modes the inhibitor is involved in hydrogen bond interactions with His12, His119, and Phe120, although through different atoms (Table 3). In addition, in molecule II, 2′,5′-ADP is involved in hydrogen bonds with Gln11 and Asn71, whereas in molecule I it forms an additional hydrogen bond with Lys41 (Table 3).
Figure 3.
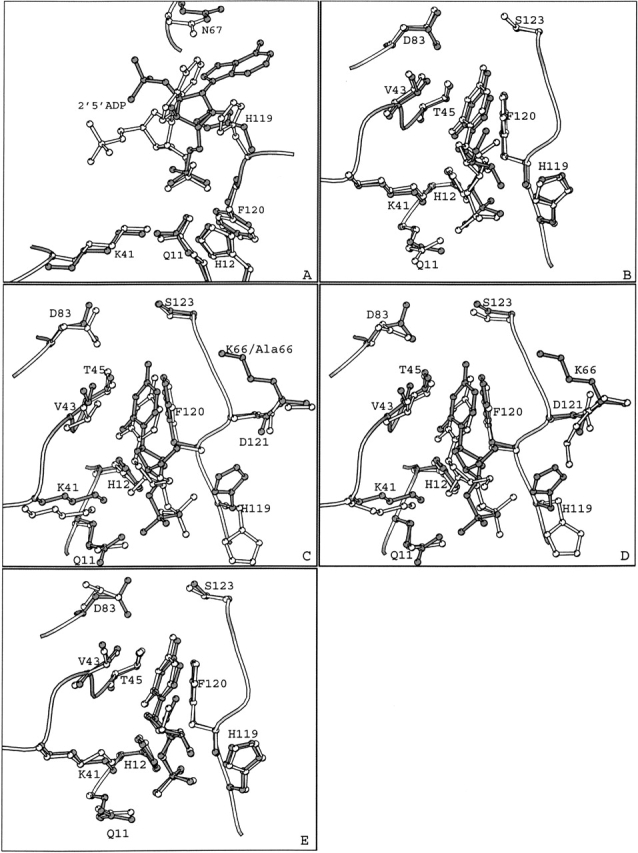
Superimposed structures of (A) the 2′,5′-ADP complex in the two RNase A molecules of the asymmetric unit (B) the RNase A–U-2′-p (gray) and the RNase A–U-3′-p complex structures (white), (C) the RNase A–U-3′-p (gray) and the K7A/R10A/K66A RNase A–U-3′-p (PDB entry 4RSK; white) complexes, (D) the RNase U–2′-p (gray) and the RNase A–2′CMP (PDB entry 1ROB; white), and (E) the RNase A–U-3′-p (gray) and the RNase A–3′CMP (PDB entry 1RPF; white) complexes.
Table 3.
Potential hydrogen bonds of 2′,5′-ADP with RNase A (present study), EDN (Leonidas et al. 2001a) and ECP (Mohan et al. 2002)
| Distance (Å) | |||||||
| 2′,5′-ADP | RNase A | EDN | ECP | RNase A mol. I | RNase A mol. II | EDN | ECP |
| O1P | His119 Nδ1 | His129 Nδ1 | Lys38 Nζ | 2.7 | — | 2.7 | 2.8 |
| O2P | Lys41 Nå | Gln14 Nɛ2 | Gln14 Nɛ2 | 2.9 | — | 3.3 | 3.0 |
| O3P | His12 Nɛ2 | His15 Nɛ2 | His15 Nɛ2 | 2.6 | — | 2.6 | 3.0 |
| O3P | Phe120 N | Leu130 N | 2.9 | — | 2.8 | — | |
| O5P | His12 Nɛ2 | — | 2.9 | — | — | ||
| O5P | Phe120 N | — | 2.8 | — | — | ||
| O6P | Gln11 Nɛ2 | Arg34 Nη2 | — | 2.9 | — | 2.7 | |
| O3′ | Leu130 O | — | — | 2.6 | — | ||
| O5′ | His119 Nδ1 | — | 2.7 | — | — | ||
| N3 | His128 Nɛ2 | — | — | — | 3.3 | ||
| N6 | Asn71 Oδ1 | — | 3.1 | — | — | ||
| N3 | Water | 2.9 | 3.2 | 2.9 | — | ||
| N3 | Water | — | 2.8 | — | — | ||
| N6 | Water | 3.2 | — | — | — | ||
| N7 | Water | — | 3.1 | — | — | ||
| O3′ | Water | 2.7 | — | — | — | ||
| O3′ | Water | 3.2 | — | — | — | ||
| O4′ | Water | — | 2.7 | — | — | ||
| O5′ | Water | — | 2.2 | — | — | ||
| O1P | Water | — | 2.6 | — | — | ||
| O2P | Water | 2.8 | 3.0 | — | 3.1 | ||
| O3P | Water | 2.9 | 2.0 | 2.8 | 2.4 | ||
| O4P | Water | 2.5 | 2.5 | 2.9 | 2.7 | ||
| O4P | Water | — | 2.6 | — | — | ||
| O5P | Water | 2.6 | 2.8 | — | — | ||
| O6P | Water | 2.2 | 2.5 | — | 2.7 | ||
| O6P | Water | — | 3.0 | — | — | ||
Hydrogen bond interactions were calculated with the program HBPLUS (McDonald and Thornton 1994). Atom names are shown in Figure 1 ▶.
Several water molecules mediate hydrogen bond interactions between 2′,5′-ADP atoms and residues Ala4, Gln11, Lys41, Pro42, Val43, Lys66, Asn67, Val118, Ph120, and Asp121 in molecule I (Fig. 2B ▶). In molecule II, Ala4, Phe8, Gln11, His12, Lys41, Lys66, Asn67, Glu111, Gly112, Asn113, Val118, and Asp121 are involved in hydrogen bonds with 2′,5′-ADP atoms, mediated by seven water molecules (Fig. 2C ▶). The inhibitor and the protein participate in 22 van der Waals contacts, in molecule I, through residues His12, Lys41, Asn67, and His119. In molecule II, there are 36 van der Waals contacts between 10 protein residues and 14 inhibitor atoms.
The binding of 5′-ADP to RNase A
The 5′-ADP molecule is bound at the P1-B2 region of the RNase A active site. The electron density is of high quality, and all atoms of 5′-ADP are well defined (full occupancy) in molecule I of the crystallographic asymmetric unit (Fig. 1C ▶). In molecule II, despite the 1.2 Å resolution data, the electron density for the inhibitor is relatively poor (occupancy 0.6). This partial binding, which has also been observed for U-2′-p and U-3′-p, as well as in previous binding studies with RNase A (Leonidas et al. 1997), might be attributed to the lattice contacts which limit access to the active site of molecule II of the asymmetric unit. Therefore, only molecule I will be considered for this complex throughout our discussion.
The conformation of 5′-ADP is similar to that of the adenosine 5′-pyrophosphate derivatives bound to RNase A (Leonidas et al. 1997, 1999; Jardine et al. 2001) and very different from all the other adenosine phosphonucleotides complexed with RNase A. The glycosyl torsion angle χ is at the unusual syn conformation, in contrast to 3′,5′-ADP and 2′,5′-ADP (Table 1; Fig. 1C ▶). The ribose is at the C2′-endo conformation and the rest of the backbone and phosphate torsion angles have values frequently observed in protein bound nucleotides (Table 1). The syn conformation is rare in protein-bound adenosine nucleotides (Moodie and Thornton 1993), and has been previously observed in adenosine 5′-pyrophosphate derivatives bound to RNase A (Leonidas et al. 1997, 1999; Jardine et al. 2001).
The adenine packs with the six-member ring against the imidazole of His119 (conformation A, χ1 = 158°), in contrast to 3′,5′-ADP and 2′,5′-ADP, and the N6 group is involved in hydrogen bond interactions with Asn67 and Asn71 (Table 4; Fig. 2D ▶). 5′-ADP binds at the RNase A active site with the β- rather than the α-phosphate group at P1, and forms hydrogen bonds with Gln11, His12, Lys41, His119, and Phe120 (Fig. 2D ▶). This mode of binding is reminiscent of the binding of the adenosine 5′-pyrophosphate derivatives to RNase A (Russo et al. 2001).
Table 4.
Potential hydrogen bonds of 5′-ADP with RNase A (present study) and EDN (Leonidas et al. 2001a)
| Distance (Å) | ||||
| 5′-ADP | RNase A | EDN | RNase A | EDN |
| O1A | Lys7 Nζ | Lys38 Nζ | 2.9 | 2.9 |
| O1A | Gln11 Nɛ2 | 3.1 | — | |
| O3A | Gln11 Nɛ2 | 3.0 | — | |
| O1B | His12 Nɛ2 | His129 Nδ1 | 2.7 | 2.5 |
| O1B | Phe120 N | 3.0 | — | |
| O2B | Lys41 Nζ | Gln14 Nɛ2 | 2.7 | 2.8 |
| O3B | His119 Nδ1 | His15 Nɛ2 | 2.6 | 2.8 |
| O3B | Leu130 N | — | 2.8 | |
| O2′ | Gln40 O | — | 3.1 | |
| O2′ | Gln40 N | — | 3.1 | |
| O5′ | Lys7 Nζ | 2.9 | — | |
| N6 | Asn67 Oδ1 | 3.3 | — | |
| N6 | Gln69 Oɛ1 | 3.3 | — | |
| N6 | Asn71 Oδ1 | 3.0 | — | |
| N1 | Water | 2.8 | — | |
| N3 | Water | 2.8 | — | |
| N7 | Water | 3.0 | — | |
| O2′ | Water | 2.8 | — | |
| O3′ | Water | 2.8 | — | |
| O3′ | Water | 2.8 | — | |
| O1A | Water | 2.7 | — | |
| O2A | Water | 2.5 | — | |
| O2A | Water | 2.9 | — | |
| O2A | Water | 2.9 | — | |
| O3A | Water | 3.2 | — | |
| O1B | Water | 2.9 | — | |
| O2B | Water | 2.9 | — | |
| O2B | Water | 2.8 | — | |
| O3B | Water | 2.4 | 3.1 | |
| O3B | Water | 3.2 | — | |
Hydrogen bond interactions were calculated with the program HBPLUS (McDonald and Thornton 1994). Atom names are shown in Figure 1 ▶.
There are 12 water molecules hydrogen bonded to 5′-ADP out of which six mediate hydrogen bond interactions to residues Glu2, Ala4, Lys7, Gln11, Arg39, Pro42, Val43, Asn67, Gln69, Glu111, Val118, Phe120, and Asp121. Ten RNase A residues and 17 atoms of 5′-ADP are involved in 38 van der Waals contacts.
The binding of U-2′-p and U-3′-p to RNase A
Both U-2′-p and U-3′-p bind to the P1-B1 region of the RNase A active site adopting similar conformations and in a manner similar to that of uridine vanadate (UVan, a transition state analog) and other uridylyl nucleotides to RNase A (Gilliland 1997; Ladner et al. 1997). The two inhibitors, are well defined within the electron density map only in RNase A molecule I of the asymmetric unit (Fig. 1D,E). The electron density for both inhibitors is poor for molecule II, like in the RNase A–5′-ADP complex; hence, our study has been focused only in the complex of the inhibitors with molecule I.
The glycosyl torsion angle of uridine, in both inhibitors, is in the anti range, as found in most free and protein-bound pyrimidine nucleotides (Moodie and Thornton 1993). Torsion angles γ and δ are in the +sc and +ac range, respectively (Table 1). The ribose rings of U-2′-p and U-3′-p adopt the C3′-endo and the C2′-endo puckering, respectively, the two furanose puckerings most frequently observed in free and bound nucleotides (Moodie and Thornton 1993). The C2′-endo puckering has also been observed in 2′-deoxyuridine in the RNase A–pdUppA-3′-p and dUppA–3′-p complexes (Leonidas et al. 1999; Jardine et al. 2001), in cytidine in the RNase A–d(CpA) complex (Zegers et al. 1994), and in thymidine in the RNase A–d(ApTpApApG) complex (Fontecilla-Camps et al. 1994), while the C3′-endo has also been found in uridine in the RNase A–UVan complex (Wladkowski et al. 1998). This is different from the ribose puckerings of uridine in the d(UpcA) (C3′-exo) complex (Gilliland 1997) and in cytidine in the 2′,5′-CpA (C4′-exo) and 3′,5′-d(CpA) (C1′-exo) complexes (Toiron et al. 1996).
In both U-2′-p and U-3′-p complexes the uridine ring is packed against the phenyl ring of Phe120 and participates in hydrogen bond interactions with Thr45, the primary functional component of this subsite (Fig. 2E,F ▶). The phosphate groups are in the P1 subsite occupying two distinct positions 2.0 Å apart (Fig. 3B ▶). However, in both complexes, the phosphate group forms hydrogen bonds with residues Gln11, Lys41, and His119 (Table 5; Fig. 2E,F ▶). In addition, the phosphate of U-2′-p forms two hydrogen bonds with residues His12 and Phe120 that might explain differences in the potency of the two uridylyl molecules (U-2′-p and U-3′-p are potent inhibitors of RNase A with Ki values of 7.1 and 82 μM, respectively; Anderson et al. 1968). There are 6 and 10 water molecules hydrogen-bonded to U-2′-p and U-3′-p, respectively (Table 5; Fig. 2E,F ▶). Among them there is a water molecule that mediates interactions between atom O4 of uracil of U-2′-p and residues Asp83 and Ser 123. The same interactions are mediated through two water molecules in the U-3′-p complex. These water molecules might correspond to those proposed previously that adjust their donor/acceptor hydrogen bonding roles depending on whether uracil or cytosine is bound at the B1 subsite (Gilliland et al. 1994).
Table 5.
Potential hydrogen bonds of U-3′-p and U-2′-p with the wild type RNase A (present study), and K7A/R10A/K66A RNase A variant (Fisher et al. 1998b) with U-3′-p in the crystal
| Distance (Å) | ||||
| Inhibitor atom | RNase A | U-3′-p | U-3′-p-variant | U-2′-p |
| N3 | Thr45 Oγ1 | 2.8 | 2.8 | 2.9 |
| O2 | Thr45 N | 2.9 | 2.8 | 3.0 |
| O2′ | Lys41 Nζ | — | 2.7 | — |
| O3′ | Lys41 Nζ | 3.2 | — | — |
| O1P | His119 Nδ1 | 2.8 | 2.7 | 2.7 |
| O2P | Lys41 Nζ | — | — | 3.1 |
| O2P | Gln11 Nɛ2 | 3.2 | — | 2.9 |
| O3p | His12 Nɛ2 | — | 3.0 | 2.6 |
| O3P | Phe120 N | — | 2.8 | 2.8 |
| O4 | Water | 3.1 | 2.9 | 2.8 |
| O4 | Water | 2.8 | — | — |
| O2′ | Water | 3.0 | — | — |
| O4′ | Water | 3.1 | — | 3.0 |
| O5′ | Water | 3.1 | 2.7 | 2.8 |
| O5′ | Water | 3.1 | — | — |
| O1P | Water | 2.6 | — | 2.7 |
| O2P | Water | 2.6 | 2.9 | 2.7 |
| O3P | Water | 2.6 | — | 3.3 |
| O3P | Water | 2.5 | — | — |
Hydrogen bond interactions were calculated with the program HBPLUS (McDonald and Thornton 1994). Atom names are shown in Figure 1D and E ▶.
Comparison of the two uridylyl complexes with the RNase A–UVan complex (Ladner et al. 1997) reveals that the uridine moieties bind almost identically in all three complexes. Furthermore, the position of the phosphate group of U-2′-p is similar to that of the vanadate ion, while in the case of U-3′-p it is in close distance (2.0 Å).
The binding of U-3′-p in the crystal of an RNase A variant, where Lys7, Arg10, and Lys66 were replaced by alanines, was studied previously (Fisher et al. 1998b). Superposition of the wild-type RNase A–U-3′-p complex structure of this study onto the K7A/R10A/K66A RNase A variant–U-3′-p complex structure reveals that U-3′-p binds in a similar mode to both enzymes (Fig. 4B ▶). However, the location of U-3′-p in the active site, in the two complexes, differs by approximately 1.0 Å (Fig. 3C ▶). In the variant complex structure His119 is in the B conformation (χ1 = −64°), in contrast to the native complex structure where it is found in conformation A. The rest of the active site residues in the two complexes superimpose well. The two uracil rings bind similarly in the B1 subsite, they are involved in stacking interactions with the phenyl ring of Phe120, and they are hydrogen-bonded to Thr45. In contrast, the position of the 3′-phosphate group is different in the two complexes (Fig. 3C ▶). The ribose of U-3′-p in the variant complex structure adopts the C3′-endo puckering (C2′-endo in the native complex structure) orienting the 2′-hydroxyl and the 3′-phosphate groups in positions 2.5 Å and 2.2 Å away from their respective positions in the wild type complex structure (Fig. 3C ▶). This difference in the ribose puckering could be attributed to steric hindrances imposed by the alternative conformations of His119. Thus, the 3′-phosphate group in the variant complex structure is placed between the side chains of His12 and His119, forming hydrogen bonds with their imidazoles and with the main chain nitrogen of Phe120. In the wild-type complex, the 3′ phosphate forms a hydrogen bond with the side chain of His119 but no hydrogen bond interactions exist with His12 and Phe120 (Table 5). Instead, there is only a water-mediated interaction of the 3′-phosphate group to the side chain of His12. In addition, the 2′-hydroxyl group in the variant complex structure is hydrogen bonded to His12 Nɛ2 and Lys41 Nζ. In the wild-type complex structure, Lys41 Nζ forms a hydrogen bond with O3′ while O2′ does not participate in any hydrogen bond interactions with the protein (Table 5). Overall, upon binding, U-3′-p forms five and six direct hydrogen bonds with the wild and variant RNase A, respectively (Table 5).
Figure 4.
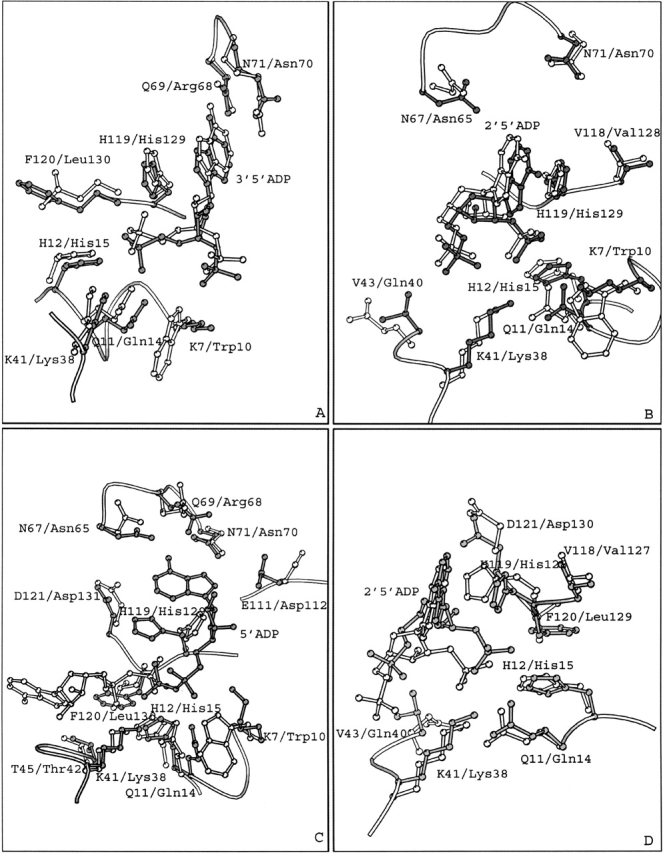
Superimposed structures of (A) the RNase A–3′,5′-ADP and EDN–3′,5′-ADP (PDB entry 1HI4) complexes; (B) the RNase A–2′,5′-ADP (molecule I) and EDN–2′,5′-ADP (PDB entry 1HI3) complexes; (C) the RNase A–5′-ADP and EDN–5′-ADP (PDB entry 1HI5) complexes, (D) the RNase A–2′,5′-ADP (molecule I) and ECP–2′,5′-ADP (PDB entry 1H1H) complexes. Residues and inhibitor molecules from the RNase A complexes are shown in gray and labeled using the one-letter code, while residues from EDN or ECP are shown in white and are labeled using the three-letter code.
Structural comparison of the RNase A K7A/R10A/K66A variant–U-3′-p complex with the wild-type RNase A–3′-CMP complex (Zegers et al. 1994) showed that the two structures are very similar, and that replacing Lys7, Arg10, and Lys66 by alanines did not affect the binding of U-3′-p (Fisher et al. 1998b). However, U-3′-p binds to the variant protein with three- to fivefold weaker affinity than the one exhibited on binding to wild-type RNase A, and this has been attributed to the long-range coulombic interactions of the mutated residues with the ligand (Fisher et al. 1998b). Our results from the wild-type complex structure also support this hypothesis, and emphasize even further the importance of the long-range coulombic interactions of residues Lys7, Arg10, and Lys66 with the ligand, because although U-3′-p binds with a weaker affinity to the variant protein, there are less hydrogen bond interactions in the wild-type complex structure than in the variant complex structure.
The different conformations adopted by the side chain of His119 in the two complex structures might also contribute to the difference in the affinity. Conformation A, found in the wild-type complex structure and also in the RNase A–UVan complex structure (Ladner et al. 1997), triggers binding of the phosphate group 2.2 Å away from the position it occupies in the variant–U-3′-p complex structure, towards the B1 subsite. It also promotes a C2′-endo puckering of the ribose, in contrast to a C3′-endo, in the variant. These conformations drive the uridine deeper, by approximately 1.0 Å, in the B1 pocket, in the native structure than in the variant structure, and allow it to form closer van der Waals contacts with the side chain of Phe120.
Superposition of the structures of U-2′-p and 2′-CMP RNase A (Lisgarten et al. 1993) complexes reveals that the nucleotide and phosphate moieties bind in similar locations to RNase A (Fig. 3D ▶). Their riboses share the same C3′-endo conformation, and both inhibitors participate in a similar hydrogen-bonding network with RNase A. Structural comparison of the structures of U-3′-p and 3′-CMP (Zegers et al. 1994) complexes shows that their binding mode to RNase A is different (Fig. 3E ▶). Their differences are similar to those observed when comparing the structures of the present RNase A–U-3′-p complex to the K7A/R10A/K66A RNase A–U-3′-p complex (Fig. 3C ▶; Fisher et al. 1998b). Thus, in contrast to the U-3′-p complex where His119 is found in conformation A, in the 3′-CMP complex His119 adopts conformation B (Fig. 3E ▶). The puckering of the riboses is also different: C2′-exo in 3′-CMP and C2′-endo in U-3′-p. The pyrimidine moieties of U-3′-p and 3′-CMP (both in anti conformation) bind in close positions (N4–O4 distance 1.0 Å) but their phosphate groups in the P1 subsite are quite apart (phosphorous-phosphorous distance: 2.4 Å). Thus, major differences are observed again in the interactions of the phosphate group in the active site, and as in the 3′-CMP complex structure, there are two additional hydrogen bonds to His12, and Phe120 and no hydrogen bonds with Gln11 when compared to the U-3′-p complex. The conformation of the side chain of Asp83 in the uridine complexes is different from its conformation in cytidine complexes (Lisgarten et al. 1993; Zegers et al. 1994). In the present uridine complexes, atom Oγ of Thr45 is hydrogen bonded to atom Oδ1 of Aps83, in agreement with previous observations with uridine vanadate (Ladner et al. 1997) and mutation results (delCardayre and Raines 1994, 1995), which indicated that this hydrogen bond strengthens preferentially the binding of uracil.
Comparison to eosinophil ribonucleases
The three central subsites, P1, B1, and B2 of the active site of RNase A, are conserved in EDN (Leonidas et al. 2001a) and ECP (Boix et al. 1999). Structural comparisons have shown that RNase A residues Gln11, His12, Lys41, and His119 of P1 are conserved as Gln14, His15, Lys38, and His129 in EDN and as Gln14, His15, Lys38, and His128 in ECP. Residues of the pyrimidine specific subsite B1 Thr45 and Phe120 in RNase A are conserved as Thr42 and Leu130 in EDN and as Thr42 and Leu129 in ECP. Furthermore, the purine binding subsite B2, constituted by residues Asn71, Glu111, and His119 in RNase A, is formed by the homologous residues Asn70, Asp112, and His128 (His129) in ECP and EDN, respectively (Boix et al. 1999; Leonidas et al. 2001a). The conformation of all the active site residues is conserved between RNase A, EDN, and ECP.
3′,5′-ADP is a potent ribonucleolytic inhibitor with a Ki value of 5 μM for RNase A (Russo et al. 1997), 32 μM for EDN (Leonidas et al. 2001a), and 6–7 μM for ECP (Mohan et al. 2002). Superimposition of the crystal structure of the RNase–3′,5′-ADP complex onto the structure of the EDN–3′,5′-ADP complex (Leonidas et al. 2001a) reveals that the inhibitor binds similarly to the active site of these two RNases, although it occupies two distinct locations approximately 1.5 Å apart (Fig. 4A ▶). The inhibitor adopts similar conformations when bound to either RNase A or EDN, adopting the energetically favored C2′-endo anti conformation (Moodie and Thornton 1993). The 5′ phosphates bind at the P1 subsite but they occupy different locations with the phosphorous atoms located 1.0 Å apart, upon superposition of the two complex structures (Fig. 4A ▶). Despite their different locations, the 5′ phosphate groups are involved in analogous interactions with residues of the P1 subsite in the two enzymes. The only difference is the atom of the inhibitor that is hydrogen-bonded to His119 (EDN: His129). In the RNase A complex it is the 5′ oxygen, while in the EDN complex it is one of the phosphate oxygens (Table 2). The adenine rings pack against the side chains of His119 or His129 with both histidines adopting conformation A (Fig. 4A ▶). Differences in the hydrogen bond interactions of two adenines include a hydrogen bond of the N1 group with Asn70 in the EDN complex, not found in the RNase A complex, and a hydrogen bond of the N6 group with Gln69 of RNase A not observed in the EDN complex (Table 2). The 3′ phosphates are located towards the N-terminal ends of RNase A and EDN, respectively (Fig. 5 ▶). Overall, the inhibitor is involved in seven hydrogen bond interactions with RNase A and EDN residues in the corresponding complexes (Table 2).
Figure 5.
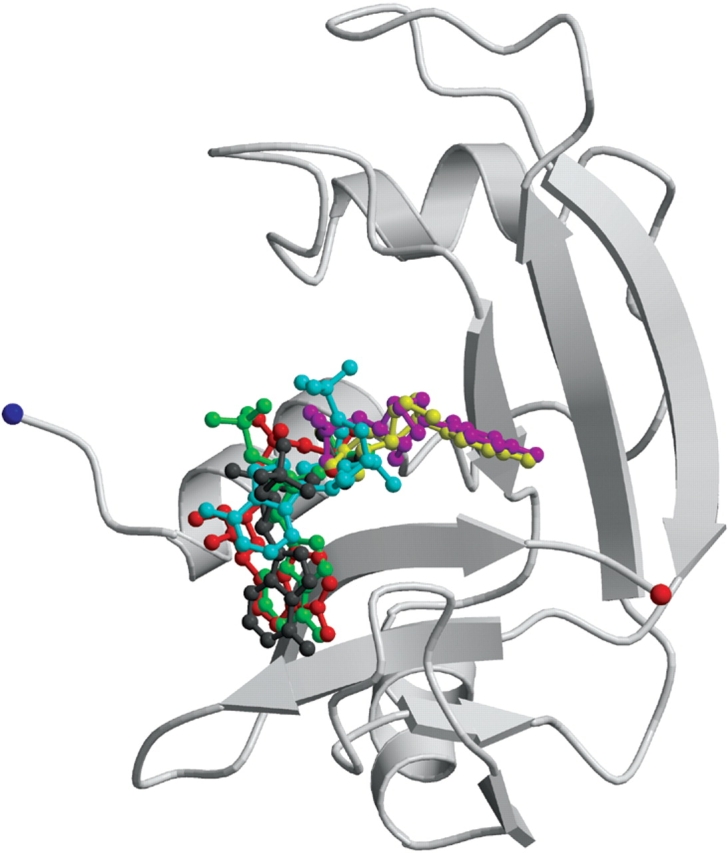
A schematic representation of RNase A in complex with 3′,5′-ADP (in green). The inhibitors 2′,5′-ADP (in gray and cyan from molecules I and II of the asymmetric unit, respectively), 5′-ADP (in red), U-3′-p (in magenta) and U-2′-p (in yellow) are also shown.
2′,5′-ADP inhibits the enzymic activities of RNase A, EDN, and ECP with Ki values of 8 μM, 64 μM (Leonidas et al. 2001a), and 6–7 μM (Mohan et al. 2002), respectively at pH 5.5–6.5. The conformation of the inhibitor when bound to RNase A, EDN, and ECP is similar (Fig. 4B,D). The backbone torsion angles γ and δ have similar values, the glycosyl torsion angles χ have values between −66° to −112°, and the nucleotide in all three complexes adopts the anti conformation. The pseudorotation angles of the ribose in 2′,5′-ADP also have similar values in all three complexes, and the ribose is found at the C2′-endo conformation.
The mode of binding of 2′,5′-ADP to RNase A is different in the two molecules of the crystallographic asymmetric unit, as mentioned above. 2′,5′-ADP binds to RNase A molecule I, as it binds to EDN and ECP (Fig. 4B,D). The 2′-phosphate occupies the P1 subsite and forms similar hydrogen bonds with protein residues in all three complexes (RNase A molecule I, EDN, and ECP; Table 3). The side chains of His119 in RNase A molecules I and II, (ECP: His128, EDN: His129) adopt different conformations. In the RNase A molecule I and in EDN, His119 adopts conformation B (Fig. 4B,D). In these complexes the adenine moiety of 2′,5′-ADP forms stacking interactions with the imidazole ring of His119 (EDN: His129; Fig. 4B ▶) and places its 2′-phosphate group in P1. In the ECP and RNase A molecule II 2′,5′-ADP complexes, His128 (RNase A: His119) is in conformation A. In the ECP–2′,5′-ADP complex the adenine moiety does not pack against the side chain of His128, but the 2′-phosphate still binds at P1 (Fig. 4D ▶; Mohan et al. 2002). It is not clear why the adenine of 2′,5′-ADP does not pack against the imidazole ring of His128 in ECP. Interestingly, the adenine ring of 2′,5′-ADP packs against the imidazole ring of His119 in the RNase A molecule II–2′,5′-ADP complex, but then it is the 5′-phosphate and not the 2′-phosphate that binds to subsite P1.
The 5′-phosphate groups of 2′,5′-ADP in the RNase A molecule I and in the EDN complexes are oriented similarly pointing to the region of subsite P2 (Lys7 in RNase A and Trp10 in EDN). However, they are not involved in any hydrogen bonding interactions with the two proteins. In the ECP–2′,5′-ADP complex (Mohan et al. 2002) the 5′-phosphate group makes a hydrogen bond interaction with Arg34, which is part of a novel P−1 subsite identified recently in EDN(Leonidas et al. 2001a) and ECP (Boix et al. 1999), and is located upstream of the cleavable phosphodiester bond (Table 3). There are also some other differences in the binding of 2′,5′-ADP to RNase A, EDN, and ECP. In the complex with RNase A molecule II, atom N6 of the adenine forms a hydrogen bond with the side chain of Asn71 and in the EDN - 2′,5′-ADP complex atom O3′ of the ribose, forms a hydrogen bond with the main chain oxygen of Leu130 (Table 3).
The RNase A inhibitor, 5′-ADP, is more potent than 3′,5′-ADP and 2′,5′-ADP, with a Ki value of 1.2 μM at pH 5.9 (Russo et al. 1997). However, in EDN, it is less potent than 3′,5′-ADP and 2′,5′-ADP (Ki = 92 μM at pH 6.5; Leonidas et al. 2001a), while in ECP, it is almost as potent as 3′,5′-ADP and 2′,5′-ADP (Ki ∼ 2–3 μM at pH 6.0; Mohan et al. 2002). The inhibitor adopts two different conformations when bound to RNase A and EDN. In RNase A the adenosine is at the syn conformation (χ = 73°), whereas in the EDN it is found at the anti conformation (χ = −100°; Leonidas et al. 2001a). In both complexes the ribose adopts the C2′-endo conformation and the pseudorotation angles v0–v5 are broadly conserved. The backbone torsion angles α, β, and δ have also similar values, whereas the γ and ζpp differ, with those in the RNase A complex in the −sc and +ap range and those in the EDN complex in the +sc and +ap range, respectively. The mode of binding of 5′-ADP to RNase A and EDN is also different (Fig. 4C ▶). In EDN, 5′-ADP is bound at the active site with the β-phosphate at subsite P1, like in RNase A, but the adenosine is located in a region away from the B2 subsite close to, but not at B1 (in RNase A the adenosine binds at B2). Thus, the inhibitor is found in two opposite directions within the active site of the two enzymes.
The active-site residue His129 in EDN is found in the B conformation, while His119 in RNase A adopts conformation A (Fig. 4C ▶). The β-phosphate groups are engaged in similar hydrogen bonding networks with RNase A and EDN, involving residues His12, His119, and Phe120 (EDN: His15, His129, and Leu130; Table 4). There are also differences in the binding of the β-phosphate group. In the EDN complex the β-phosphate makes a hydrogen bond to Gln14 (RNase A: Gln11), whereas in the RNase A complex it is hydrogen-bonded to Lys41 (EDN: Lys38). The α-phosphate group makes a single hydrogen bond interaction with Lys38 (RNase A: Lys41) in the EDN complex, while in the RNase A complex forms four hydrogen bonds.
In the EDN complex the adenine does not form any direct hydrogen bonding interaction with the protein, in contrast to the RNase A complex (Table 4). The binding of the adenosine in EDN is stabilized through two hydrogen bonds of the ribose with Gln40 and through van der Waals interactions of the adenine ring with Lys38, Gln40, and His82 (Leonidas et al. 2001a). The difference in the potency of 5′-ADP against RNase A and EDN (5′-ADP exhibits a 75 times lower Ki in RNase A than in EDN) might be attributed to differences in the polar interactions made by 5′-ADP in the two complexes.
Conclusions
The binding of five different phosphonucleotide inhibitors to RNase A was studied. Each inhibitor binds at the catalytic site by anchoring a phosphate group at the P1 subsite and adopts a completely different conformation (Fig. 5 ▶). The order of potency against RNase A for the five inhibitors studied is 5′-ADP > 3′,5′-ADP > U-2′-p ≥ 2′,5′-ADP > U-3′-p. The occluded surface area of RNase A, calculated with the program GRASP (Nicholls and Honig 1991), by 5′-ADP, 3′,5′-ADP, U-2′-p, 2′,5′-ADP, and U-3′-p, is 638 Å2, 576 Å2, 500 Å2, 455 Å2 (molec I), 577 Å2 (molec II), and 497 Å2, respectively. Its size follows the potency of the inhibitors against the activity of RNase A. The extensive number of interactions of 5′-ADP with RNase A can explain its higher potency. Although 3′,5′-ADP is a fragment of the physiological substrate of RNase A (RNA) and its binding mode should represent the binding of RNA in the active site, it is involved in fewer interactions with RNase A than 5′-ADP because only one of its phosphate groups is involved in hydrogen bond interactions with the enzyme. The diversity in the binding mode of 2′,5′-ADP in RNase A (it is found in two different binding modes) may explain its high affinity for RNase A despite the smaller number of direct hydrogen bonds with RNase A in comparison to those of 3′,5′-ADP. Significant differences on the phosphate binding of U-2′-p and U-3′-p in subsite P1 (two distinct locations), may contribute to the difference in the potency between the two inhibitors (U-2′-p has a 10 fold lower Ki for RNase A than U-3′-p; Anderson et al. 1968). The position in the U-2′-P complex allows the phosphate group to form more interactions with P1 residues than in the U-3′-p complex. Interestingly, similar difference in the locations of the phosphate exists also between the RNase A–2′CMP and the RNase A–3′CMP complexes (Howlin et al. 1987; Zegers et al. 1994) where 2′CMP has a 10-fold lower Ki for RNase than 3′CMP (Anderson et al. 1968).
The RNase A complex structures, described in this work, suggest how further development of tight binding inhibitors of RNases could be achieved through structure driven design. The high resolution of the crystal structures in the present study, allowed the identification of a large number of water molecules in the active site with bound inhibitors. Several of these water molecules mediate interactions between the inhibitors, and the protein and their positions could be the starting point for the design of more potent inhibitors. The adenine of 5′-ADP is involved in water-mediated interactions through two water molecules with the main chain of Asn67 and the side chain of Gln69. The 3′ hydroxyl group of 2′,5′-ADP in its complex with RNase A molecule II, is involved in water-mediated interactions with Glu111 and Val118. Two water molecules link the 5′-phosphate group in the same complex with the main-chain nitrogen of Ala4. The uracyl of U-2′-p is involved in water-mediated interactions with Ser123 and the side chain of Asp83. These water-mediated interactions are also conserved in the U-3′-p complex. All these water molecules indicate positions with high potential to form hydrogen bonds with the protein. Introduction of various substituents to the respective lead molecules could be used to decorate these ligands with groups that will allow them to reach those water positions and exploit their interactions with the protein, thus increasing their potency. The water network of the RNase A active site is partially conserved in the EDN active site as revealed by comparison of the atomic resolution structure of EDN (Swaminathan et al. 2002) with the structures reported here, further emphasizing its importance in the structure-based design of EDN inhibitors.
The structural comparison of the binding of analogous inhibitors in RNase A, EDN, and ECP has also implications for further design efforts of potent specific ribonucleolytic inhibitors of EDN and ECP. Interactions at P1 are broadly conserved for the three adenylic and the two uridylyl complexes with RNase A, for the three adenylic with EDN (Leonidas et al. 2001a), and for 2′,5′-ADP with ECP (Mohan et al. 2002). Subsite P1 shows a clear preference for 5′ phosphate groups over 3′ because the binding of 3′,5′-ADP in RNase A (both molecules) and EDN is very similar with the 5′-phosphate group always bound at P1. This preference is also supported by the binding mode observed by different 3′,5′-ADP derivatives to RNase A, which is very similar to that for 3′,5′-ADP. However, it is not clear whether a 5′- over a 2′-phosphate group is preferable, as the enzyme binds both phosphate groups equally well in different complexes. For example, in the case of 2′,5′-ADP in RNase A molecule II, the 5′- is preferred while in RNase molecule I the 2′-phosphate group binds at P1.
The adenine base occupies a different location in each adenylic complex with RNase A, EDN, and ECP. The position observed in the 3′,5′-ADP complex and in the 2′,5′-ADP complex (RNase A molecule II) seems to be a substrate analogous position because it has also been observed in previous RNase A and EDN nucleotide complexes (Leonidas et al. 2001a). The additional positions observed in the RNase A–5′-ADP, the RNase A molecule I–2′,5′-ADP, and the ECP–2′,5′-ADP complexes seem to represent equivalent locations within the B2 subsite that can accommodate an adenine base, and in each case there are sufficient protein interactions to support the binding. Moreover, it seems that there is a correlation between the binding of a specific phosphate group (5′-or 2′-) and the position occupied by the adenine. Thus, when the 5′-phosphate group binds to P1, the nucleotide adopts a conformation that allows packing of the five-member ring of adenine against the imidazole of His119 (3′,5′-ADP in RNase A, and in EDN; 2′,5′-ADP in RNase A molecule II). In contrast, when the 2′-phosphate is bound to subsite P1 the nucleotide adopts various conformations placing the adenine in different locations in RNase A, EDN, and ECP.
The binding mode of 5′-ADP to RNase A confirms the preference of subsite P1 for the β- rather than the α-phosphate group when a pyrophosphate group is present in the inhibitor. This preference has been previously shown in complexes of RNase A with ppA-2′-p, ppA-3′-p, dUppA, and pdUppA-3′-p (Leonidas et al. 1997, 1999; Jardine et al. 2001). The binding of the β-phosphate seems to force the nucleotide to adopt the unusual syn conformation and to place the six-member ring against the imidazole of His119. The only case where it adopts the most preferred conformation anti (Moodie and Thornton 1993) is in the EDN–5′-ADP complex (Leonidas et al. 2001a), and the adenosine is placed outside subsite B2 to an opposite direction, towards subsite B1. All these observations reveal a high flexibility of subsite B2, which can accommodate an adenine base in a variety of locations.
In contrast, subsite B1 seems to be more confined, and bound uridines are always placed in almost identical positions with the uridine ring being packed against the phenyl ring of Phe120 and the 2′-hydroxyl group in hydrogen bonding distance from the side chain atoms of Thr45. This has been observed in the current complexes of RNase A with U-2′-p and U-3′-p as well as in RNase A in complex with other uridylyl nucleotides (Leonidas et al. 1999; Jardine et al. 2001). However, the phosphate group binds to P1 despite its position in the ligand (either 2′- or 3′-). The binding of either the 2′- or the 3′-phosphate group to P1 does not seem to affect the binding mode of uridine to subsite B1, as shown in the RNase A complexes with U-2′-p and U-3′-p in contrast to adenylic inhibitors.
Comparative structural analysis of different complexes of nucleotide inhibitors to RNase A, EDN and ECP shows that the interactions at the P1 subsite are the main driving force for the binding of all the phosphonucleotides to RNases. Our results suggest that the specificity of P1 surpasses the specificity of the other subsites, and therefore, subsite P1 should be the target for the design of new RNase inhibitors. We also showed that each inhibitor does not necessarily bind to RNase A, EDN, and ECP in the same mode as in the case of 2′,5′-ADP and 5′-ADP, and that subtle differences in the enzymic active sites can lead to profound alterations in the binding modes of an inhibitor to these RNases. These findings emphasize further the importance of obtaining direct structural information on the binding of each new inhibitor with RNase A and each RNase homolog in the process of structure-based inhibitor design.
Materials and methods
Bovine pancreatic RNase A (type XII-A) and phosphonucleotides were obtained from Sigma. Crystals of RNase A were grown at 16°C using the hanging drop vapor diffusion technique as described previously (Leonidas et al. 1997) ; they belong to space group C2, with two molecules per asymmetric unit and cell dimensions a = 99.7 Å, b =32.5 Å, c = 72.2 Å, and β = 90.03°. Crystals of the inhibitor complexes were obtained by soaking the RNase A crystals in 20 mM sodium citrate, pH 5.5, 25% PEG 4000, and 50 mM, 2′,5′-ADP, 3′,5′-ADP, U-2′-p, U-3′-p, or 5 mM 5′-ADP for at least 4 h prior to data collection. Diffraction data for the 3′,5′-ADP, 2′ UMP, and 3′ UMP complexes, were collected to 1.5 Å resolution at 100 K using an R-AXIS IV image plate mounted on a RIGAKU RU-H3RHB rotating anode X-ray source with CuKα radiation. Data for the 5′-ADP and 2′,5′-ADP complexes were collected to 1.2 Å resolution at 100 K at The European Synchrotron Radiation Facility (ESRF) at Grenoble (France) on station ID14-2 (λ = 0.933 Å) using an ADSC Q4 CCD detector, and at the Protein Diffraction Beamline (λ = 1.0006 Å) at Elettra-Trieste (Italy) using a MAR345 image plate, respectively. Three different data sets (589 frames in total) were collected for the 2′,5′-ADP complex, to 1.2 Å, 1.48 Å, and to 2.3 Å resolution, to ensure completeness at the low resolution shell (92% for the 30.0–3.26 Å resolution shell). Similarly for the 5′-ADP complex, two different data sets (to 1.2 Å and 2.6 Å, 376 frames in total) were collected to ensure completeness at the low resolution shell (30.0–3.26 Å: 87.8%). Raw data were processed using the HKL program suite (Otwinowski and Minor 1997), and intensities were truncated to amplitudes by the TRUNCATE (French and Wilson 1978) program. Phases were obtained using the structure of free RNase A from monoclinic crystals (Leonidas et al. 1997) as a starting model. Alternate cycles of manual building with the program O (Jones et al. 1991), and refinement by torsion angle dynamics, conjugate gradient minimization, restrained individual B-factor refinement, and bulk solvent correction using the maximum likelihood target function as implemented in the program CNS v1.1 (Brünger et al. 1998) improved the model. Ligand molecules were included during the final stages of the refinement by use of their respective coordinates from the Protein Data Bank (ID codes: 2′,5′-ADP, 1HI3; 3′,5′-ADP, 1HI4; 5′-ADP, 1HI5 [Leonidas et al. 2001a]; U-3′-p, 4RSK [Fisher et al. 1998b]). A model of U-2′-p, was generated using the program SYBYL (Tripos Associates Inc.). Furthermore, the 1.2 Å resolution data from the 2′,5′-ADP and 5′-ADP complexes enabled the anisotropic refinement of temperature factors with the program REFMAC (Murshudov et al. 1997). Details of data processing and refinement statistics are provided in Table 6.
Table 6.
Crystallographic statistics
| RNase A complex | 2′,5′-ADP | 3′,5′-ADP | 5′-ADP | U-2′-p | U-3′-p |
| Resolution (Å) | 30–1.2 | 30–1.5 | 30–1.2 | 30–1.5 | 30–1.5 |
| (Outermost shell) (Å) | 1.22–1.20 | 1.55–1.50 | 1.22–1.20 | 1.55–1.50 | 1.55–1.50 |
| Reflections measured | 949,552 | 423,949 | 623,242 | 278,544 | 239,794 |
| Unique reflections | 67,242 | 36,948 | 70,118 | 37,206 | 37,930 |
| Rsymma | 0.038 | 0.062 | 0.053 | 0.052 | 0.061 |
| (Outermost shell) | 0.226 | 0.369 | 0.130 | 0.390 | 0.294 |
| Completeness (outermost shell) (%) | 92.9 | 97.9 | 96.5 | 98.3 | 99.4 |
| 71.2 | 95.5 | 96.9 | 95.6 | 99.6 | |
| <I/σI> (outermost shell) | 30.9 (10.8) | 15.1 (3.6) | 20.9 (5.8) | 14.1 (2.5) | 15.4 (2.6) |
| Rcrystb | 0.194 | 0.229 | 0.190 | 0.211 | 0.220 |
| (Outermost shell) | 0.261 | 0.446 | 0.195 | 0.442 | 0.412 |
| Rfreec | 0.228 | 0.271 | 0.219 | 0.24 | 0.240 |
| (Outermost shell) | 0.277 | 0.452 | 0.202 | 0.446 | 0.436 |
| Number of solvent molecules | 558 | 389 | 540 | 399 | 415 |
| R.m.s. deviation from ideality | |||||
| in bond lengths (Å) | 0.011 | 0.004 | 0.011 | 0.004 | 0.004 |
| in angles (°) | 1.7 | 1.2 | 1.8 | 1.3 | 1.3 |
| Average B factor | |||||
| Protein atoms (Å2) (molecule I/molecule II) | 18.6/16.7 | 18.8/23.6 | 18.2/17.7 | 19.0/18.7 | 19.0/20.2 |
| Solvent molecules (Å2) | 30.9 | 33.0 | 33.1 | 30.7 | 30.8 |
| Ligand atoms (Å2) (molecule I/molecule II) | 17.8/31.1 | 22.4/34.4 | 13.6/21.3 | 18.0/30.6 | 19.9/26.8 |
aRsymm = ∑h∑i│I(h) − Ii(h)/∑h∑iIi(h) where Ii(h) and I(h) are the ith and the mean measurements of the intensity of reflection h.
bRcryst = ∑h│Fo − Fc│/∑hFo, where Fo and Fc are the observed and calculated structure factors amplitudes of reflection h, respectively.
cRfree is equal to Rcryst for a randomly selected 5% subset of reflections not used in the refinement (Brünger 1992).
The atomic coordinates and diffraction data for the five RNase A complexes have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University (http://www.rcsb.org; PDB ID Codes are 1O0F, 1O0O, 1O0H, 1O0M, and 1O0N for the 3′,5′-ADP, 2′,5′-ADP, 5′-ADP, U-2′-p, and U-3′-p RNase A complexes, respectively). All figures were prepared with the program Molscript (Kraulis 1991), and rendered with RASTER3D (Merritt and Bacon 1997).
Acknowledgments
We thank Prof. G. Archontis, Department of Physics, University of Cyprus, for useful discussions and critical reading of the manuscript. We also acknowledge the assistance of the staff at Elettra, ESRF, and CDSB during the X-ray data collection. This work was supported in part by the Greek GSRT, through a "Career development program for Greek speaking scientists" (to D.D.L and M.V.), Wellcome Trust (UK) Programme Grant (067288) to K.R.A., and a Joint Research and Technology project between Greece and Cyprus (2000–2002; to N.G.O.). This work was also supported by grants from E.U. through the Access to Research Infrastructure action of the Improving Human Potential program for work at the Elettra Synchrotron Light Source (contract HPRI-CT-1999-00033), ESRF (contract HPRI-CT-1999-00022), and CDSB (contract HPRI-CT-1999-00097) to D.D.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RNase A, bovine pancreatic Ribonuclease A
Ang, angiogenin
EDN, eosinophil derived neurotoxin
ECP, eosinophil cationic protein
3′,5′-ADP, adenosine-3′,5′-diphosphate, 2′,5′-ADP, adenosine-2′,5′-diphosphate, 5′-ADP, adenosine-5′-diphosphate, pdUppA-3′-p, 5′-phospho-2′-deoxyuridine 3-pyrophosphate (P′→5′) adenosine 3′-phosphate
ppA-3′-p, 5′-diphosphoadenosine 3′-phosphate
ppA-2′-p, 5′-diphosphoadenosine 2′-phosphate
dUppA-3′-p
2′-deoxyuridine 3′-pyrophosphate (P′→5′) adenosine
PEG, poly(ethylene glycol)
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03196603.
References
- Altona, C. and Sundaralingam, M. 1972. Conformational analysis of the sugar ring in nucleosides and nucleotides. A new description using the concept of pseudorotation. J. Am. Chem. Soc. 94 8205–8212. [DOI] [PubMed] [Google Scholar]
- Anderson, D.G., Hammes, G.G., and Walz, F.G. 1968. Binding of phosphate ligands to Ribonuclease A. Biochemistry 7 1637–1645. [DOI] [PubMed] [Google Scholar]
- Berisio, R., Lamzin, V.S., Sica, F., Wilson, K.S., Zagari, A., and Mazzarella, L. 1999. Protein titration in the crystal state. J. Mol. Biol. 292 845–854. [DOI] [PubMed] [Google Scholar]
- Berisio, R., Sica, F., Lamzin, V.S., Wilson, K.S., Zagari, A., and Mazzarella, L. 2002. Atomic resolution structures of ribonuclease A at six pH values. Acta Crystallogr. D 58 441–450. [DOI] [PubMed] [Google Scholar]
- Boix, E., Leonidas, D.D., Nikolovski, Z., Nogues, M.V., Cuchillo, C.M., and Acharya, K.R. 1999. Crystal structure of eosinophil cationic protein at 2.4 Å resolution. Biochemistry 38 16794–16801. [DOI] [PubMed] [Google Scholar]
- Borkakoti, N., Moss, D.A., and Palmer, R.A. 1982. Ribonuclease A: Least squares refinement of structure at 1.45 Å resolution. Acta Crystallogr. B 38 2210–2217. [Google Scholar]
- Brünger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- D’Alessio, G. and Riordan, J.F. 1997. Ribonucleases: Structure and functions. Academic Press, New York.
- delCardayre, S.B. and Raines, R.T. 1994. Structural determinants of enzymatic processivity. Biochemistry 33 6031–6037. [DOI] [PubMed] [Google Scholar]
- ———. 1995. A residue to residue hydrogen bond mediates the nucleotide specificity of ribonuclease A. J. Mol. Biol. 252 328–336. [DOI] [PubMed] [Google Scholar]
- deMel, V.S.J., Doscher, M.S., Martin, P.D., and Edwards, B.F.P. 1994. The occupancy of two distinct conformations by active-site histidine-119 in crystals of Ribonuclease is modulated by pH. FEBS Lett. 349 155–160. [DOI] [PubMed] [Google Scholar]
- Domachowske, J.B., Bonville, C.A., Dyer, K.D., and Rosenberg, H.F. 1998a. Evolution of antiviral activity in the ribonuclease A gene superfamily: Evidence for a specific interaction between eosinophil-derived neurotoxin (EDN/RNase 2) and respiratory syncytial virus. Nucleic Acids Res. 26 5327–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domachowske, J.B., Dyer, K.D., Bonville, C.A., and Rosenberg, H.F. 1998b. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J. Infect. Dis. 177 1458–1464. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of Molscript that includes greatly enhanced coloring capabilities. J. Mol. Graphics Modelling 15 132–134. [DOI] [PubMed] [Google Scholar]
- Fedorov, A.A., Joseph-McCarthy, D., Fedorov, E., Sirakova, D., Graf, I., and Almo, S.C. 1996. Ionic interactions in crystalline bovine pancreatic ribonuclease A. Biochemistry 35 15962–15979. [DOI] [PubMed] [Google Scholar]
- Fett, J.W., Strydom, D.J., Lobb, R.R., Alderman, E.M., Bethune, J.L., Riordan, J.F., and Vallee, B.L. 1985. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma-cells. Biochemistry 24 5480–5486. [DOI] [PubMed] [Google Scholar]
- Fisher, B.M., Ha, J.H., and Raines, R.T. 1998a. Coulombic forces in protein–RNA interactions: Binding and cleavage by ribonuclease A and variants at Lys7, Arg10, and Lys66. Biochemistry 37 12121–12132. [DOI] [PubMed] [Google Scholar]
- Fisher, B.M., Schultz, L.W., and Raines, R.T. 1998b. Coulombic effects of remote subsites on the active site of ribonuclease A. Biochemistry 37 17386–17401. [DOI] [PubMed] [Google Scholar]
- Fontecilla-Camps, J.C., de Llorens, R., le Du, M.H., and Cuchillo, C.M. 1994. Crystal structure of ribonuclease A•d(ApTpApApG) complex. J. Biol. Chem. 269 21526–21531. [DOI] [PubMed] [Google Scholar]
- French, S. and Wilson, K.S. 1978. On the treatment of the negative intensity observations. Acta Crystallogr. A 34 517–525. [Google Scholar]
- Gilliland, G. 1997. Crystallographic studies of Ribonuclease complexes. In Ribonucleases: Structures and functions. (eds. G. D’Alessio and J.F. Riordan), pp. 305–341. Academic Press, New York.
- Gilliland, G.L., Dill, J., Pechik, I., Svensson, L.A., and Sjolin, L. 1994. The active site of bovine pancreatic ribonuclease: An example of solvent modulated specificity. Protein Pept. Lett. 1 60–65. [Google Scholar]
- Gleich, G.J. and Adolphson, C.R. 1986. The eosinophil leukocyte: Structure and function. Adv. Immunol. 39 177–253. [DOI] [PubMed] [Google Scholar]
- Howlin, B., Harris, G.W., Moss, D.S., and Palmer, R.A. 1987. X-ray refinement study on the binding of cytidylic acid (2′-CMP) to ribonuclease A. J. Mol. Biol. 196 159–164. [DOI] [PubMed] [Google Scholar]
- Howlin, B., Moss, D.S., and Harris, G.W. 1989. Segmented anisotropic refinement of bovine ribonuclease A by the application of rigid-body tls model. Acta Crystallogr. A 45 851–861. [DOI] [PubMed] [Google Scholar]
- IUPAC-IUB, Joint Commission on Biochemical Nomenclature. 1983. Symbols for specifying the conformation of polysaccharide chains. Recommendations 1981. Eur. J. Biochem. 131 5–7. [DOI] [PubMed] [Google Scholar]
- Jardine, A.M., Leonidas, D.D., Jenkins, J.L., Park, C., Raines, R.T., Acharya, K.R., and Shapiro, R. 2001. Cleavage of 3,′5-pyrophosphate-linked dinucleotides by Ribonuclease A and Angiogenin. Biochemistry 40 10262–10272. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT—A program to produce both detailed & schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Ladner, J.E., Wladkowski, B.D., Svensson, L.A., Sjolin, L., and Gilliland, G.L. 1997. X-ray structure of a ribonuclease A—uridine complex at 1.3 Å resolution. Acta Crystallogr. D 53 290–301. [DOI] [PubMed] [Google Scholar]
- Leonidas, D.D., Shapiro, R., Irons, L.I., Russo, N., and Acharya, K.R. 1997. Crystal structures of ribonuclease A complexes with 5′-diphosphoadenosine 3′-phosphate and 5′-diphosphoadenosine 2′-phosphate at 1.7 Å resolution. Biochemistry 36 5578–5588. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Toward rational design of ribonuclease inhibitors: High-resolution crystal structure of a ribonuclease A complex with a potent 3′,5′-pyrophosphate-linked dinucleotide inhibitor. Biochemistry 38 10287– 10297. [DOI] [PubMed] [Google Scholar]
- Leonidas, D.D., Boix, E., Prill, R., Suzuki, M., Turton, R., Minson, K., Swaminathan, G.J., Youle, R.J., and Acharya, K.R. 2001a. Mapping the ribonucleolytic active site of eosinophil-derived neurotoxin (EDN). High resolution crystal structures of EDN complexes with adenylic nucleotide inhibitors. J. Biol. Chem. 276 15009–15017. [DOI] [PubMed] [Google Scholar]
- Leonidas, D.D., Chavali, G.B., Jardine, A.M., Li, S., Shapiro, R., and Acharya, K.R. 2001b. Binding of phosphate and pyrophosphate ions at the active site of human angiogenin as revealed by X-ray crystallography. Protein Sci. 10 1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisgarten, J.N., Gupta, V., Maes, D., Wyns, L., Zegers, I., Palmer, R.A., Dealwis, C.G., Aguilar, C.F., and Hemmings, A.M. 1993. Structure of the crystalline complex of cytidylic acid (2′-CMP) with ribonuclease at 1.6 Å resolution. Conservation of solvent sites in RNase-A high resolution structures. Acta Crystallogr. D 49 541–547. [DOI] [PubMed] [Google Scholar]
- McDonald, I.K. and Thornton, J.M. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238 777–793. [DOI] [PubMed] [Google Scholar]
- McPherson, A., Brayer, G.D., and Morrison, R.D. 1986. Crystal structure of RNase A complexed with d(pA)4. J. Mol. Biol. 189 305–327. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Macromol. Crystallogr. B277 505–524. [DOI] [PubMed] [Google Scholar]
- Mohan, C.G., Boix, E., Evans, H.R., Nikolovski, Z., Nogues, M.V., Cuchillo, C.M., and Acharya, K.R. 2002. The crystal structure of eosinophil cationic protein in complex with 2′,5′-ADP at 2.0 Å resolution reveals the details of the ribonucleolytic active site. Biochemistry 41 12100–12106. [DOI] [PubMed] [Google Scholar]
- Moodie, S.L. and Thornton, J.M. 1993. A study into the effects of protein binding on nucleotide conformation. Nucleic Acids Res. 21 1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the Maximum-Likelihood Method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Newton, D.L., Nicholls, P., Rybak, S.M., and Youle, R.J. 1994. Expression and characterization of recombinant human eosinophil-derived neurotoxin and eosinophil-derived neurotoxin-anti-transferrin receptor sFv. J. Biol. Chem. 269 26739–26745. [PubMed] [Google Scholar]
- Nicholls, A. and Honig, B. 1991. A rapid finite difference algorithm, utilising successive over relaxation to solve the Poisson-Boltzmann equation. J. Comput. Chem. 12 435–445. [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods in enzymology (eds. C.W.J. Carter and R.M. Sweet), pp. 307–326. Academic Press, New York. [DOI] [PubMed]
- Raines, R.T. 1998. Ribonuclease A. Chem. Rev. 98 1045–1065. [DOI] [PubMed] [Google Scholar]
- Rugeles, M.T., Trubey, C.M., Bedoya, V.I., Pinto, L.A., Oppenheim, J.J., Rybak, S.M., and Shearer, G.M. 2003. Ribonuclease is partly responsible for the HIV-1 inhibitory effect activated by HLA alloantigen recognition. AIDS 17 481–486. [DOI] [PubMed] [Google Scholar]
- Russo, A., Acharya, K.R., and Shapiro, R. 2001. Small molecule inhibitors of RNase A and related enzymes. Methods Enymol. 341 629–648. [DOI] [PubMed] [Google Scholar]
- Russo, N. and Shapiro, R. 1999. Potent inhibition of mammalian ribonucleases by 3′,5′-pyrophosphate-linked nucleotides. J. Biol. Chem. 274 14902–14908. [DOI] [PubMed] [Google Scholar]
- Russo, N., Shapiro, R., and Vallee, B.L. 1997. 5′-diphosphoadenosine 3′-phosphate is a potent inhibitor of bovine pancreatic ribonuclease A. Biochem. Biophys. Res. Commun. 231 671–674. [DOI] [PubMed] [Google Scholar]
- Shapiro, R. 1998. Structural features that determine the enzymatic potency and specificity of human Angiogenin: Thr-80 and residues 58–70 and 116–123. Biochemistry 37 6847–6856. [DOI] [PubMed] [Google Scholar]
- Shapiro, R. and Vallee, B.L. 1989. Site-directed mutagenesis of Histidine-13 and Histidine-114 of human Angiogenin–Alanine derivatives inhibit Angiogenin-induced angiogenesis. Biochemistry 28 7401–7408. [DOI] [PubMed] [Google Scholar]
- Shapiro, R., Fox, E.A., and Riordan, J.F. 1989. Role of lysines in human angiogenin—Chemical modification and site-directed mutagenesis. Biochemistry 28 1726–1732. [DOI] [PubMed] [Google Scholar]
- Sorrentino, S., Glitz, D.G., Hamann, K.J., Loegering, D.A., Checkel, J.A., and Gleich, G.J. 1992. Eosinophil-derived neurotoxin and human liver ribonuclease: Identity of structure and linkage of neurotoxicity to nuclease activity. J. Biol. Chem. 267 14859–14865. [PubMed] [Google Scholar]
- Swaminathan, G.J., Holloway, D.E., Veluraja, K., and Acharya, K.R. 2002. Atomic resolution (0.98 Å) structure of eosinophil-derived neurotoxin. Biochemistry 41 3341–3352. [DOI] [PubMed] [Google Scholar]
- Toiron, C., Gonzalez, C., Bruix, M., and Rico, M. 1996. Three-dimensional structure of the complexes of ribonuclease A with 2′,5′-CpA and 3′,5′-d(CpA) in aqueous solution, as obtained by NMR and restrained molecular dynamics. Protein Sci. 5 1633–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladkowski, B.D., Svensson, L.A., Sjolin, L., Ladner, J.E., and Gilliland, G.L. 1998. Structure (1.3 Å) and charge states of a Ribonuclease A-uridine vanadate complex: Implications for the phosphate ester hydrolysis mechanism. J. Am. Chem. Soc. 120 5488–5498. [Google Scholar]
- Zegers, I., Maes, D., Dao-Thi, M.-H., Poortmans, F., Palmer, R., and Wyns, L. 1994. The structures of RNase A complexed with 3′CMP and d(CpA): Active site conformation and conserved water molecules. Protein Sci. 31 2322–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]