

Abstract
Streptococcus pneumoniae is a major human pathogen that causes high mortality and morbidity and has developed resistance to many antibiotics. We show that the gene product from SP1603, identified from S. pneumoniae TIGR4, is a CMP kinase that is essential for bacterial growth. It represents an attractive drug target for the development of a novel antibiotic to overcome the problems of drug resistance development for this organism. Here we describe the three-dimensional solution structure of the S. pneumoniae CMP kinase as determined by NMR spectroscopy. The structure consists of eight α-helices and two β-sheets that fold into the classical core domain, the substrate-binding domain, and the LID domain. The three domains of the protein pack together to form a central cavity for substrate-binding and enzymatic catalysis. The S. pneumoniae CMP kinase resembles the fold of the Escherichia coli homolog. An insertion of one residue is observed at the β-turn in the substrate-binding domain of the S. pneumoniae CMP kinase when compared with the E. coli homolog. Chemical shift perturbations caused by the binding of CMP, CDP, and ATP revealed that CMP or CDP binds to the junction between the core and substrate-binding domains, whereas ATP binds to the junction between the core and LID domains. From NMR relaxation studies, we determined that the loops in the LID domain are highly mobile. These mobile loops could aid in the closing/opening of the LID domain during enzyme catalysis.
Keywords: Streptococcus pneumoniae CMP kinase, cytidylate kinase, cytidine monophosphate kinase, nucleoside monophosphate kinase, NMR structure, structural genomics
Streptococcus pneumoniae is a major human pathogen that can cause potentially life-threatening community-acquired invasive diseases such as pneumonia, meningitis, and otitis media. S. pneumoniae has developed resistance to the antibiotics that are commonly used for its treatment (Schutze et al. 1994; Baquero 1995). One approach that is being used to address this problem is a full genome search for novel protein targets from this organism.
From a genome-wide survey of the uncharacterized proteins from S. pneumoniae, we have identified several structural genomics targets. One of the targets is a protein corresponding to SP1603 in the recently determined complete genome sequence of a virulent isolate of S. pneumoniae (Tettelin et al. 2001) and to SPR1456 in the genome sequence of an avirulent strain of S. pneumoniae R6 (Hoskins et al. 2001). This protein has about 50% sequence homology with bacterial CMP kinases and thus could potentially be a CMP kinase. CMP kinase catalyzes the phosphoryl transfer from ATP to CMP and dCMP, resulting in the formation of nucleotide diphosphates. Therefore, CMP kinase plays a key role in cellular nucleic acid synthesis.
Several crystal structures of CMP/UMP kinases have been determined including CMP kinase from Escherichia coli (Briozzo et al. 1998; Bertrand et al. 2002), UMP/CMP kinase from Dictyostelium discoideum (Scheffzek et al. 1996; Schlichting and Reinstein 1997), and UMP kinase from Saccharomyces cerevisiae (Müller-Dieckmann and Schulz 1994, 1995). The crystal structures of E. coli CMP kinase resemble those of other NMP kinases. They share the common features of a central five-stranded β-sheet connected by α-helices (Briozzo et al. 1998), a fingerprint sequence of Gly-x-x-Gly-x-Gly-Lys (P-loop), and an anion hole in the central cavity for substrate binding. However, E. coli CMP kinase differs from other NMP kinases by the presence of a long insertion of ∼40 residues in the NMP binding domain. These inserted residues form a three-stranded β-sheet and two α-helices, which undergo conformational rearrangement on CDP binding (Briozzo et al. 1998). There are no crystal structures reported so far for CMP/UMP kinases in complex with ATP.
Here we demonstrate that SP1603 is an essential CMP kinase for bacterial growth in S. pneumoniae. This S. pneumoniae CMP kinase thus represents an attractive drug target for developing novel antibiotics to overcome the problems of drug resistance for this organism. In order to aid in the discovery of S. pneumoniae CMP kinase inhibitors, we determined the three-dimensional structure of this enzyme by heteronuclear multidimensional NMR spectroscopy (Clore and Gronenborn 1998). Using chemical shift perturbation techniques (Zuiderweg 2002) and NMR relaxation methods (Kay 1998), we have also mapped out the substrate-binding sites and determined the backbone mobility.
Results and Discussion
Essentiality and function of the gene encoding SP1603 in S. pneumoniae
Insertion–duplication mutagenesis (IDM) was used to assess the essentiality of the gene encoding SP1603 in S. pneumoniae. As described previously (Yu et al. 2001), IDM takes advantage of the highly efficient natural genetic transformation system in S. pneumoniae. The mutagenic plasmids were prepared by using the E. coli plasmid pEVP3 that contains a chloramphenicol-resistance gene and that does not replicate in S. pneumoniae. The pEVP3 knockout constructs contained a 300-bp internal 5′ portion of the gene encoding SP1603 or, as a control, a 300-bp internal 5′ portion of the nonessential competence gene comD. If a gene is not essential for bacterial growth, the bacterial cell can tolerate insertion of the plasmid into the target gene and chloramphenicol-resistant transformants are observed. If the gene is essential for growth, the disruption of the target gene by the plasmid insertion is not tolerated and chloramphenicol-resistant transformants are not observed. The transformation with the nonessential control comD-pEVP3 knockout construct yielded >6 × 104 chloramphenicol-resistant transformants per microgram of plasmid DNA in a standard 1-mL transformation. Extremely small chloramphenicol-resistant transformants were observed in multiple experiments with the SP1603-pEVP3 knockout construct. The frequency of transformation was also reduced by threefold compared with the nonessential control construct. Thus, inactivation of the gene encoding SP1603 significantly decreased the viability and growth rate of the cells. Therefore, these data indicate that SP1603 gene is essential for the growth of S. pneumoniae.
Protein sequence comparisons reveal that SP1603 shares about 50% sequence homology with bacterial CMP kinases, indicating that this protein could potentially be a CMP kinase. To test if the protein has CMP kinase activity, we prepared a solution containing SP1603 in 50 mM Tris, 50 mM KCl, 5 mM MgCl2 (pH 7.4) and added substrates CMP and ATP to the protein solution. We then assayed this solution by HPLC and found that CMP and ATP are efficiently converted into CDP and ADP, respectively, in the reaction mixture. Thus, these experiments establish unequivocally that SP1603 is a CMP kinase.
Besides the essentiality of the CMP kinase in S. pneumoniae presented here, the CMP/UMP kinases from other microorganisms have also been reported recently to be essential in S. cerevisiae (Liljelund and Lacroute 1986; Liou et al. 2002), Bacillus subtilis (Kobayashi et al. 2003), and Haemophilus influenzae (Akerley et al. 2002). However, the E. coli CMP kinase is not essential for bacterial growth (Bertrand et al. 2002).
Assignments
The 15N/1H HSQC spectrum of the uniformly 15N-labeled S. pneumoniae CMP kinase is shown in Figure 1 ▶. The NMR construct that was used in this study for the S. pneumoniae CMP kinase contains 236 residues including an N-terminal His tag (13 residues) with a molecular weight of 26.3 kD. The 15N/1H cross peaks in the HSQC spectrum are well dispersed for this relatively large protein. The backbone 1HN, 13C, and 15N resonances and side-chain methyl resonances of the protein were nearly completely assigned (99%) from an analysis of several heteronuclear multidimensional NMR spectra using a uniformly 15N-, 13C-, and 2H-labeled protein with selectively protonated methyl groups of Val, Leu, and Ile (δ1) (Yamazaki et al. 1994; Goto et al. 1999; Medek et al. 2000). 1Hα resonances were assigned from a 15N-edited TOCSY spectrum using a uniformly 15N-labeled protein (Clore and Gronenborn 1994) and from HCACO and HACA(CO)NH spectra with a uniformly 15N- and 13C-labeled protein (Grzesiek and Bax 1993; Feng et al. 1996). The other side-chain resonances of the protein were assigned from 3D HN(CA)CB, HN(COCA)CB, C(CO)NH-TOCSY, H(CCO)NH-TOCSY, 13C-edited 3D HCCH-TOCSY and NOESY, and 15N-edited 3D TOCSY and NOESY spectra (Fesik and Zuiderweg 1988; Clore and Gronenborn 1994).
Figure 1.

15N/1H HSQC spectrum of the uniformly 15N-labeled S. pneumoniae CMP kinase recorded at DRX500 NMR spectrometer. The folded cross peaks along the 15N-dimension are indicated with asterisks.
Structure determination
The three-dimensional structure of the S. pneumoniae CMP kinase was determined using a distance geometry/simulated annealing protocol (Nilges et al. 1988) with the CNX program (Accelrys Inc.) from a total of 3426 NMR-derived distance and torsional angle restraints (Table 1). A superposition of the 20 low-energy NMR structures is shown in Figure 2 ▶. The protein structure is well defined by the NMR data with an rmsd about the mean coordinate positions of 1.04 ± 0.21 Å for the backbone atoms and 1.46 ± 0.19 Å for all heavy atoms for the residues 16–234 (Table 1). If these structures are superimposed by using only the residues in the core and substrate-binding domains (Fig. 3 ▶), the rmsd about the mean coordinate positions becomes 0.74 ± 0.15 Å for the backbone atoms and 1.13 ± 0.12 Å for all heavy atoms (Fig. 2 ▶; Table 1). The residues located on the loops between β1 and α1 (residues 23–28) and between α7 and β8 (residues 199–210; Figs. 2, 3 ▶ ▶) are less well defined.
Table 1.
Structural statistics and root-mean-square deviation for 20 structures of S. pneumoniae CMP kinase
| Structural statisticsa | ≤SA> | ≤ >r >r
|
| Rmsd from experimental distance restraints (Å)b | ||
| All (3100) | 0.014 ± 0.001 | 0.014 |
| Intraresidue (906) | 0.016 ± 0.001 | 0.016 |
| Sequential (777) | 0.017 ± 0.001 | 0.017 |
| Medium range (597) | 0.011 ± 0.001 | 0.010 |
| 0 Long range (712) | 0.010 ± 0.002 | 0.010 |
| Hydrogen bond (108) | 0.012 ± 0.001 | 0.011 |
| Rmsd from experimental torsional angle restraints (deg)c | ||
| φ and ξ angles (326) | 0.24 ± 0.02 | 0.25 |
| CNX potential energies (kcal mole−1) | ||
| Etot | 144 ± 5 | 140 |
| Ebond | 6 ± 0.3 | 6 |
| Eang | 85 ± 2 | 84 |
| Eimp | 7 ± 0.6 | 7 |
| Erepel | 12 ± 1 | 12 |
| Enoe | 33 ± 3 | 30 |
| Ecdih | 1 ± 0.2 | 1 |
| Rmsd from idealized geometry | ||
| Bonds (Å) | 0.00 ± 0.00 | 0.00 |
| Angles (deg) | 0.29 ± 0.00 | 0.29 |
| Impropers (deg) | 0.16 ± 0.01 | 0.15 |
| Cartesian coordinate rmsd (Å) | N, Cα, and C′ | All heavy |
≤SA> vs. ≤ >d >d
|
1.04 ± 0.21 | 1.46 ± 0.19 |
≤SA> vs. ≤ >e >e
|
0.74 ± 0.15 | 1.13 ± 0.12 |
a Where ≤SA> is the ensemble of 20 NMR-derived solution structures of S. pneumoniae CMP kinase; ≤ > is the mean atomic structure; ≤
> is the mean atomic structure; ≤ >r is the energy-minimized average structure. The CNX Frepel function was used to simulate van der Waals interactions using a force constant of 4.0 kcal mole−1 Å−4 with the atomic radii set to 0.8 times their CHARMM values (Brooks et al. 1983).
>r is the energy-minimized average structure. The CNX Frepel function was used to simulate van der Waals interactions using a force constant of 4.0 kcal mole−1 Å−4 with the atomic radii set to 0.8 times their CHARMM values (Brooks et al. 1983).
b Distance restraints were used with a square-well potential (Fnoe = 50 kcal mole−1 Å−2). Hydrogen bonds were given bounds of 1.8–2.4 Å (H−O) and 2.7–3.3 Å (N−O). No distance restraint was violated by more than 0.3 Å in any of the final structures.
c Torsional restraints were applied with values derived from an analysis of the C′, N, Cα, Hα, and Cβ chemical shifts using the TALOS program (Cornilescu et al. 1999). A force constant of 200 kcal mole−1 rad−2 was applied for all torsional restraints.
d Rmsd for the residues 16–234.
e Rmsd for the residues 16–161 and 208–234 in the core and substrate-binding domains.
Figure 2.

Stereoview of the backbone (N, Cα, C′) of 20 superimposed NMR-derived structures of S. pneumoniae CMP kinase (residues 14–236). The residues from the core domain and the substrate-binding domain (residues 16–161 and 208–234) were used here for the superposition. The N and C termini are labeled N and C, respectively.
Figure 3.

Ribbon plot (Carson 1987) depicting the averaged minimized NMR structure of S. pneumoniae CMP kinase (residues 14–236). The α-helices, β-strands, and loops are colored red, green, and gray, respectively.
Description of the structure
Figure 3 ▶ depicts the ribbon structure of the S. pneumoniae CMP kinase. Like other CMP kinases, the structure of S. pneumoniae CMP kinase consists of a core domain, a substrate-binding domain, and a small LID domain. The core domain is composed of a five-stranded parallel β-sheet and three α-helices (α1, α8, and part of α5) that are packed on both sides of the β-sheet. The substrate-binding domain contains a three-stranded antiparallel β-sheet and four α-helices (α2–α4 and part of α5). The LID domain, which acts like a lid over the substrate phosphate groups, consists of two long α-helices. The three domains of the CMP kinase pack together to form a central cavity where the substrates bind for the enzymatic catalysis (Fig. 3 ▶).
The substrate-binding domain packs tightly against the core domain. The two domains share a common α5 helix and their packing is defined by many interdomain long-range NOEs. However, the LID domain does not pack tightly to the other two domains. In particular, no NOEs were observed between the residues located at the C terminus of α6, the N terminus of α7, and the loop between α6 and α7 of the LID domain and the residues on the other two domains. Therefore, although the LID domain by itself is well defined by the NMR data with an rmsd about the mean coordinate positions of 0.58 ± 0.12 Å for the backbone atoms for the residues 162–201, its packing against the other two domains is less well defined (Fig. 2 ▶).
Comparison to other CMP kinases
The structures of the E. coli CMP kinase when free and complexed with CDP have been determined by X-ray crystallography (Briozzo et al. 1998). The S. pneumoniae and E. coli CMP kinases share a sequence homology of 47% (Fig. 4 ▶). The structure-based sequence alignment indicates that there is an one-residue insertion in the β-turn between β3 and β4 strands in the S. pneumoniae CMP kinase (Figs. 3, 4 ▶ ▶). Interestingly, the insertion at this position was not predicted using the GCG sequence alignment program (Accelrys Inc).
Figure 4.

Sequence alignment of the CMP kinases from S. pneumoniae and E. coli. The sequences are aligned using the three-dimensional structures of the proteins (Briozzo et al. 1998). The arrows and rectangles indicate the secondary structures of β-strands and α-helices, respectively. The dots in the sequence indicate gaps in the alignment. The rectangular open box indicates the phosphate-binding loop (P-loop).
The structure of S. pneumoniae CMP kinase resembles the E. coli CMP kinase crystal structure (Fig. 5A ▶; Briozzo et al. 1998) with an rmsd of 2.9 Å for the superposition of 202 pairs of Cα atoms (of 209 common residues). Residues 180–192 (E. coli CMP kinase numbering) located in the α7 helix and the loop between α7 and β8 are not determined in the crystal structure of E. coli CMP kinase. The residues around the β-turn between β3 and β4 strands are more highly charged in the S. pneumoniae CMP kinase than the E. coli homolog (Fig. 4 ▶). This could be the reason for the more open structure of this β-hairpin of S. pneumoniae CMP kinase (Fig. 5A ▶). The small difference in the orientation of the helix α6 between the two structures could be caused by the crystal contacts of the LID domain, as discussed previously (Briozzo et al. 1998).
Figure 5.
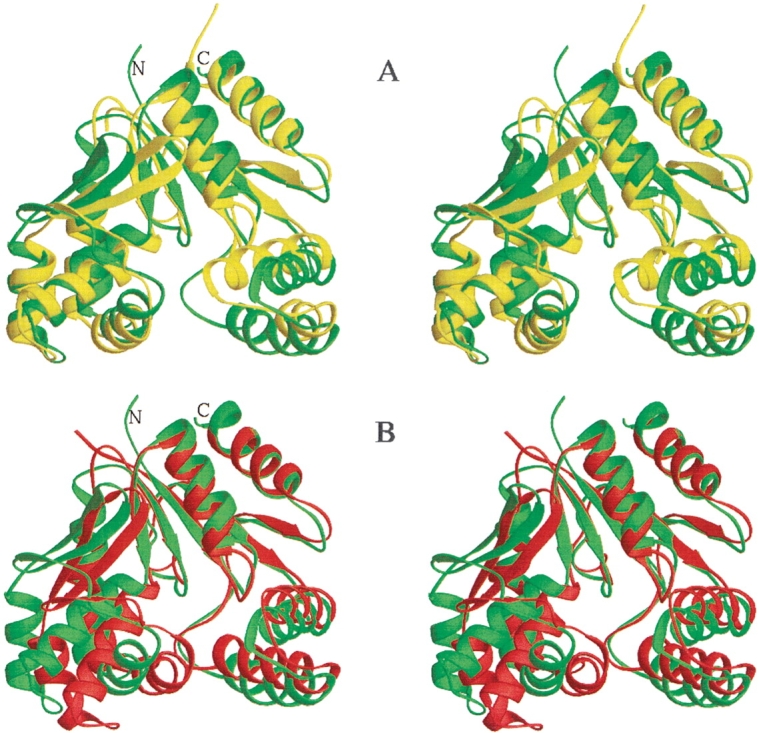
Stereoviews of the ribbon plots (Carson 1987) comparing the structures of the different CMP kinases. (A) Comparison of S. pneumoniae CMP kinase (residues 14–236) structure (green) with the free E. coli CMP kinase crystal structure (yellow; Briozzo et al. 1998). (B) Comparison of S. pneumoniae CMP kinase (residues 14–236) structure (green) with the E. coli CMP kinase crystal structure in complex with CDP (red; Briozzo et al. 1998). The structures were superimposed by using 202 pairs of Cα atoms in A and 76 pairs of Cα atoms of the core domains in B. It is noted that 13 residues in the α7 and the loop between α7 and β8 are not determined in the free E. coli CMP kinase crystal structure and that there is an insertion in the β-turn between the β3 and β4 in the S. pneumoniae CMP kinase.
When the S. pneumoniae CMP kinase structure is compared with the structure of the E. coli CMP kinase complexed with CDP (Fig. 5B ▶), the substrate-binding domain was found to be rotated by ∼17 degrees toward the CDP-binding cavity. The cytosine base of CDP binds near the ends of β2 and β6, and the two phosphates point toward the phosphate-binding loop (P-loop) located between β1 and α1 in the E. coli CMP kinase structure.
Substrate-binding sites
Titration of CMP into a solution of the S. pneumoniae CMP kinase caused several backbone amides in β2 to be broadened beyond detection and perturbed the chemical shifts of many backbone CαH and NH groups (Fig. 6A ▶). The side chains that were affected by the binding of CMP are located mainly in the interface between the core domain and substrate-binding domain (Fig. 6A ▶). Titration of CDP into the protein caused similar chemical shift perturbations. These data indicate that CMP or CDP binds to the junction between the core domain and substrate-binding domain of the S. pneumoniae CMP kinase. These results are consistent with the crystal structure of the E. coli CMP kinase/CDP complex (Briozzo et al. 1998). The broadening of the backbone amide cross peaks of the β2 strand may be caused by the motion (rotation) of the substrate-binding domain on the binding of CDP or CMP (Fig. 5B ▶).
Figure 6.

Ribbon plots depicting the chemical shift perturbations of S. pneumoniae CMP kinase (residues 14–236) by the binding of CMP (A) and ATP (B). The ribbons are color coded on the basis of different degrees of chemical shift perturbations. Magenta indicates where the backbone NHs are broadened beyond detection by the binding of the substrate. Yellow indicates where the chemical shifts are perturbed by ≥0.3 ppm for the backbone 13Cα and/or 15N atoms and/or by ≥0.05 ppm for the Hα and/or NH protons. The side chains with their chemical shifts perturbed by ≥0.3 ppm for the carbon atoms and/or by ≥0.05 ppm for the protons are shown in ball-and-stick mode.
Titration of ATP into the S. pneumoniae CMP kinase protein shows a distinct set of chemical shift perturbations, indicating that ATP binds to a different site on the protein. ATP binding caused chemical shift perturbation and peak broadening for residues that are located in the junction between the core domain and the LID domain (Fig. 6B ▶). The side chains that were perturbed by the binding of ATP include Ile 21, Pro 24, Ser 27, Ile 157, Leu 159, Thr 216, and Ile 221. This site is likely to be the binding site of the adenine ring of ATP. In this binding mode, the conserved phosphate-binding loop (P-loop) located between β1 and α1 on the core domain would be in position to bind to the triphosphate group of ATP. Mutations of the Lys residue in the P-loop to either Met or Glu resulted in a dramatic decrease in kcat/Km in Arabidopsis UMP/CMP kinase (Zhou and Thornburg 1998), indicating that this residue plays an important role in both the binding of ATP and enzyme catalysis.
So far, no structures of the CMP kinase/ATP complex have been reported. The crystal structures of the D. discoideum UMP/CMP kinase complexed with the bisubstrate inhibitor UP5A (Scheffzek et al. 1996) and the yeast adenylate kinase complexed with the bisubstrate inhibitor AP5A (Abele and Schulz 1995) or the ATP analog (Schlauderer et al. 1996) showed that the ATP portion of the inhibitor binds to the junction between the core domain and the LID domain. The folds of the substrate-binding domain of the D. discoideum UMP/CMP kinase and yeast adenylate kinase are, however, very different from those of the S. pneumoniae or E. coli CMP kinase. Our chemical shift mapping data for S. pneumoniae CMP kinase are consistent with the ATP binding site observed previously for the homologs in the other organisms.
Backbone dynamics
Structural mobility plays an important role for enzyme catalysis (Eisenmesser et al. 2002; Hammes-Schiffer 2002). To study the dynamics of the S. pneumoniae CMP kinase, we measured 15N relaxation parameters (Stone et al. 1992; Yu et al. 1996). The plot of the measured 15N-heteronuclear NOEs as a function of residue number is shown in Figure 7 ▶. The heteronuclear NOEs for most residues of the protein were determined to be in the range of 0.75 to 0.85, indicating that internal motions on the fast (picosecond) timescale are restricted. Exceptions were found for the N-terminal 17 residues (including 13 His tag residues), where the heteronuclear NOEs were significantly reduced. In addition, the NOEs for the residues 64–67, 135–136, 178–182, and 201–207 located in the loops between α2 and α3, α5 and β6, α6 and α7, and α7 and β8, respectively, were all significantly reduced. The β-turn residues (residues 86–87) between β3 and β4 were also fairly mobile as evidenced by the significantly reduced NOEs. The α-helices (α7 and the C-terminal part of α6) in the LID domain also have slightly smaller heteronuclear NOEs than average. Interestingly, the high mobility of the loop between α7 and β8 as observed here for the S. pneumoniae CMP kinase is correlated with the structural disorder of the corresponding region in the E. coli CMP kinase (Fig. 5A ▶; Briozzo et al. 1998).
Figure 7.

Plot of the measured heteronuclear 15N{1H} NOEs of the backbone amides and their uncertainties at 14.1T magnetic field as a function of residue number. Residues for which no results are shown correspond to the N-terminal His tag, six proline residues, and the overlapped residues in the 15N/1H HSQC spectra.
The highly mobile loops observed in the LID domain could serve as flexible hinges that help close the LID domain on the binding of ATP. This interpretation is consistent with the recent time-resolved fluorescence studies, showing that the enzyme adopts a closed conformation on the binding of an ATP analog probe (Sierra et al. 2000). Because the binding of CDP to E. coli CMP kinase caused a rotation of the substrate-binding domain toward the central cavity (Fig. 5B ▶; Briozzo et al. 1998), it is possible that the central cavity could be completely closed transiently on the binding of both substrates of CMP and ATP during catalysis.
Conclusion
S. pneumoniae SP1603 protein is one of the many structural genomics targets selected from a genome-wide survey of this organism. We have found that SP1603 is an essential CMP kinase and shares no sequence homology with the human ortholog. Thus, it represents a potential novel therapeutic target for the development of new antibacterial agents. The solution structure of this CMP kinase has been determined by NMR spectroscopy. It is composed of eight α-helices and two β-sheets that fold into the classical core domain, the substrate-binding domain, and the LID domain. Each individual domain of the protein is well defined by the NMR data. However, the orientation of the LID domain with respect to the core and substrate-binding domains is less well defined. This is consistent with the greater mobility of the loops in the LID domain, in particular, the long flexible loop that links the LID domain to the core domain. This is also consistent with the lack of observable NOEs between the residues located at the tip of the LID domain and the residues on the other two domains. The highly mobile loops observed in the LID domain could play an important role in aiding the closing/opening of the LID domain during the cycle of enzyme catalysis. Using this structure, a structure-based drug design effort could be initiated to design novel antibiotics against this target.
Materials and methods
NMR sample preparation
The validation of essentiality of SP1603 was carried out according to the previously described procedure (Yu et al. 2001). SP1603 was cloned into a modified pET15b vector with an N-terminal His tag and expressed in E. coli BL21 (DE3) cells. Uniformly 15N- and 15N, 13C-labeled proteins were prepared for the NMR experiments by growing bacteria containing 15NH4Cl with or without [U-13C]-glucose. A uniformly 15N-, 13C-labeled sample with selectively protonated methyl groups of Val, Leu, and Ile (δ1) was also prepared by growing cells in 100% 2H2O and by supplementing media with [U-13C] α-ketobutyrate (50 mg/L), [U-13C] α-ketoisovalerate (100 mg/L), [U-13C, 2H] glucose (3 g/L), and 15NH4Cl (1 g/L) (Goto et al. 1999; Hajduk et al. 2000; Medek et al. 2000).
The labeled recombinant proteins were purified by affinity chromatography on a nickel-IDA column (Invitrogen). The purified protein with uncleaved N-terminal His tag was dialyzed against a buffer containing 20 mM sodium phosphate and 50 mM (NH4)2SO4 (pH 7.0) and concentrated to 1 mM for NMR studies.
NMR spectroscopy and structure determination
The NMR spectra were collected at 30°C on a Bruker DRX500, DRX600, or DRX800 NMR spectrometer. The 1H, 15N, and 13C resonances of the backbone were assigned using triple resonance experiments (HNCA, HN(CO)CA, HN(CA)CB, HN(COCA)CB, HNCO, and HN(CA)CO [Yamazaki et al. 1994]) using the uniformly 15N-, 13C-, and 2H-labeled sample with selectively protonated methyl groups of Val, Leu, and Ile (δ1). 1Hα resonances were assigned from a 15N-edited TOCSY spectrum using a uniformly 15N-labeled protein (Clore and Gronenborn 1994) and from HCACO and HACA(CO)NH spectra with a uniformly 15N- and 13C-labeled protein (Grzesiek and Bax 1993; Feng et al. 1996). The side-chain signals were assigned from 3D H(CCO) NH-TOCSY, C(CO)NH-TOCSY, HCCH-TOCSY, and 15N-edited TOCSY experiments (Clore and Gronenborn 1994). 15N heteronuclear NOEs were collected and analyzed as previously described (Stone et al. 1992; Yu et al. 1996). Two duplicate data sets of heteronuclear NOEs were collected and used to calculate their average values and standard deviations.
Structures of the S. pneumoniae CMP kinase were generated using a distance-geometry/simulated annealing protocol (Nilges et al. 1988) with the CNX program (Accelrys Inc.). Structure calculations for the CMP kinase used a total of 2992 proton–proton distances derived from 3D 15N- and 13C-resolved NOESY spectra (Fesik and Zuiderweg 1988) collected with a mixing time of 80 msec. The NOE-derived distance restraints were given upper bounds of 2.7, 3.3, 4.0, and 5.0 Å based on the measured NOE intensities. One hundred eight hydrogen bonds from the α-helices and β-sheets were included in the structural calculations. Three hundred twenty-six φ and ξ angular restraints derived from an analysis of the C′, N, Cα, Hα, and Cβ chemical shifts using the TALOS program (Cornilescu et al. 1999) were also included in the structural calculations.
Protein Data Bank accession number
The coordinates for the S. pneumoniae CMP kinase structures have been deposited in the Protein Data Bank under the identifier code 1Q3T.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
NMR, nuclear magnetic resonance
NOE, nuclear Overhauser effect
HSQC, heteronuclear single-quantum coherence
rmsd, root-mean-square deviation
TIGR, The Institute for Genomic Research
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03256803.
References
- Abele, U. and Schulz, G.E. 1995. High-resolution structures of adenylate kinase from yeast ligated with inhibitor AP5A, showing the pathway of phosphoryl transfer. Protein Sci. 4 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerley, B.J., Rubin, E.J., Novick, V.L., Amaya, K., Judson, N., and Mekalanos, J.J. 2002. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. 99 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero, F. 1995. Pneumococcal resistance to β-lactam antibiotics. Microb. Drug Resist. 1 115–120. [DOI] [PubMed] [Google Scholar]
- Bertrand, T., Briozzo, P., Assairi, L., Ofiteru, A., Bucurenci, N., Munier-Lehmann, H., Golinelli-Pimpaneau, B., Barzu, O., and Gilles, A.-M. 2002. Sugar specificity of bacterial CMP kinases as revealed by crystal structures and mutagenesis of Escherichia coli enzyme. J. Mol. Biol. 315 1099–1110. [DOI] [PubMed] [Google Scholar]
- Briozzo, P., Golinelli-Pimpaneau, B., Gilles, A.-M., Gaucher, J.-F., Burlacu-Miron, S., Sakamoto, H., Janin, J., and Barzu, O. 1998. Structures of Escherichia coli CMP kinase alone and in complex with CDP: A new fold of the nucleoside monophosphate binding domain and insights into cytosine nucleotide specificity. Structure 6 1517–1527. [DOI] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Carson, M. 1987. Ribbon models of macromolecules. J. Mol. Graph. 5 103–106. [Google Scholar]
- Clore, G.M. and Gronenborn, A.M. 1994. Multidimensional heteronuclear magnetic resonance of proteins. Methods Enzymol. 239 349–363. [DOI] [PubMed] [Google Scholar]
- ———. 1998. NMR structure determination of proteins and protein complexes larger than 20 kDa. Curr. Opin. Chem. Biol. 2 564–570. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- Eisenmesser, E.Z., Bosco, D.A., Akke, M., and Kern, D. 2002. Enzyme dynamics during catalysis. Science 295 1520–1523. [DOI] [PubMed] [Google Scholar]
- Feng, W., Rios, C.B., and Montelione, G.T. 1996. Phase labeling of C-H and C-C spin-system topologies: Application in PFG-HACANH and PFG-HACA(CO)NH triple-resonance experiments for determining backbone resonance assignments in proteins. J. Biomol. NMR 8 98–104. [DOI] [PubMed] [Google Scholar]
- Fesik, S.W. and Zuiderweg, E.R.P. 1988. Heteronuclear three-dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR spectra. J. Magn. Reson. 78 588–593. [Google Scholar]
- Goto, N.K., Gardner, K.H., Mueller, G.A., Willis, R.C., and Kay, L.E. 1999. A robust and cost-effective method for the production of Val, Leu, Ile (δ1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR 13 369–374. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1993. The origin and removal of artifacts in 3D HCACO spectra of proteins uniformly enriched with 13C. J. Magn. Reson. B 102 103–106. [Google Scholar]
- Hajduk, P.J., Augeri, D.J., Mack, J., Mendoza, R., Yang, J., Betz, S.F., and Fesik, S.W. 2000. NMR-based screening of proteins containing 13C-labeled methyl groups. J. Am. Chem. Soc. 122 7898–7904. [Google Scholar]
- Hammes-Schiffer, S. 2002. Impact of enzyme motion on activity. Biochemistry 41 13335–13343. [DOI] [PubMed] [Google Scholar]
- Hoskins, J., Alborn Jr., W.E., Arnold, J., Blaszczak, L.C., Burgett, S., DeHoff, B.S., Estrem, S.T., Fritz, L., Fu, D.-J., Fuller, W., et al. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183 5709–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, L.E. 1998. Protein dynamics from NMR. Biochem. Cell Biol. 76 145–152. [DOI] [PubMed] [Google Scholar]
- Kobayashi, K., Ehrlich, S.D., Albertini, A., Amati, G., Andersen, K.K., Arnaud, M., Asai, K., Ashikaga, S., Aymerich, S., Bessieres, P., et al. 2003. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. 100 4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljelund, P. and Lacroute, F. 1986. Genetic characterization and isolation of the Saccharomyces cerevisiae gene coding for uridine monophosphokinase. Mol. Gen. Genet. 205 74–81. [DOI] [PubMed] [Google Scholar]
- Liou, J.-Y., Dutschman, G.E., Lam, W., Jiang, Z., and Cheng, Y.-C. 2002. Characterization of human UMP/CMP kinase and its phosphorylation of D- and L-form deoxycytidine analogue monophosphates. Cancer Res. 62 1624–1631. [PubMed] [Google Scholar]
- Medek, A., Olejniczak, E.T., Meadows, R.P., and Fesik, S.W. 2000. An approach for high-throughput structure determination of proteins by NMR spectroscopy. J. Biomol. NMR 18 229–238. [DOI] [PubMed] [Google Scholar]
- Müller-Dieckmann, H.-J. and Schulz, G.E. 1994. The structure of uridylate kinase with its substrates, showing the transition state geometry. J. Mol. Biol. 236 361–367. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Substrate specificity and assembly of the catalytic center derived from two structures of ligated uridylate kinase. J. Mol. Biol. 246 522–530. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Clore, G.M., and Gronenborn, A.M. 1988. Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical simulated annealing calculations. FEBS Lett. 229 317–324. [DOI] [PubMed] [Google Scholar]
- Scheffzek, K., Kliche, W., Wiesmuller, L., and Reinstein, J. 1996. Crystal structure of the complex of UMP/CMP kinase from Dictyostelium discoideum and the bisubstrate inhibitor P1–(5′-adenosyl) P5–(5′-uridyl) pentaphosphate (UP5A) and Mg2+ at 2.2 Å: Implications for water-mediated specificity. Biochemistry 35 9716–9727. [DOI] [PubMed] [Google Scholar]
- Schlauderer, G.J., Proba, K., and Schulz, G.E. 1996. Structure of a mutant adenylate kinase ligated with an ATP-analogue showing domain closure over ATP. J. Mol. Biol. 256 223–227. [DOI] [PubMed] [Google Scholar]
- Schlichting, I. and Reinstein, J. 1997. Structures of active conformations of UMP kinase from Dictyostelium discoideum suggest phosphoryl transfer is associative. Biochemistry 36 9290–9296. [DOI] [PubMed] [Google Scholar]
- Schutze, G.E., Kaplan, S.L., and Jacobs, R.F. 1994. Resistant pneumococcus: A worldwide problem. Infection 22 233–237. [DOI] [PubMed] [Google Scholar]
- Sierra, I.M.L., Gallay, J., Vincent, M., Bertrand, T., Briozzo, P., Barzu, O., and Gilles, A.-M. 2000. Substrate-induced fit of the ATP binding site of cytidine monophosphate kinase from Escherichia coli: Time-resolved fluorescence of 3′-anthraniloyl-2′-deoxy-ADP and molecular modeling. Biochemistry 39 15870–15878. [DOI] [PubMed] [Google Scholar]
- Stone, M.J., Fairbrother, W.J., Palmer III, A.G., Reizer, J., Saier Jr., M.H., and Wright, P.E. 1992. Backbone dynamics of the Bacillus subtilis glucose permease IIA domain determined from 15N NMR relaxation measurements. Biochemistry 31 4394–4406. [DOI] [PubMed] [Google Scholar]
- Tettelin, H., Nelson, K.E., Paulsen, I.T., Eisen, J.A., Read, T.D., Peterson, S., Heidelberg, J., DeBoy, R.T., Haft, D.H., Dodson, R.J., et al. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293 498–506. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T., Lee, W., Arrowsmith, C.H., Muhandiram, D.R., and Kay, L.E. 1994. A suite of triple-resonance NMR experiments for the backbone assignment of 15N, 13C, 2H-labeled proteins with high sensitivity. J. Am. Chem. Soc. 116 11655–11666. [Google Scholar]
- Yu, L., Zhu, C.-X., Tse-Dinh, Y.-C., and Fesik, S.W. 1996. Backbone dynamics of the C-terminal domain of Escherichia coli topoisomerase I in the absence and presence of single-stranded DNA. Biochemistry 35 9661–9666. [DOI] [PubMed] [Google Scholar]
- Yu, L., Gunasekera, A.H., Mack, J., Olejniczak, E.T., Chovan, L.E., Ruan, X., Towne, D.L., Lerner, C.G., and Fesik, S.W. 2001. Solution structure and function of a conserved protein SP14.3 encoded by an essential Streptococcus pneumoniae gene. J. Mol. Biol. 311 593–604. [DOI] [PubMed] [Google Scholar]
- Zhou, L. and Thornburg, R. 1998. Site-specific mutations of conserved residues in the phosphate-binding loop of the Arabidopsis UMP/CMP kinase alter ATP and UMP binding. Arch. Biochem. Biophys. 358 297–302. [DOI] [PubMed] [Google Scholar]
- Zuiderweg, E.R.P. 2002. Mapping protein–protein interactions in solution by NMR spectroscopy. Biochemistry 41 1–7. [DOI] [PubMed] [Google Scholar]