

Abstract
We have investigated the organization and dynamics of the functionally important tryptophan residues of erythroid spectrin in native and denatured conditions utilizing the wavelength-selective fluorescence approach. We observed a red edge excitation shift (REES) of 4 nm for the tryptophans in the case of spectrin in its native state. This indicates that tryptophans in spectrin are localized in a microenvironment of restricted mobility, and that the regions surrounding the spectrin tryptophans offer considerable restriction to the reorientational motion of the water dipoles around the excited state tryptophans. Interestingly, spectrin exhibits a REES of 3 nm even when denatured in 8 M urea. This represents the first report of a denatured protein displaying REES. Observation of REES in the denatured state implies that some of the structural and dynamic features of this microenvironment around the spectrin tryptophans are retained even when the protein is denatured. Fluorescence quenching data of denatured spectrin support this conclusion. In addition, we have deduced the organization and dynamics of the hydrophobic binding site of the polarity-sensitive fluorescent probe PRODAN that binds erythroid spectrin with high affinity. When bound to spectrin, PRODAN exhibits a REES of 9 nm. Because PRODAN binds to a hydrophobic site in spectrin, such a result would directly imply that this region of spectrin offers considerable restriction to the reorientational motion of the solvent dipoles around the excited state fluorophore. The results of our study could provide vital insight into the role of tryptophans in the stability and folding of spectrin.
Keywords: Spectrin, REES, denaturation, tryptophan, PRODAN, wavelength-selective fluorescence
Spectrin is the major constituent protein of the erythrocyte cytoskeleton that forms a filamentous network on the cytoplasmic face of the membrane by providing a scaffold for a variety of proteins (Bennett and Gilligan 1983). The interaction of the spectrin-based protein network with the cytoplasmic membrane controls the elasticity of the bilayer membrane. The inherent flexibility of spectrin is believed to be a major factor that contributes to the elastic deformability displayed by red cells during their passage through the circulatory system. Several blood diseases are associated with erythrocyte deformation and defects in spectrin, for example, various types of hereditary hemolytic anemia involve mutations in spectrin (Delaunay and Dhermy 1993; Wichterle et al. 1996; Gallagher et al. 1997). A spectrin-based network has recently been implicated as a membrane protein-sorting machine (Beck and Nelson 1996) and in maintenance of Golgi structure and organization (De Matteis and Morrow 2000). In addition, spectrin has been reported to be involved in the maintenance of dynamic (phase-state) asymmetry in erythrocyte membranes (Williamson et al. 1982) and to have a chaperone-like activity (Chakrabarti and Bhattacharya 1999; Chakrabarti et al. 2001).
Spectrin is a large dimeric amphiphilic protein having hydrophobic stretches in its polypeptide sequence. It is an elongated heterodimer with two subunits (α and β with mol wt of 240 and 220 kD). The two subunits are homologous with about 30% identity and are aligned in the highly elongated, worm-like heterodimer in an antiparallel side-to-side orientation to give a flexible 100-nm rod-shaped molecule with the amino and carboxy termini toward the ends of the rods. These heterodimers associate head-to-head to form 200-nm tetramers and higher order oligomers. The primary sequence of spectrin is comprised of a series of contiguous motifs called "spectrin repeats" (typically 106 amino acid repeating sequences) that are characteristic of all members of the spectrin family of proteins (Pascual et al. 1997). Besides these features, spectrin exhibits additional structural motifs. These include an actin-binding domain, a pleckstrin homology (PH) domain, a Src homology 3 (SH3) domain, and a calmodulin-like domain. These structural features allow spectrin to take part in a number of physiological events through protein–protein interactions. The ability of spectrin to expand and contract has been attributed to its modular structure made of repeats (Grum et al. 1999).
The spectrin dimer has a number of tryptophan residues. There are 42 tryptophans in each of the α and β subunits in the spectrin dimer (Sahr et al. 1990; Winkelmann et al. 1990). These tryptophans are distributed over the entire spectrin molecule. It is noteworthy that the typically 106 amino acid long repeat units in spectrin have tryptophans strongly conserved at the 45th residue and partially conserved at the 11th residue. Careful examination shows that there are 41 tryptophans in 23 repeat motifs in the α subunit while there are 35 tryptophans in 17 repeat motifs in the β subunit of the spectrin dimer. Taken together, the tryptophans in these positions (repeat motifs) represent more than 90% of the total tryptophans in the spectrin dimer. In addition, there are five tryptophans in the actin binding domain at the amino terminus and two tryptophans at the carboxy terminus in the β subunit of the spectrin dimer.
Some of the conserved tryptophans have been shown to promote folding of spectrin domains (MacDonald et al. 1994) and contribute to their thermodynamic stability (Subbarao and MacDonald 1994; Pantazatos and MacDonald 1997). The fact that tryptophans are distributed over the entire molecule and yet are localized in the same position in each domain makes them convenient intrinsic fluorescence reporter groups for monitoring conformational changes in spectrin that contribute to its elastic deformability exhibited in physiological situations (Subbarao and MacDonald 1994). We have recently utilized the intrinsic tryptophan fluorescence of spectrin to monitor interaction of spectrin with micellar detergents (Ray and Chakrabarti 2003).
Many of these tryptophans are at or in the vicinity of hydrophobic patches in spectrin, which can bind hydrophobic ligands such as fatty acids and phospholipids and cause quenching of tryptophan fluorescence (Sikorski et al. 1987; Kahana et al. 1992). The hydrophobic binding site in spectrin is crucial because this region is believed to facilitate interaction of spectrin with membranes. One of us has previously shown that the hydrophobic fluorescent probe 6-propionyl-2-dimethylaminonaphthalene (PRODAN; Weber and Farris 1979), which shows polarity-sensitive fluorescence, binds erythroid spectrin with a high affinity (Chakrabarti 1996). In addition, we have recently shown that the widely used hydrophobic fluorescent probe pyrene also binds to spectrin with high affinity and estimated the apparent dielectric constant of the binding site to be ∼7 from analysis of the ratios of pyrene vibronic band intensities (Haque et al. 2000).
In this article, we have investigated the organization and dynamics of the functionally important tryptophan residues of erythroid spectrin in native and denatured conditions and PRODAN bound to spectrin utilizing the wavelength-selective fluorescence approach. Wavelength-selective fluorescence comprises a set of approaches based on the red edge effect in fluorescence spectroscopy, which can be used to directly monitor the environment and dynamics around a fluorophore in a complex biological system (Mukherjee and Chattopadhyay 1995; Demchenko 2002; Chattopadhyay 2003; Raghuraman et al. 2003). A shift in the wavelength of maximum fluorescence emission toward higher wavelengths, caused by a shift in the excitation wavelength toward the red edge of absorption band, is termed red edge excitation shift (REES). This effect is mostly observed with polar fluorophores in motionally restricted media such as very viscous solutions or condensed phases where the dipolar relaxation time for the solvent shell around a fluorophore is comparable to or longer than its fluorescence lifetime (Galley and Purkey 1970; Lakowicz and Keating-Nakamoto 1984; Demchenko 1988b, 2002; Mukherjee and Chattopadhyay 1995; Chattopadhyay 2002, 2003; Raghuraman et al. 2003). REES arises from slow rates of solvent relaxation (reorientation) around an excited state fluorophore, which is a function of the motional restriction imposed on the solvent molecules in the immediate vicinity of the fluorophore. Utilizing this approach, it becomes possible to probe the mobility parameters of the environment itself (which is represented by the relaxing solvent molecules) using the fluorophore merely as a reporter group. Further, since the ubiquitous solvent for biological systems is water, the information obtained in such cases will come from the otherwise "optically silent" water molecules. The unique feature about REES is that while all other fluorescence techniques (such as fluorescence quenching, energy transfer, and polarization measurements) yield information about the fluorophore (either intrinsic or extrinsic) itself, REES provides information about the relative rates of solvent (water in biological systems) relaxation dynamics, which is not possible to obtain by other techniques. This makes the use of REES in particular and the wavelength-selective fluorescence approach in general extremely useful, because hydration plays a crucial modulatory role in a large number of important cellular events (Haussinger 1996; Nishimura et al. 1997; Mentré 2001) including protein folding, lipid–protein interactions, and ion transport. An in-depth discussion of the photophysical framework for REES and wavelength-selective fluorescence approach is provided in recent reviews (Chattopadhyay 2003; Raghuraman et al. 2003).
We have previously shown that REES and related techniques (wavelength-selective fluorescence approach) serve as a powerful tool to monitor organization and dynamics of probes and peptides bound to membranes (Chattopadhyay and Mukherjee 1993, 1999a,b; Chattopadhyay and Rukmini 1993; Mukherjee and Chattopadhyay 1994; Ghosh et al. 1997; Chattopadhyay et al. 1997; Kelkar et al. 2003) and membrane-mimetic media such as micelles and reverse micelles (Rawat et al. 1997; Rawat and Chattopadhyay 1999; Chattopadhyay et al. 2002). In addition, we have previously used the wavelength-selective fluorescence approach to analyze the organization and dynamics of tryptophans in the soluble hemolytic protein α-toxin (Raja et al. 1999) and the cytoskeletal protein tubulin, which is a component of the microtubular network in eukaryotes (Guha et al. 1996).
Results
Urea denaturation of spectrin monitored by fluorescence and circular dichroism
Figure 1A ▶ shows the fluorescence emission spectra of native spectrin and spectrin denatured with 8 M urea. As shown in the figure, tryptophans in native dimeric spectrin exhibit an emission maximum at 338 nm in agreement with previous literature (Subbarao and MacDonald 1994). Figure 1a ▶ also shows the fluorescence emission spectrum of spectrin denatured with 8 M urea, which displays a red shift, and the emission maximum is shifted to 347 nm. This red shift can be attributed to increased exposure of spectrin tryptophans to water upon denaturation in 8 M urea. Tryptophan in water exhibits an emission maximum at 355 nm (Eftink 1991a). The emission maximum of 347 nm for denatured spectrin therefore indicates that the tryptophans are not completely exposed to water even when denatured in 8 M urea but are shielded from the bulk water to a considerable extent (see later). To confirm the changes in secondary structure of spectrin upon denaturation with urea, we examined the far-UV CD spectra of native and denatured spectrin. The CD spectra of native and urea denatured spectrin are shown in Figure 1B ▶. The backbone CD spectrum of native spectrin is characteristic of a protein with predominantly α-helical structure, as reported earlier (Majee et al. 1999). When denatured in urea, the CD spectrum of spectrin exhibited considerable loss of helicity, indicating loss of secondary structure elements.
Figure 1.

(A) Fluorescence emission spectra and (B) far-UV CD spectra of spectrin in native (—) and urea denatured (- - -) state. For fluorescence measurements, the concentration of native spectrin was 0.6 μM, while that of denatured spectrin was 0.4 μM, and the excitation wavelength was 280 nm. The spectrin concentration was 0.35 μM for CD measurements. Spectrin was denatured in 8 M urea. The fluorescence spectra are intensity-normalized at the emission maximum. See Materials and Methods for other details.
The urea denaturation curves for spectrin as monitored by fluorescence emission maximum and CD spectroscopy measurements are shown in Figure 2A ▶. The denaturation profiles show that the denaturation process is complete at 8 M urea, and that there is good agreement between these two techniques. The overall shape of the denaturation curve suggests that urea-induced denaturation is predominantly a two state process, as indicated previously (Pantazatos and MacDonald 1997). The denaturation curve monitored by fluorescence intensity ratio at 337 and 350 nm (F337/F350), shown in Figure 2B ▶, supports this conclusion. This, however, does not completely rule out the possibility that some regions of the multidomain protein spectrin may retain some structure even under these conditions.
Figure 2.

Urea denaturation curves of spectrin as monitored by fluorescence and CD measurements: (A) Fluorescence emission maximum (open circles) and ellipticity at 222 nm (filled circles), and (B) fluorescence emission maximum (open circles), and fluorescence emission intensity ratio (filled circles) at 337 and 350 nm (F337/F350) are plotted as a function of urea concentration. The excitation wavelength was 295 nm for fluorescence measurements. The concentration of spectrin was 0.05 μM for fluorescence measurements and 0.24 μM for CD measurements. See Materials and Methods for other details.
Red edge excitation shift of spectrin tryptophans in native and denatured states
The shifts in the maxima of fluorescence emission of spectrin as a function of excitation wavelength are shown in Figure 3 ▶. (We have used the term maximum of fluorescence emission in a somewhat wider sense here. In every case, we have monitored the wavelength corresponding to maximum fluorescence intensity, as well as the center of mass of the fluorescence emission. In most cases, both these methods yielded the same wavelength. In cases where minor discrepancies were found, the center of mass of emission has been reported as the fluorescence maximum.) Upon excitation at 280 nm, tryptophans in native dimeric spectrin exhibit an emission maximum at 338 nm as described above. As the excitation wavelength is changed from 280 nm to 307 nm, the emission maximum of native spectrin is shifted from 338 nm to 342 nm, which corresponds to a REES of 4 nm. It is possible that there could be further red shift when native spectrin is excited beyond 307 nm. We found it difficult to work in this wavelength range because of very low signal-to-noise ratio and artifacts due to the solvent Raman peak that some times remained even after background subtraction. Such a shift in the wavelength of emission maximum with change in excitation wavelength is characteristic of the red edge effect and indicates that the tryptophans in native dimeric spectrin are localized in a motionally restricted environment. Spectrin is a multitryptophan protein, and therefore, the red edge shift may be indicative of the average environment experienced by the tryptophans. Nevertheless, such a result would directly imply that the regions surrounding at least some of the spectrin tryptophans offer considerable restriction to the reorientational motion of the solvent (water) dipoles around the excited state tryptophans. This is significant, because some of the functionally important spectrin tryptophans are localized in the invariant region and are shielded from the bulk (characterized by fast solvent reorientational motion) solvent (MacDonald et al. 1994; Pantazatos and MacDonald 1997; also see acrylamide quenching results later). Further, many of these tryptophans are at or in the vicinity of hydrophobic patches in spectrin, which can bind hydrophobic ligands (Sikorski et al. 1987; Kahana et al. 1992). As mentioned earlier, the estimated apparent dielectric constant of the hydrophobic binding site is ∼7 (Haque et al. 2000). The low dielectric characteristics coupled with the presence of restricted water molecules contribute in making some of these regions ideal environments for exhibiting REES and related effects (Mukherjee and Chattopadhyay 1995; Chattopadhyay 2002).
Figure 3.

Effect of changing excitation wavelength on the wavelength of maximum emission for native (open circles) and denatured (filled circles) spectrin. The concentration of native spectrin was 0.6 μM, while that of denatured spectrin was 0.4 μM. Spectrin was denatured in 8 M urea. REES data for NATA in 8 M urea (open triangles) is shown as a control. The concentration of NATA used was 33 μM. See Materials and Methods for other details.
The fluorescence emission spectrum of spectrin denatured with 8 M urea displays a red shift, and the emission maximum is shifted to 347 nm (see Fig. 1A ▶). Analysis of REES effect in spectrin denatured with 8 M urea provides interesting results. Figure 3 ▶ shows that as the excitation wavelength is changed from 280 nm to 307 nm, the emission maximum of the tryptophans in denatured spectrin is shifted from 347 nm to 350 nm, which corresponds to a REES of 3 nm. This is surprising, because tryptophans in denatured proteins generally do not exhibit REES effects due to fast solvent relaxation in the denatured state. This result therefore is in contrast to earlier studies where it has been shown that emission maximum for tryptophans in denatured proteins such as tubulin do not exhibit excitation wavelength dependence (Guha et al. 1996). To the best of our knowledge, this is the first report of a denatured protein exhibiting REES. This result, along with the emission maximum of 347 nm for the denatured protein, indicates that the tryptophan microenvironment in spectrin is characterized by unique structural and dynamic features that are maintained to a considerable extent even when denatured with 8 M urea. Further, this could suggest that solvent (water) molecules may have a possible structural role in the overall stability of spectrin as has been shown earlier for other proteins (Fischer et al. 1994; Kandori et al. 1995; Sankararamakrishnan and Sansom 1995; Nishimura et al. 1997). These results are further supported by analysis of quenching of spectrin tryptophan fluorescence by the water soluble quencher acrylamide (see later).
Because the bulk physical properties (such as viscosity) of the solution change in the presence of 8 M urea, we monitored REES of the model tryptophan derivative N-acetyl-L-tryptophanamide (NATA) in 8 M urea as a control experiment. Figure 3 ▶ shows that NATA in 8 M urea does not exhibit REES, and its emission maximum remains invariant at 352 nm irrespective of the excitation wavelength when the excitation wavelength was varied from 280–307 nm. Another concern arises due to the possibility of any artifacts induced by aggregation of spectrin in the denatured state. To address this issue, we performed the REES experiments with denatured spectrin after centrifugation at high speed. In most cases, we obtained a small pellet but the supernatant gave identical REES results in all cases. This rules out any possible complication due to aggregation.
Tryptophan polarization changes with excitation and emission wavelengths
In addition to the shift in emission maximum on red edge excitation, fluorescence polarization is also known to be dependent on excitation wavelength in motionally restricted media (Mukherjee and Chattopadhyay 1995, and references therein). Due to strong dipolar interactions with the surrounding solvent molecules, there is a decreased rotational rate of the fluorophore in the solvent relaxed state. On red edge excitation, a selective excitation of this subclass of fluorophore occurs. Because of strong interactions with the polar solvent molecules in the excited state, one may expect these "solvent relaxed" fluorophores to rotate more slowly, thereby increasing the polarization.
The excitation polarization spectra (i.e., a plot of steady-state polarization versus excitation wavelength) of native spectrin and spectrin denatured with 8 M urea are shown in Figure 4 ▶. The polarization of spectrin tryptophans undergoes considerable change upon altering the excitation wavelength, with a sharp increase toward the red edge of the absorption band and a characteristic dip at 290 nm. Such an increase in polarization upon red edge excitation for peptides and proteins containing tryptophans (Weber 1960; Guha et al. 1996) as well as other aromatic fluorophores (Valeur and Weber 1978; Chattopadhyay and Mukherjee 1993), especially in media of reduced mobility has been previously reported. Interestingly, Figure 4 ▶ also shows that the polarization of denatured spectrin shows an excitation wavelength dependence although to a lesser extent. This reinforces our earlier conclusion that the spectrin tryptophans are in a motionally restricted region (due to the presence of slow reorienting solvent shell) not only in its native state but also when denatured in 8 M urea.
Figure 4.

Fluorescence polarization of native (open circles) and denatured (filled circles) spectrin as a function of excitation wavelength. Polarization values were recorded keeping emission wavelength constant at 340 nm. The data points shown are the means ± standard errors of at least three independent measurements. All other conditions are as in Figure 3 ▶. See Materials and Methods for other details.
It is known that tryptophan has two overlapping So → S1 electronic transitions (1La and 1Lb) which are almost perpendicular to each other (Song and Kurtin 1969; Yamamoto and Tanaka 1972; Eftink 1991a; Albinsson and Norden 1992). Both So → 1La and So → 1Lb transitions occur in the 260–300 nm range. In nonpolar solvents, 1La has higher energy than 1Lb. However, in polar solvents, the energy level of 1La is lowered, making it the lowest energy state. This inversion is believed to occur because 1La transition has higher dipole moment (as it is directed through the ring– NH group), and can have dipole–dipole interactions with polar solvent molecules. Irrespective of whether 1La or 1Lb is the lowest S1 state, equilibration between these two states is believed to be very fast (of the order of 10−12 sec), so that only emission from the lower S1 state is observed (Ruggiero et al. 1990). In a motionally restricted polar environment, absorption at the red edge photoselects the lowest energy S1 (1La in this case), and thus the polarization is high because only depolarization due to small angular differences between the absorption and emission transition moments and solvent reorientation, if any, occurs. Excitation at the shorter wavelengths, however, populates both 1La and 1Lb states. Equilibration between these two states produces a depolarization due to the approximately 90° angular difference between 1La and 1Lb moments. Thus, near 290 nm, there is a sharp dip in polarization due to maximal absorption by the 1Lb state. Figure 4 ▶ shows such a characteristic dip around 290 nm in the excitation polarization spectrum of spectrin in both cases. Thus, the sharp increase in polarization toward the red edge of the absorption band is probably because the extent of depolarization in spectrin is reduced at the red edge not only due to decreased rotational rate of the fluorophore in the solvent relaxed state, but also due to photoselection of the predominantly 1La transition, which in turn, reduces the contribution to depolarization because of 1Lb → 1La equilibration.
For fluorophores incorporated in motionally restricted media, fluorescence polarization is also known to be dependent on emission wavelength. Under such conditions, a steady and significant decrease in polarization is observed with increasing emission wavelength (Mukherjee and Chattopadhyay 1995, and references therein). Figure 5 ▶ shows the variation in steady-state polarization of spectrin tryptophans in the native state as a function of wavelength across its emission spectrum. As seen from the figure, there is a considerable decrease in polarization with increasing emission wavelength for spectrin in its native state. The lowest polarization is observed toward the red edge where the solvent relaxed emission predominates. More interestingly, and in agreement with Figures 3 and 4 ▶ ▶, spectrin denatured with 8 M urea displays a pronounced variation in fluorescence polarization as a function of increasing emission wavelength, indicating that the rigidity of the environment around the tryptophans is maintained even after denaturation with urea. This result is in sharp contrast to our earlier studies where it was shown that fluorescence polarization of tryptophans in denatured proteins such as tubulin do not exhibit emission wavelength dependence and remain practically invariant (Guha et al. 1996). Taken together, these results reinforce the presence of ordered, motionally restricted solvent molecules in the vicinity of spectrin tryptophans, which are resistant to denaturation by urea.
Figure 5.

Fluorescence polarization of native (open circles) and denatured (filled circles) spectrin as a function of emission wavelength. The excitation wavelength was 280 nm in all cases. The data points shown are the means ± standard errors of at least three independent measurements. All other conditions are as in Figure 3 ▶. See Materials and Methods for other details.
Acrylamide quenching of spectrin tryptophan fluorescence
Acrylamide quenching of tryptophan fluorescence is widely used to monitor tryptophan environments in proteins (Eftink 1991b). Figure 6A ▶ shows a representative Stern-Volmer plot of acrylamide quenching of spectrin tryptophans in native and denatured state. The slope (KSV) of such a plot is related to the accessibility (degree of exposure) of the tryptophans. The quenching parameters obtained by analyzing the Stern-Volmer plot are shown in Table 1. The Stern-Volmer constant (KSV) for acrylamide quenching of native spectrin was found to be 4.40 M−1. The value of KSV was found to increase for spectrin denatured with 8 M urea to 8.60 M−1, indicating increased exposure upon denaturation. However, this is considerably low in comparison to the value of KSV for fully exposed tryptophans as in the case of N-acetyl-L-tryptophanamide (NATA; 13.42 M−1) in 8 M urea (see Table 1). We measured KSV for NATA in 8 M urea (as a control) for comparison with KSV of denatured spectrin because the bulk physical properties (such as viscosity) of the solution may change KSV. That this is actually the case is shown by the value of KSV of NATA in water, which is 17.52 M−1, as shown in Table 1 in agreement with an earlier report (Eftink and Ghiron 1976). The bimolecular quenching constants (kq) for acrylamide quenching, which are more accurate measures of the degree of exposure (because kq takes into account differences in fluorescence lifetime), are also shown in Table 1. The kq values are in overall agreement with Stern-Volmer constants implying that the conclusions derived by the analysis of Stern-Volmer constants are not influenced by changes in lifetime.
Figure 6.
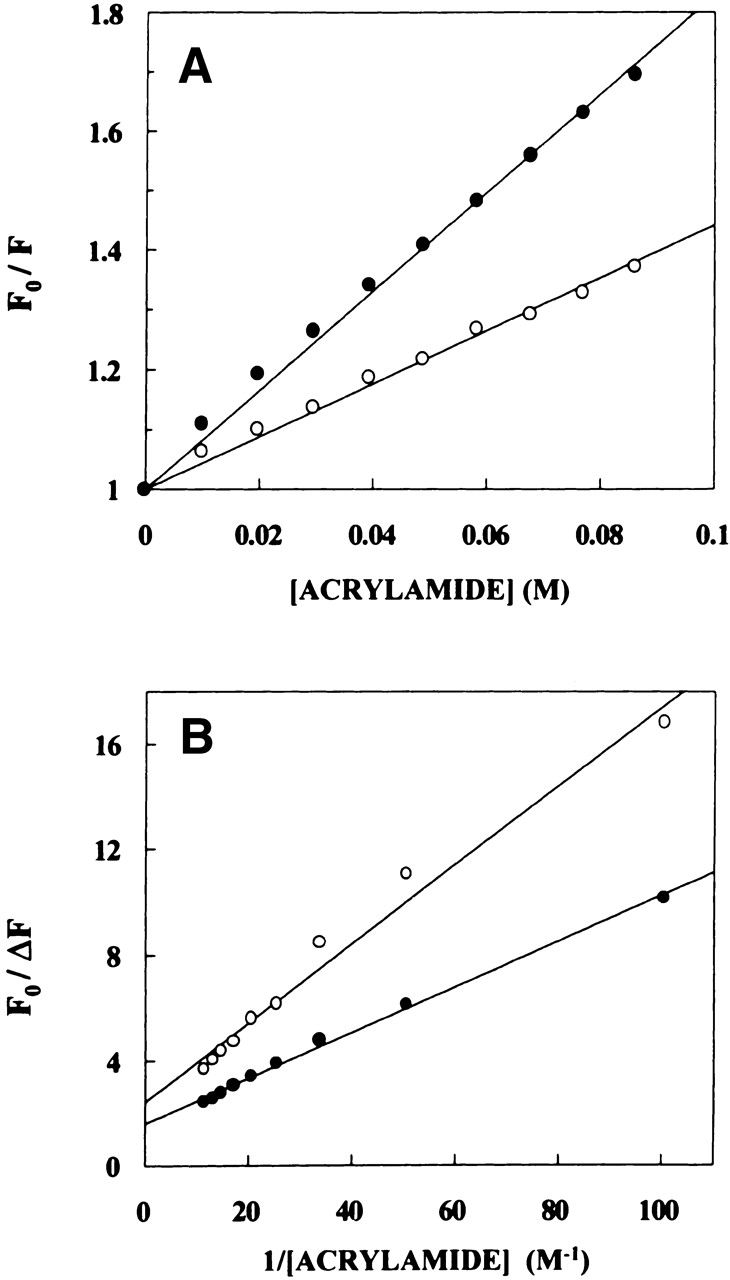
Representative data for (A) Stern-Volmer (B) Lehrer analysis of acrylamide quenching of native (open circles) and denatured (filled circles) spectrin fluorescence. Fo is the fluorescence in the absence of quencher, F is the corrected fluorescence in the presence of quencher, and δF = Fo − F. The concentration of spectrin used was 0.1 μM. The excitation wavelength was fixed at 295 nm, and emission was monitored at 337 nm in all cases. See Materials and Methods for other details.
Table 1.
Acrylamide quenching of tryptophan fluorescence
| System | KSVa (M−1) | kq (×10−9)b (M−1s−1) | fac |
| Spectrin (native) | 4.40 ± 0.04 | 1.29 | 0.40 ± 0.01 |
| Spectrin (denatured) | 8.60 ± 0.30 | 2.02 | 0.65 ± 0.04 |
| NATA (water) | 17.52 ± 0.30 | 5.84 | |
| NATA (8 M Urea) | 13.42 ± 0.30 | 3.33 |
The concentration of spectrin and NATA used was 0.1 and 33 μM, respectively.
a Calculated using equation 3. The quenching parameter shown represents mean ± standard error of three independent measurements while quenching data shown in Figure 6A ▶ are from representative experiments.
b Calculated using mean fluorescence lifetimes from Table 3 and using equation 3.
c Calculated using equation 4. The quenching parameter shown represents mean ± standard error of three independent measurements while quenching data shown in Figure 6B ▶ are from representative experiments.
The above results indicate that even when denatured with 8 M urea, the spectrin tryptophans are not fully exposed and are considerably shielded from the bulk aqueous environment. These results, along with the REES results for denatured spectrin mentioned earlier, reinforce our conclusion that the tryptophan microenvironment in spectrin is characterized by unique structural and dynamic features, which are maintained to a considerable extent even when denatured with 8 M urea. This is further supported by Lehrer analysis (Lehrer 1971) of the quenching data (see Fig. 6B ▶). The Lehrer analysis shows that only 65% of the tryptophan fluorescence was accessible (see Table 1) even upon denaturation indicating a significant fraction of spectrin tryptophans are not accessible to the aqueous quencher.
Wavelength-selective fluorescence of PRODAN bound to spectrin
As mentioned earlier, many of the tryptophan residues in spectrin are at or in the vicinity of hydrophobic patches in spectrin, which can bind hydrophobic ligands such as fatty acids and phospholipids and cause quenching of tryptophan fluorescence (Sikorski et al. 1987; Kahana et al. 1992). The hydrophobic patches are thought to be important for interaction of spectrin with membranes. To explore the organization and dynamics of these functionally important hydrophobic sites characterized by a low dielectric constant (Haque et al. 2000), we performed wavelength-selective fluorescence experiments utilizing the fluorescence of PRODAN bound to spectrin. It was previously shown that the hydrophobic, polarity-sensitive fluorescent probe PRODAN binds erythroid spectrin with a high affinity (Kapp = 2 × 106 M−1) and with a molar stoichiometry of 1 : 1 (Chakrabarti 1996). PRODAN and its derivatives have earlier been successfully used as hydrophobic markers to estimate the polarity of the heme-binding pocket in apomyoglobin (Macgregor and Weber 1986).
An important criterion for a fluorophore to be able to exhibit REES is that it should be polar; that is, it should have a permanent dipole moment in the ground state. In addition, there should be a change in the dipole moment upon excitation, so as to cause the solvent dipoles to reorient in response to this altered dipole moment to attain an energetically favorable orientation (Mukherjee and Chattopadhyay 1995). For a totally nonpolar fluorophore, there will be no change in dipole moment upon excitation, and the process of solvent reorientation becomes irrelevant, because it is the change in dipole moment that triggers the solvent reorientation. We have previously shown that the dipole moment of the 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) group changes by ∼4 Debye upon excitation (Mukherjee et al. 1994), and this contributes to REES effects of NBD-labeled lipids in membranes (Chattopadhyay and Mukherjee 1993, 1999a), micelles (Rawat et al. 1997; Rawat and Chattopadhyay 1999) and reverse micelles (Chattopadhyay et al. 2002). PRODAN fluorescence is very sensitive to the polarity of the environment. Further, its dipole moment changes by ∼5–8 Debye upon excitation (Balter et al. 1988; Samanta and Fessenden 2000). A change in dipole moment of this magnitude, along with its hydrogen bonding capability (Samanta and Fessenden 2000) makes PRODAN a suitable probe for REES effects to characterize the hydrophobic binding sites in spectrin.
The shifts in the maxima of fluorescence emission of PRODAN bound to spectrin as a function of excitation wavelength are shown in Figure 7 ▶. To avoid any complications due to ground state heterogeneity, we chose conditions in which PRODAN was completey bound to spectrin (Chakrabarti 1996). REES obtained under such conditions can therefore be attributed to the organization and dynamics of the hydrophobic site in spectrin where PRODAN binds. PRODAN shows an emission maximum of 520 nm in water (Chakrabarti 1996), which exhibits a blue shift of 90 nm when bound to spectrin, indicating the nonpolar environment of the binding site. Figure 7 ▶ shows that when excited at 360 nm, PRODAN bound to spectrin displays a blue-shifted emission maximum at 430 nm. As the excitation wavelength is changed from 360 nm to 410 nm, the emission maxima of spectrin-bound PRODAN is shifted from 430 nm to 439 nm, which amounts to a REES of 9 nm. Observation of REES for spectrin-bound PRODAN suggests that when bound to spectrin, PRODAN is in an environment where its mobility is considerably reduced. Because PRODAN binds to the hydrophobic site in spectrin, such a result would directly imply that this region of spectrin offers considerable restriction to the reorientational motion of the solvent dipoles around the excited state fluorophore.
Figure 7.

Effect of changing excitation wavelength on the wavelength of maximum emission for PRODAN bound to spectrin. The concentration of PRODAN was 0.5 μM, and that for spectrin was 1.5 μM. See Materials and Methods for other details.
In addition to the dependence of fluorescence emission maxima on the excitation wavelength, fluorescence polarization of PRODAN bound to spectrin also shows characteristic dependence on excitation and emission wavelengths. The excitation polarization spectra of PRODAN bound to spectrin (shown in Fig. 8 ▶) shows that the polarization of spectrin-bound PRODAN undergoes considerable change upon altering the excitation wavelength from 360 nm to 400 nm, with a sharp increase toward the red edge of the absorption band. Further, fluorescence polarization of PRODAN bound to spectrin demonstrates typical dependence on emission wavelength (shown in Fig. 9 ▶). The variation in steady-state polarization of PRODAN bound to spectrin as a function of emission wavelength from 420 nm to 460 nm across the emission spectrum of PRODAN is shown in Figure 9 ▶. As is apparent from the figure, there is a decrease in polarization with increasing emission wavelength. Spectrin-bound PRODAN thus shows all the signatures of a restricted fluorophore in a motionally confined region of the protein (Mukherjee and Chattopadhyay 1995).
Figure 8.

Fluorescence polarization of PRODAN bound to spectrin as a function of excitation wavelength. Polarization values were recorded keeping emission wavelength constant at 433 nm. The data points shown are the means ± standard errors of at least three independent measurements. All other conditions are as in Figure 7 ▶. See Materials and Methods for other details.
Figure 9.

Fluorescence polarization of PRODAN bound to spectrin as a function of emission wavelength. The excitation wavelength was 360 nm in all cases. The data points shown are the means ± standard errors of at least three independent measurements. All other conditions are as in Figure 7 ▶. See Materials and Methods for other details.
Time-resolved fluorescence of spectrin tryptophans in native and denatured states and PRODAN bound to spectrin
Fluorescence lifetime serves as a sensitive indicator for the local environment and polarity in which a given fluorophore is placed (Prendergast 1991). Table 2 shows the tryptophan lifetimes for spectrin in native and denatured states. As can be seen from the table, all the fluorescence decays for tryptophans in native and denatured spectrin fit a biexponential function. The mean fluorescence lifetimes of spectrin tryptophans were calculated using equation 6 and are shown in Table 3.
Table 2.
Lifetimes of spectrin tryptophans, NATA, and PRODAN
| System | α1 | τ1 (ns) | α2 | τ2 (ns) |
| Spectrina (native) | 0.73 | 1.48 | 0.27 | 4.98 |
| Spectrina (denatured) | 0.82 | 2.85 | 0.18 | 6.91 |
| NATAb (water) | 1.00 | 3.00 | ||
| NATAb (8 M Urea) | 1.00 | 4.03 | ||
| PRODANc (water) | 1.00 | 1.54 | ||
| PRODANd (bound to spectrin) | 0.77 | 4.41 | 0.23 | 8.08 |
The concentration of native and denatured spectrin was 0.6 and 0.4 μM, respectively. The concentration of NATA was 100 μM. PRODAN in water was used at a concentration of 1 μM. The concentration of PRODAN was 0.5 μM and spectrin was 1.5 μM for PRODAN bound to spectrin.
a Excitation at 297 nm; emission monitored at 340 nm.
b Excitation at 297 nm; emission monitored at 330 nm.
c Excitation at 360 nm; emission monitored at 520 nm.
d Excitation at 360 nm; emission monitored at 430 nm.
Table 3.
Mean fluorescence lifetimes for spectrin tryptophans, NATA and PRODAN
| System | Mean fluorescence lifetime (ns) |
| Spectrin (native) | 3.42 |
| Spectrin (denatured) | 4.26 |
| NATA (water) | 3.00 |
| NATA (8 M Urca) | 4.03 |
| PRODAN (water) | 1.54 |
| PRODAN (bound to spectrin) | 5.71 |
Mean fluorescence lifetimes were calculated using equation 6.
In general, tryptophan lifetimes are known to be reduced when exposed to polar environments (Kirby and Steiner 1970; Ho and Stubbs 1992). Because denaturation of a folded protein usually involves exposure of some of the otherwise buried tryptophans to water, it may be expected that the mean lifetimes for tryptophans in native proteins would show a reduction when denatured. Based on such consideration alone, a lower value for mean fluorescence lifetime would be expected for tryptophans in the denatured state compared to the value obtained in the native state. However, there are other factors that need to be considered while interpreting changes in fluorescence lifetime upon denaturation. An important factor is the viscosity of the medium, which increases considerably when denaturation is carried out in the presence of 8 M urea. An increase in medium viscosity generally brings about an increase in fluorescence lifetime (Tredwell and Keary 1979; Krishna and Periasamy 1998). This would, therefore, lead to an increase in lifetime upon denaturation by 8 M urea. In a control experiment, we performed time-resolved fluorescence measurements of the model tryptophan derivative NATA in water and in 8 M urea. The fluorescence lifetimes of NATA in these conditions are shown in Table 3. The lifetime of NATA is found to be ∼34% more in 8 M urea when compared to the value in water. This increase in lifetime could be attributed to the increased solution viscosity of 8 M urea. Interestingly, the mean fluorescence lifetime for spectrin tryptophans is longer (by ∼25%) in denatured spectrin (4.26 ns) than what is obtained in native spectrin (3.42 ns). This could be due to a combination of the two factors (exposure to polar environment and increase in solution viscosity) mentioned above. The mean fluorescence lifetimes of a number of representative proteins in the native and denatured state has earlier been analyzed by Swaminathan et al. (1994). The results show that the mean fluorescence lifetime may either increase or decrease upon denaturation.
Moreover, lifetime of protein tryptophans in the folded state has previously been shown to be shorter due to local interactions involving various residues, which are spatially close in the native conformation of the protein. This has been shown to be true for the hemolytic peptide melittin in which the steric interaction between the closely spaced Trp-19 and Lys-23 is known to shorten the tryptophan lifetime (Weaver et al. 1992). An alternate explanation for the increased tryptophan lifetime in denatured spectrin could still be loss of such local interactions upon denaturation resulting in an increased lifetime.
Table 2 also shows the lifetimes of PRODAN in water and when bound to spectrin. The fluorescence decay of PRODAN in water fits to a monoexponential function with a lifetime of 1.54 ns in agreement with previous literature value (Krasnowska et al. 1998). The fluorescence decay of PRODAN bound to spectrin, on the other hand, fits to a biexponential function. The mean fluorescence lifetimes of spectrin-bound PRODAN was calculated using equation 6 and is shown in Table 3. As can be seen from the table, the mean lifetime of PRODAN is increased to 5.71 ns upon binding to spectrin. We attribute this increase in mean fluorescence lifetime of PRODAN to the nonpolar nature of the binding site because fluorescence lifetime of PRODAN is known to increase with decreasing polarity (Krasnowska et al. 1998; Sengupta et al. 2000). This reinforces our earlier conclusion that PRODAN binding site in spectrin is very hydrophobic (Chakrabarti 1996), characterized by a low dielectric constant (Haque et al. 2000).
Discussion
The dynamic properties of the protein matrix surrounding a given amino acid residue or a fluorophore either covalently attached or partitioned from the aqueous phase can be examined from the rate at which this matrix responds to (or relaxes around) the newly created excited state dipole moment of the fluorophore. In other words, the magnitude of REES could be utilized to estimate the relative rigidity of the region of the protein surrounding the fluorophore. Early pioneering work in the application of this effect to proteins was carried out by Demchenko and coworkers (Demchenko 1988b, 2002). In one of their early studies (Demchenko 1982), it was shown that the fluorescence emission spectra of 2-(p-toluidinylnaphthalene)-6-sulfonate (TNS) associated with proteins such as β-lactoglobulin, β-casein, and bovine and human serum albumins depend on the excitation wavelength used, giving rise to REES of the order of 10 nm in all cases. The fact that this effect was actually a result of the slow rate of solvent dipolar relaxation was confirmed when a similar effect was observed in case of the same fluorophore in glucose glass, and in glycerol at 1°C, but not in liquid solutions (Demchenko 1982). Such red edge effects have also been observed with other proteins bound to TNS (Lakowicz and Keating-Nakamoto 1984; Demchenko 1985; Albani 1992), 2′-(N,N-dimethyl)amino-6-naphthoyl-4-trans-cyclohexanoic acid (DANCA; Macgregor and Weber 1986), or NBD (MacPhee et al. 1999) as well as utilizing the intrinsic tryptophan fluorescence of proteins and peptides (Demchenko 1988a; Wasylewski et al. 1992; Guha et al. 1996; Pattnaik et al. 1997; Santos et al. 1998; Raja et al. 1999). Very fast solvent relaxation (femtosecond) has recently been monitored in proteins (Pal et al. 2002).
In this article, we have utilized the wavelength-selective fluorescence approach to monitor the organization and dynamics of the functionally important tryptophan residues of erythroid spectrin in native and denatured conditions and of PRODAN bound to spectrin. We observe a REES of 4 nm for the tryptophans in the case of native spectrin. This result indicates that the tryptophans in native dimeric spectrin are localized in a motionally restricted environment and that the regions surrounding at least some of the spectrin tryptophans offer considerable restriction to the reorientational motion of the solvent (water) dipoles around the excited state tryptophans. This is in agreement with an earlier report (Subbarao and MacDonald 1994) in which it was suggested that the tryptophan environments in spectrin are heterogeneous, and there are significant numbers of tryptophans that are in crevices that are shielded from bulk water. Although it is difficult to accurately estimate the number of such tryptophan residues, it is very likely that these are the tryptophans contributing to the REES of spectrin due o the presence of restricted water molecules in their vicinity.
It has previously been shown that tryptophan residues in denatured proteins do not show REES, because the tryptophans are exposed to water and, therefore, they do not offer any restriction to the solvent (water) dipoles around them in the excited state (Demchenko 1988a). For example, we have shown this to be true for the cytoskeletal protein tubulin in 8 M urea whose emission wavelength remains constant at 353 nm when excited in the wavelength range of 280–310 nm (Guha et al. 1996). This is particularly significant because tubulin is also a large protein (such as spectrin), and has multiple tryptophan residues. It is against this backdrop that our observation of REES of denatured spectrin assumes significance. Our results show that tryptophans in spectrin denatured with urea, show a REES of 3 nm, suggesting that the tryptophans are shielded from bulk solvent even when denatured. The tryptophan microenvironment in spectrin, therefore, is characterized by unique structural and dynamic features that are maintained to a significant extent even when denatured with urea. As mentioned earlier, these results therefore represent the first report of a denatured protein exhibiting REES. In view of the fact that REES of protein tryptophans was first described more than 2 decades ago for human serum albumin (Demchenko 1981; recently reviewed in Demchenko 2002), and no work has yet claimed REES of a denatured protein, this represents a major leap in this area.
This above conclusion is reinforced by acrylamide quenching experiments in which the accessibilities of the spectrin tryptophans are determined in native and denatured conditions. Experiments in which fluorescence polarization of spectrin was monitored as a function of excitation and emission wavelengths provide data supporting this general conclusion. The importance of these results is brought out by the observation that tryptophan residues have earlier been shown to be important for folding (MacDonald et al. 1994) and thermodynamic stability (Pantazatos and MacDonald 1997) of spectrin repeats. Interestingly, it has very recently been reported that tryptophan residues can stabilize native-like structures in a denatured protein (Klein-Seetharaman et al. 2002). This points out the crucial structural role played by water molecules in the overall stability of the spectrin as has been shown earlier for other proteins (Fischer et al. 1994; Kandori et al. 1995; Sankararamakrishnan and Sansom 1995; Nishimura et al. 1997). These results assume special significance in light of the fact that interaction of water molecules with cytoskeletal elements has been recently implicated as a major factor in maintaining the organization and function of the cytoskeleton (Leterrier 2001).
The hydrophobic, polarity-sensitive fluorescent probe PRODAN binds erythroid spectrin with a high affinity (Chakrabarti 1996). When bound to spectrin, PRODAN exhibits a REES of 9 nm, indicating that PRODAN is in an environment where its mobility is considerably reduced. Because PRODAN binds to the hydrophobic site in spectrin, such a result would directly imply that this region of spectrin offers considerable restriction to the reorientational motion of the solvent dipoles around the excited state fluorophore. These results are supported by studies in which fluorescence polarization of spectrin-bound PRODAN was monitored as a function of excitation and emission wavelengths. These hydrophobic sites could play a pivotal role in the recently reported chaperone-like activity of spectrin (Chakrabarti and Bhattacharya 1999; Chakrabarti et al. 2001) because one of the primary roles of a chaperone is to prevent the aggregation of denatured proteins via exposed hydrophobic sites.
It has been realized for some time now that denatured proteins often have a significant amount of residual structure and play an important role in folding and stability of proteins (Dill and Shortle 1991; Shortle 1993; Swaminathan et al. 1994; Garcia et al. 1998). Denatured states of proteins are of interest not only in the context of the energetics of protein folding but also for their potential contribution to the understanding of the functional aspects of the proteins. The denatured state of a protein appears to be the form recognized by chaperones, by protein complexes that initiate transport across biological membranes, and by a variety of protease systems responsible for intracellular protein turnover (Dill and Shortle 1991; Shortle 1993). As a result, a comprehensive understanding of these cellular processes requires some degree of structural information about the nonnative states of proteins encountered in these situations. In this context, the recently reported group of proteins, termed "natively unfolded proteins" (Uversky 2002), which have disordered structures in their native states assume importance. It is for this reason that new approaches have recently been developed to monitor structural details of denatured proteins. Recent advances in NMR spectroscopy is particularly useful in this regard because some of these methods do not rely on the presence of stable structure (Neri et al. 1992; Bhavesh et al. 2001).
The residual structural integrity that remains even after spectrin is treated with denaturing concentrations of urea can be considered as a hallmark of a cytoskeletal protein whose main function is to provide a stable scaffold to the cell membrane. The residual structure could serve as a nucleation point for refolding of spectrin. In fact, it has recently been shown that urea-denatured peptide repeats of spectrin can be renatured suggesting that unfolding could be reversed (MacDonald and Pozharski 2001). In summary, our results provide novel information into the dynamics of spectrin tryptophans and hydrophobic site, which could be important in protein–protein interaction and signal transduction (Ziemnicka-Kotula et al. 1998) and interaction of spectrin with membranes (Beck and Nelson 1996; Ray and Chakrabarti 2003).
Materials and methods
Materials
Tris, KCl, phenylmethylsulfonyl fluoride (PMSF), dithiothreitol (DTT), SDS, EDTA, NATA, and ultrapure grade urea were from Sigma Chemical Company. Ultrapure grade acrylamide was from Invitrogen Life Technologies. PRODAN was purchased from Molecular Probes. The purity of acrylamide was checked from its absorbance using its molar extinction coefficient (ɛ) of 0.23 M−1cm−1 at 295 nm and optical transparency beyond 310 nm (Eftink 1991c). Concentration of PRODAN in methanolic stock solution was calculated from its molar extinction coefficient (ɛ) of 18,000 M−1cm−1 at 360 nm (Weber and Farris 1979). All other chemicals used were of the highest purity available. Solvents used were of spectroscopic grade. Water was purified through a Millipore Milli-Q system and used throughout.
Isolation and purification of spectrin
Clean, white ghosts from goat blood were prepared by hypotonic lysis in 5 mM phosphate, 1 mM EDTA containing 20 μg/mL of PMSF at pH 8.0 (lysis buffer) following the procedure of Dodge and coworkers (Dodge et al. 1963). Spectrin dimers were purified as described earlier (Dodge et al. 1963; Gratzer 1985). After washing the membranes thoroughly in lysis buffer, the band 6-depleted ghosts were resuspended in 20 volumes of spectrin removal buffer (0.2 mM sodium phosphate, 0.1 mM EDTA, 0.2 mM DTT, 20 μg/mL PMSF, pH 8.0) and incubated at 37°C for 30 min. Crude spectrin was collected in the supernatant after centrifugation. Spectrin was then purified after concentration by 30% ammonium sulfate precipitation followed by chromatography on Sepharose CL-4B. Spectrin was stored in buffer containing 5 mM sodium phosphate, 1 mM EDTA, 20 mM KCl, and 0.2 mM DTT, pH 8.0. Before performing any experiments involving fluorescence, spectrin was dialyzed extensively against buffer containing 10 mM Tris, 20 mM NaCl, pH 7.8 to remove DTT. The purity of the preparation was checked by 7.5% SDS polyacrylamide gel electrophoresis under reducing conditions showing the characteristic bands of spectrin dimer (α chain of 240 and β chain of 220 kD) after Coomassie blue staining. Concentration of spectrin was determined spectrophotometrically using an absorbance of 10.7 at 280 nm for 1% spectrin solution (Gratzer 1985) and by Lowry’s method (Lowry et al. 1951).
Sample preparations and steady-state fluorescence measurements
Spectrin solution was made in 5 mM sodium phosphate, 1 mM EDTA, 20 mM KCl, pH 8.0. Spectrin was denatured in 8 M urea by incubating spectrin in urea solution for 2 h prior to spectroscopic measurements. For studies on spectrin-bound PRODAN, PRODAN was incubated for 3 h with three times molar excess of spectrin.
Steady-state fluorescence measurements were performed with a Hitachi F-4010 spectrofluorometer using 1-cm pathlength quartz cuvettes. Excitation and emission slits with a nominal bandpass of 5 nm were used for all measurements. All spectra were recorded using the correct spectrum mode. Background intensities of samples in which fluorophores were omitted were negligible in most cases and were subtracted from each sample spectrum to cancel out any contribution due to the solvent Raman peak and other scattering artifacts. Fluorescence polarization measurements were performed using an Hitachi polarization accessory. Polarization values were calculated from the equation (Chen and Bowman 1965):
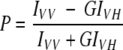 |
(1) |
where IVV and IVH are the measured fluorescence intensities (after appropriate background subtraction) with the excitation polarizer vertically oriented and emission polarizer vertically and horizontally oriented, respectively. G is the grating correction factor and is equal to IHV/IHH. All experiments were done with multiple sets of samples and average values of polarization are shown in the figures. The standard errors in all cases were less than 3%. The spectral shifts obtained with different sets of samples were identical in most cases. In other cases, the values were within ±1 nm of the ones reported.
Fluorescence quenching measurements
Acrylamide quenching experiments of tryptophan fluorescence were carried out by measurement of fluorescence intensity after serial addition of small aliquots of freshly prepared stock solution of 2 M acrylamide in water to a stirred sample followed by incubation for 3 min in the sample compartment in the dark. For samples containing spectrin, the excitation wavelength used was 295 nm, and emission was monitored at 337 nm. The excitation wavelength used was 295 nm, and emission was collected as 350 nm for NATA. The fluorescence intensities obtained were corrected for dilution. Corrections for inner filter effect were made using the following equation (Lakowicz 1999):
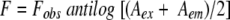 |
(2) |
where F is the corrected fluorescence intensity and Fobs is the background subtracted fluorescence intensity of the sample (also corrected for dilution). Aex and Aem are the measured absorbance at the excitation and emission wavelengths. The absorbances of the samples were measured using a Hitachi U-2000 UV-visible absorption spectrophotometer. Quenching data were analyzed by fitting to the Stern-Volmer equation (Lakowicz 1999):
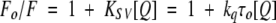 |
(3) |
where Fo and F are the fluorescence intensities in the absence and presence of the quencher, respectively, [Q] is the molar quencher concentration and KSV is the Stern-Volmer quenching constant. The Stern-Volmer quenching constant KSV is equal to kqτo, where kq is the bimolecular quenching constant and τo is the lifetime of the fluorophore in the absence of quencher. To quantitate the fraction of fluorophores accessible to the quencher, the quenching data were also analyzed using the modified equation by Lehrer (1971):
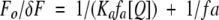 |
(4) |
where δF = Fo − F, Ka is the Stern-Volmer quenching constant for the accessible fraction, and fa is the fraction of initial fluorescence accessible to the quencher. Acrylamide quenching of NATA fluorescence was analyzed as described previously (Eftink and Ghiron 1976).
Time-resolved fluorescence measurements
Fluorescence lifetimes were calculated from time-resolved fluorescence intensity decays using a Photon Technology International LS-100 luminescence spectrophotometer in the time-correlated single photon counting mode. This machine uses a thyratron-gated nanosecond flash lamp filled with nitrogen as the plasma gas (16 ± 1 inches of mercury vacuum) and is run at 22–25 kHz. Lamp profiles were measured at the excitation wavelength using Ludox (colloidal silica) as the scatterer. To optimize the signal to noise ratio, 5000 photon counts were collected in the peak channel. The excitation wavelengths used were 297 nm for tryptophans and 360 nm for PRODAN, which correspond to peaks in the spectral output of the nitrogen flash lamp. Emission wavelength was set at 340 nm for spectrin tryptophans and 330 nm for NATA. Emission was monitored at 430 nm for PRODAN bound to spectrin while the emission wavelength was set at 520 nm for free PRODAN. All experiments were performed using excitation and emission slits with a nominal bandpass of 4 nm or less. The sample and the scatterer were alternated after every 10% acquisition to ensure compensation for shape and timing drifts occurring during the period of data collection. The data stored in a multichannel analyzer was routinely transferred to an IBM PC for analysis. Intensity decay curves so obtained were fitted as a sum of exponential terms:
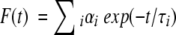 |
(5) |
where αi is a preexponential factor representing the fractional contribution to the time-resolved decay of the component with a lifetime τi. The decay parameters were recovered using a nonlinear least-squares iterative fitting procedure based on the Marquardt algorithm (Bevington 1969). The program also includes statistical and plotting subroutine packages (O’Connor and Phillips 1984). The goodness of the fit of a given set of observed data and the chosen function was evaluated by the reduced χ2 ratio, the weighted residuals (Lampert et al. 1983), and the autocorrelation function of the weighted residuals (Grinvald and Steinberg 1974). A fit was considered acceptable when plots of the weighted residuals and the autocorrelation function showed random deviation about zero with a minimum χ2 value not more than 1.5. Mean (average) lifetimes ≤τ> for biexponential decays of fluorescence were calculated from the decay times and preexponential factors using the following equation (Lakowicz 1999):
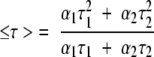 |
(6) |
Circular dichroism (CD) measurement
Far-UV CD measurements of the native and denatured spectrin were carried out at room temperature (25°C) on a JASCO J-720 spectropolarimeter, which was calibrated with (+)-10-camphorsulfonic acid (Chen and Yang 1977). The spectra were scanned in a cylindrical quartz optical cell with a pathlength of 0.1 cm. All spectra were recorded in 0.5-nm wavelength increments with a 2-sec response and a band width of 1 nm using a scan rate of 20 nm/min. Each spectrum is the average of five continuous scans. All spectra were corrected for background by subtraction of appropriate blanks without spectrin. The spectra were subjected to a moderate degree of noise-reduction analysis by smoothing, making sure that the overall shape of the spectrum remains unaltered (Chakrabarti and Basak 1996; Haque et al. 1999).
Acknowledgments
This work was supported by a grant (00-141 RG/BIO/AS) from The Third World Academy of Sciences, Trieste, Italy, to A.C. and by the Council of Scientific and Industrial Research and Department of Atomic Energy, Government of India. S.S.R. and S.R. thank the Council of Scientific & Industrial Research, Government of India, for the award of Senior Research Fellowships. D.A.K. thanks the University Grants Commission for the award of a Senior Research Fellowship. We thank anonymous reviewers for helpful suggestions and H. Raghuraman for useful discussions. We thank Y.S.S.V. Prasad and G.G. Kingi for technical help and members of the Chattopadhyay laboratory for critically reading the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DANCA, 2′-(N,N-dimethyl)amino-6-naphthoyl-4-trans-cyclohexanoic acid
DTT, dithiothreitol
EDTA, ethylenediaminetetraacetic acid
NATA, N-acetyl-L-tryptophanamide
NBD, 7-nitrobenz-2-oxa-1,3-diazol-4-yl
PMSF, phenylmethylsulfonyl fluoride
PRODAN, 6-propionyl-2-dimethylaminonaphthalene
REES, red edge excitation shift
SDS, sodium dodecyl sulfate
TNS, 2-(p-toluidinylnaphthalene)-6-sulfonate
TRIS, tris-(hydroxymethyl)aminomethane
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03302003.
References
- Albani, J. 1992. Motions studies of the human α 1-acid glycoprotein (orosomucoid) followed by red-edge excitation spectra and polarization of 2-p-toluidinylnaphthalene-6-sulfonate (TNS) and of tryptophan residues. Biophys. Chem. 44 129–137. [DOI] [PubMed] [Google Scholar]
- Albinsson, B. and Norden, B. 1992. Excited-state properties of the indole chromophore: Electronic transition moment directions from linear dichroism measurements: Effect of methyl and methoxy substituents. J. Phys. Chem. 96 6204–6212. [Google Scholar]
- Balter, A., Nowak, W., Pawelkiewicz, W., and Kowalczyk, A. 1988. Some remarks on the interpretation of the spectral properties of prodan. Chem. Phys. Lett. 143 565–570. [Google Scholar]
- Beck, K.A. and Nelson, W.J. 1996. The spectrin-based membrane skeleton as a membrane protein-sorting machine. Am. J. Physiol. 270 C1263–C1270. [DOI] [PubMed] [Google Scholar]
- Bennett, V. and Gilligan, M.D. 1993. The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annu. Rev. Cell Biol. 9 27–66. [DOI] [PubMed] [Google Scholar]
- Bevington, P.R. 1969. Data reduction and error analysis for the physical sciences. McGraw-Hill, New York.
- Bhavesh, N.S., Panchal, S.C., and Hosur, R.V. 2001. An efficient high-throughput resonance assignment procedure for structural genomics and protein folding research by NMR. Biochemistry 40 14727–14735. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, A. 1996. Fluorescence of spectrin-bound prodan. Biochem. Biophys. Res. Commun. 226 495–497. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, A. and Basak, S. 1996. Structural alterations of horseradish peroxidase in the presence of low concentrations of guanidinium chloride. Eur. J. Biochem. 241 462–467. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, A. and Bhattacharya, S. 1999. Spectrin exhibits chaperone-like activity. Curr. Sci. 77 812–813. [Google Scholar]
- Chakrabarti, A., Bhattacharya, S., Ray, S., and Bhattacharyya, M. 2001. Binding of a denatured heme protein and ATP to erythroid spectrin. Biochem. Biophys. Res. Commun. 282 1189–1193. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A. 2002. Application of the wavelength-selective fluorescence approach to monitor membrane organization and dynamics. In Fluorescence: Spectroscopy, imaging and probes (eds. R. Kraayenhof et al.), pp. 211–224. Springer-Verlag, Heidelberg, Germany.
- ———. 2003. Exploring membrane organization and dynamics by the wavelength-selective fluorescence approach. Chem. Phys. Lipids 122 3–17. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A. and Mukherjee, S. 1993. Fluorophore environments in membrane-bound probes: A red edge excitation shift study. Biochemistry 32 3804–3811. [DOI] [PubMed] [Google Scholar]
- ———. 1999a. Red edged excitation shift of a deeply embedded membrane probe: Implications in water penetration in the bilayer J. Phys. Chem. B 103 8180–8185. [Google Scholar]
- ———. 1999b. Depth-dependent solvent relaxation in membranes: Wavelength-selective fluorescence as a membrane dipstick. Langmuir 15 2142–2148. [Google Scholar]
- Chattopadhyay, A. and Rukmini, R. 1993. Restricted mobility of the sole tryptophan in membrane-bound mellitin. FEBS Lett. 335 341–344. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A., Mukherjee, S., Rukmini, R., Rawat, S.S., and Sudha, S. 1997. Ionization partitioning, and dynamics of tryptophan octyl ester: Implications for membrane-bound tryptophan residues. Biophys. J. 73 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay, A., Mukherjee, S., and Raghuraman, H. 2002. Reverse micellar organization and dynamics: A wavelength-selective fluorescence approach. J. Phys. Chem. B 106 13002–13009. [Google Scholar]
- Chen, R.F. and Bowman, R.L. 1965. Fluorescence polarization: Measurement with ultraviolet-polarizing filters in a spetrophotofluorometer. Science 147 729–732. [DOI] [PubMed] [Google Scholar]
- Chen, G.C. and Yang, J.T. 1977. Two-point calibration of circular dichrometer with d-10-camphorsulfonic acid. Anal. Lett. 10 1195–1207. [Google Scholar]
- Delaunay, J. and Dhermy, D. 1993. Mutations involving the spectrin heterodimer contact site: Clinical expression and alterations in specific function. Semin. Hematol. 30 21–33. [PubMed] [Google Scholar]
- De Matteis, M.A. and Morrow, J.S. 2000. Spectrin tethers and mesh in the biosynthetic pathway. J. Cell Sci. 113 2331–2343. [DOI] [PubMed] [Google Scholar]
- Demchenko, A.P. 1981. Dependence of human serum albumin fluorescence spectrum on excitation wavelength. Ukr. Biochim. Zh. 53 22–27. [PubMed] [Google Scholar]
- ———. 1982. On the nanosecond mobility in proteins. Edge excitation fluorescence red shift of protein-bound 2-(p-toluidinylnaphthalene)-6-sulfonate. Biophys. Chem. 15 101–109. [DOI] [PubMed] [Google Scholar]
- ———. 1985. Fluorescence molecular relaxation studies of protein dynamics: The probe binding site of melittin is rigid on the nanosecond time scale. FEBS Lett. 182 99–102. [Google Scholar]
- ———. 1988a. Red-edge-excitation fluorescence spectroscopy of single-tryptophan proteins. Eur. Biophys. J. 16 121–129. [DOI] [PubMed] [Google Scholar]
- ———. 1988b. Site-selective excitation: A new dimension in protein and membrane spectroscopy. Trends Biochem. Sci. 13 374–377. [DOI] [PubMed] [Google Scholar]
- ———. 2002. The red-edge effects: 30 years of exploration. Luminescence 17 19–42. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. and Shortle, D. 1991. Denatured states of proteins. Annu. Rev. Biochem. 60 795–825. [DOI] [PubMed] [Google Scholar]
- Dodge, J.T., Mitchell, C., and Hanahan, D.J. 1963. The preparation and chemical characteristics of hemoglobin free ghosts of human erythrocytes. Arch. Biochem. Biophys. 100 119–130. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1991a. Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 35 127–205. [DOI] [PubMed] [Google Scholar]
- ———. 1991b. Fluorescence quenching: Theory and applications. In Topics in fluorescence spectroscopy (ed. J.R. Lakowicz), Vol. 2, pp. 53–126. Plenum Press, New York.
- ———. 1991c. Fluorescence quenching reactions: Probing biological macromolecular structure. In Biophysical and biochemical aspects of fluorescence spectroscopy (ed. T.G. Dewey), pp. 1–41. Plenum Press, New York.
- Eftink, M.R. and Ghiron, C.A. 1976. Fluorescence quenching of indole and model micelle systems. J. Phys. Chem. 80 486–493. [Google Scholar]
- Fischer, W.B., Sonar, S., Marti, T., Khorana, H.G., and Rothschild, K.J. 1994. Detection of a water molecule in the active-site of bacteriorhodopsin: Hydrogen bonding changes during the primary photoreaction. Biochemistry 33 12757–12762. [DOI] [PubMed] [Google Scholar]
- Gallagher, P.G., Petruzzi, M.J., Weed, S.A., Zhang, Z., Marchesi, S.L., Mohandas, N., Morrow, J.S., and Forget, B.G. 1997. Mutation of a highly conserved residue of βI spectrin associated with fatal and near-fatal neonatal hemolytic anemia. J. Clin. Invest. 99 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galley, W.C. and Purkey, R.M. 1970. Role of heterogeneity of the solvation site in electronic spectra in solution. Proc. Natl. Acad. Sci. 67 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, P., Mérola, F., Receveur, V., Blandin, P., Minard, P., and Desmadril, M. 1998. Steady state and time-resolved fluorescence study of residual structures in an unfolded form of yeast phosphoglycerate kinase. Biochemistry 37 7444–7455. [DOI] [PubMed] [Google Scholar]
- Ghosh, A., Rukmini, R., and Chattopadhyay, A. 1997. Modulation of tryptophan environment in membrane-bound melittin by negatively charged phospholipids: Implications in membrane organization and function. Biochemistry 36 14291–14305. [DOI] [PubMed] [Google Scholar]
- Gratzer, W.B. 1985. Preparation of spectrin. Methods Enzymol. 85 745–480. [DOI] [PubMed] [Google Scholar]
- Grinvald, A. and Steinberg, I.Z. 1974. On the analysis of fluorescence decay kinetics by the method of least-squares. Anal. Biochem. 59 583–598. [DOI] [PubMed] [Google Scholar]
- Grum, V.L., Dongning, L., MacDonald, R.I., and Mondragón, A. 1999. Structures of two repeats of spectrin suggest models of flexibility. Cell 98 523–535. [DOI] [PubMed] [Google Scholar]
- Guha, S., Rawat, S.S., Chattopadhyay, A., and Bhattacharyya, B. 1996. Tubulin conformation and dynamics: A red edge excitation shift study. Biochemistry 35 13426–13433. [DOI] [PubMed] [Google Scholar]
- Haque, M.E., Debnath, D., Basak, S., and Chakrabarti, A. 1999. Structural changes of horseradish peroxidase in presence of low concentrations of urea. Eur. J. Biochem. 259 269–274. [DOI] [PubMed] [Google Scholar]
- Haque, M.E., Ray, S., and Chakrabarti, A. 2000. Polarity estimate of the hydrophobic binding sites in erythroid spectrin: A study by pyrene fluorescence. J. Fluorescence 10 1–6. [Google Scholar]
- Haussinger, D. 1996. The role of cellular hydration in the regulation of cell function. Biochem. J. 313 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, C. and Stubbs, C.D. 1992. Hydration at the membrane protein–lipid interface. Biophys. J. 63 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahana, E., Pinder J.C., Smith, K.S., and Gratzer, W.B. 1992. Fluorescence quenching of spectrin and other red cell membrane cytoskeletal proteins. Relation to hydrophobic binding sites. Biochem. J. 282 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandori, H., Yamazaki, Y., Sasaki, J., Needleman, R., Lanyi, J.K., and Maeda, A. 1995. Water-mediated proton transfer in proteins: An FTIR study of Bacteriorhodopsin. J. Am. Chem. Soc. 117 2118–2119. [Google Scholar]
- Kelkar, D.A., Ghosh, A., and Chattopadhyay, A. 2003. Modulation of fluorophore environment in host membranes of varying charge. J. Fluorescence (in press).
- Kirby, E.P. and Steiner, R.F. 1970. The influence of solvent and temperature upon fluorescence of indole derivatives. J. Phys. Chem. 74 4480–4490. [Google Scholar]
- Klein-Seetharaman, J., Oikawa, M., Grimshaw, S.B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L.J., Dobson, C.M., and Schwalbe, H. 2002. Long-range interactions within a nonnative protein. Science 295 1719–1722. [DOI] [PubMed] [Google Scholar]
- Krasnowska, E.K., Gratton, E., and Parasassi, T. 1998. Prodan as a membrane surface fluorescence probe: Partitioning between water and phospholipid phases. Biophys. J. 74 1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, M.M.G. and Periasamy, N. 1998. Fluorescence of organic dyes in lipid membranes: Site of solubilization and effects of viscosity and refractive index on lifetimes. J. Fluorescence 8 81–91. [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy. Kluwer-Plenum, New York.
- Lakowicz, J.R. and Keating-Nakamoto, S. 1984. Red-edge excitation of fluorescence and dynamic properties of proteins and membranes. Biochemistry 23 3013–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert, R.A., Chewter, L.A., Phillips, D., O’Connor, D.V., Roberts, A.J., and Meech, S.R. 1983. Standards for nanosecond fluorescence decay time measurements. Anal. Chem. 55 68–73. [Google Scholar]
- Lehrer, S.S. 1971. Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10 3254–3263. [DOI] [PubMed] [Google Scholar]
- Leterrier, J.-F. 2001. Water and the cytoskeleton. Cell. Mol. Biol. 47 901–923. [PubMed] [Google Scholar]
- Lowry, O.H., Rosebrough, N.J., Farr, A.L., and Randall, R.J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 265– 275. [PubMed] [Google Scholar]
- MacDonald, R.I. and Pozharski, E.V. 2001. Free energies of urea and of thermal unfolding show that two tandem repeats of spectrin are thermodynamically more stable than a single repeat. Biochemistry 40 3974–3984. [DOI] [PubMed] [Google Scholar]
- MacDonald, R.I., Musacchio, A., Holmgren, R.A., and Saraste, M. 1994. Invariant tryptophan at a shielded site promotes folding of the conformational unit of spectrin. Proc. Natl. Acad. Sci. 91 1299–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor, R.B. and Weber, G. 1986. Estimation of the polarity of the protein interior by optical spectroscopy. Nature 319 70–73. [DOI] [PubMed] [Google Scholar]
- MacPhee, C.E., Howlett, G.J., Sawyer, W.H., and Clayton, A.H.A. 1999. Helix–helix association of a lipid-bound amphipathic α-helix derived from apolipoprotein C-II. Biochemistry 38 10878–10884. [DOI] [PubMed] [Google Scholar]
- Majee, S., Dasgupta, D., and Chakrabarti, A. 1999. Interaction of the DNA-binding antitumor antibiotics, chromomycin and mithramycin with erythroid spectrin. Eur. J. Biochem. 260 619–626. [DOI] [PubMed] [Google Scholar]
- Mentré, P., ed. 2001. Water in the cell. Cell. Mol. Biol. 47: 709–970. [PubMed]
- Mukherjee, S. and Chattopadhyay, A. 1994. Motionally restricted tryptophan environments at the peptide–lipid interface of gramicidin channels. Biochemistry 33 5089–5097. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Wavelength-selective fluorescence as a novel tool to study organization and dynamics in complex biological systems. J. Fluorescence 5 237–246. [DOI] [PubMed] [Google Scholar]
- Mukherjee, S., Chattopadhyay, A., Samanta, A., and Soujanya, T. 1994. Dipole moment change of NBD group upon excitation studied using solvatochromic and quantum chemical approaches: Implications in membrane research. J. Phys. Chem. 98 2809–2812. [Google Scholar]
- Neri, D., Billeter, M., Wider, G., and Wuthrich, K. 1992. NMR determination of residual structure in a urea-denatured protein, the 434-repressor. Science 257 1559–1563. [DOI] [PubMed] [Google Scholar]
- Nishimura, S., Kandori, H., and Maeda, A. 1997. Transmembrane signaling mediated by water in bovine rhodopsin. Photochem. Photobiol. 66 796–801. [DOI] [PubMed] [Google Scholar]
- O’Connor, D.V. and Phillips, D. 1984. Time-correlated single photon counting, pp. 180–189. Academic Press, London.
- Pal, S.K., Peon, J., and Zewail, A.H. 2002. Biological water at the protein surface: Dynamical solvation probed directly with femtosecond resolution. Proc. Natl. Acad. Sci. 99 1763–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazatos, D.P. and MacDonald, R.I. 1997. Site-directed mutagenesis of either the highly conserved Trp-22 or the moderately conserved Trp-95 to a large, hydrophobic residue reduces the thermodynamic stability of a spectrin repeating unit. J. Biol. Chem. 272 21052–21059. [DOI] [PubMed] [Google Scholar]
- Pascual, J., Pfuhl, M., Walther, D., Saraste, M., and Nilges, M. 1997. Solution structure of the spectrin repeat: A left-handed antiparallel triple-helical coiled-coil. J. Mol. Biol. 273 740–751. [DOI] [PubMed] [Google Scholar]
- Pattnaik, B.R., Ghosh, S., and Rajeswari, M.R. 1997. Selective excitation of tryptophans in OmpF: A fluorescence emission study. Biochem. Mol. Biol. Int. 42 173–181. [DOI] [PubMed] [Google Scholar]
- Prendergast, F.G. 1991. Time-resolved fluorescence techniques: Methods and applications in biology. Curr. Opin. Struct. Biol. 1 1054–1059. [Google Scholar]
- Raghuraman, H., Kelkar, D.A., and Chattopadhyay, A. 2003. Novel insights into membrane protein structure and dynamics utilizing the wavelength-selective fluorescence approach. Proc. Ind. Natl. Sci. Acad. A 69 25–35. [Google Scholar]
- Raja, S.M., Rawat, S.S., Chattopadhyay, A., and Lala, A.K. 1999. Localization and environment of tryptophans in soluble and membrane-bound states of a pore-forming toxin from Staphylococcus aureus. Biophys. J. 76 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat, S.S. and Chattopadhyay, A. 1999. Structural transitions in the micellar assembly: A fluorescence study. J. Fluorescence 9 233–244. [Google Scholar]
- Rawat, S.S., Mukherjee, S., and Chattopadhyay, A. 1997. Micellar organization and dynamics: A wavelength-selective fluorescence approach. J. Phys. Chem. B 101 1922–1929. [Google Scholar]
- Ray, S. and Chakrabarti, A. 2003. Erythroid spectrin in miceller detergents. Cell Motil. Cytoskeleton 54 16–28. [DOI] [PubMed] [Google Scholar]
- Ruggiero, A.J., Todd, D.C., and Fleming, G.R. 1990. Subpicosecond fluorescence anisotropy studies of tryptophan in water. J. Am. Chem. Soc. 112 1003–1014. [Google Scholar]
- Sahr, K.E., Laurila, P., Kotula, L., Scarpa, A.L., Coupal, E., Leto, T.L., Linnenbach, A.J., Winkelmann, J.C., Speicher, D.W., Marchesi, V.T., et al. 1990. The complete cDNA and polypeptide sequences of human erythroid α-spectrin. J. Biol. Chem. 265 4434–4443. [PubMed] [Google Scholar]
- Samanta, A. and Fessenden R.W. 2000. Excited state dipole moment of PRODAN as determined from transient dielectric loss measurements. J. Phys. Chem. A 104 8972–8975. [Google Scholar]
- Sankararamakrishnan, R. and Sansom, M.S.P. 1995. Water-mediated conformational transitions in nicotinic receptor M2 helix bundles: A molecular dynamics study. FEBS Lett. 377 377–382. [DOI] [PubMed] [Google Scholar]
- Santos, N.C., Prieto, M., and Castanho, M.A.R.B. 1998. Interaction of the major epitope region of HIV protein gp41 with membrane model systems. A fluorescence spectroscopy study. Biochemistry 37 8674–8682. [DOI] [PubMed] [Google Scholar]
- Sengupta, B., Guharay, J., and Sengupta, P.K. 2000. Characterization of the fluorescence properties of prodan in different reverse micellar environments. Spectrochim. Acta A 56 1433–1441. [DOI] [PubMed] [Google Scholar]
- Shortle, D. 1993. Denatured states of proteins and their roles in folding and stability. Curr. Opin. Struct. Biol. 3 66–74. [Google Scholar]
- Sikorski, A.F., Michalak, K., and Bobrowska, M. 1987. Interaction of spectrin with phospholipids. Quenching of spectrin intrinsic fluorescence by phospholipid suspensions. Biochim. Biophys. Acta 904 55–60. [DOI] [PubMed] [Google Scholar]
- Song, P.-S. and Kurtin, W.E. 1969. Photochemistry of the model phototropic system involving flavines and indoles. III. A spectroscopic study of the polarized luminescence of indoles. J. Am. Chem. Soc. 91 4892–4906. [Google Scholar]
- Subbarao, N.K. and MacDonald, R.C. 1994. Fluorescence studies of spectrin and its subunits. Cell Motil. Cytoskeleton 29 72–81. [DOI] [PubMed] [Google Scholar]
- Swaminathan, R., Krishnamoorthy, G., and Periasamy, N. 1994. Similarity of fluorescence lifetime distributions for single tryptophan proteins in the random coil state. Biophys. J. 67 2013–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tredwell, C.J. and Keary, C.M. 1979. Picosecond time resolved fluorescence lifetimes of the polymethine and related dyes. Chem. Phys. 43 307–316. [Google Scholar]
- Uversky, V.N. 2002. What does it mean to be natively unfolded? Eur. J. Biochem. 269 2–12. [DOI] [PubMed] [Google Scholar]
- Valeur, B. and Weber, G. 1978. A new red-edge effect in aromatic molecules: Anomaly of apparent rotation revealed by fluorescence polarization. J. Chem. Phys. 69 2393–2400. [Google Scholar]
- Wasylewski, Z., Koloczek, H., Wasniowska, A., and Slizowska, K. 1992. Red-edge excitation fluorescence measurements of several two-tryptophan-containing proteins. Eur. J. Biochem. 206 235–242. [DOI] [PubMed] [Google Scholar]
- Weaver, A.J., Kemple, M.D., Brauner, J.W., Mendelsohn, R., and Prendergast, F.G. 1992. Fluorescence, CD, attenuated total reflectance (ATR) FTIR, and 13C NMR characterization of the structure and dynamics of synthetic melittin and melittin analogues in lipid environments. Biochemistry 31 1301–1313. [DOI] [PubMed] [Google Scholar]
- Weber, G. 1960. Fluorescence-polarization spectrum and electronic energy transfer in proteins. Biochem. J. 75 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, G. and Farris, F.J. 1979. Synthesis and spectral properties of a hydrophobic fluorescent probe: 6-propionyl-2–(dimethylamino)naphthalene. Biochemistry 18 3075–3078. [DOI] [PubMed] [Google Scholar]
- Wichterle, H., Hanspal, M., Palek, J., and Jarolim, P. 1996. Combination of two mutant α spectrin alleles underlies a severe spherocytic hemolytic anemia. J. Clin. Invest. 98 2300–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, P., Bateman, J., Kozarsky, K., Mattocks, K., Hermanowicz, N., Choe, H.-R., and Schlegel, R.A. 1982. Involvement of spectrin in the maintenance of phase-state asymmetry in the erythrocyte membrane. Cell 30 725–733. [DOI] [PubMed] [Google Scholar]
- Winkelmann, J.C., Chang, J.-G., Tse, W.T., Scarpa, A.L., Marchesi, V.T., and Forget, B.G. 1990. Full-length sequence of the cDNA for human erythroid β-spectrin. J. Biol. Chem. 265 11827–11832. [PubMed] [Google Scholar]
- Yamamoto, Y. and Tanaka, J. 1972. Polarized absorption spectra of crystals of indole and its related compounds. Bull. Chem. Soc. Jpn. 45 1362–1366. [Google Scholar]
- Ziemnicka-Kotula, D., Xu, J., Gu, H., Potempska, A., Kim, K.S., Jenkins, E.C., Trenkner, E., and Kotula, L. 1998. Identification of a candidate human spectrin Src homology 3 domain-binding protein suggests a general mechanism of association of tyrosine kinases with the spectrin-based membrane skeleton. J. Biol. Chem. 273 13681–13692. [DOI] [PubMed] [Google Scholar]