

Abstract
The crystal structures of lactose repressor protein (LacI) provide static endpoint views of the allosteric transition between DNA- and IPTG-bound states. To obtain an atom-by-atom description of the pathway between these two conformations, motions were simulated with targeted molecular dynamics (TMD). Strikingly, this homodimer exhibited asymmetric dynamics. All asymmetries observed in this simulation are reproducible and can begin on either of the two monomers. Asymmetry in the simulation originates around D149 and was traced back to the pre-TMD equilibrations of both conformations. In particular, hydrogen bonds between D149 and S193 adopt a variety of configurations during repetitions of this process. Changes in this region propagate through the structure via noncovalent interactions of three interconnected pathways. The changes of pathway 1 occur first on one monomer. Alterations move from the inducer-binding pocket, through the N-subdomain β-sheet, to a hydrophobic cluster at the top of this region and then to the same cluster on the second monomer. These motions result in changes at (1) side chains that form an interface with the DNA-binding domains and (2) K84 and K84’, which participate in the monomer–monomer interface. Pathway 2 reflects consequent reorganization across this subunit interface, most notably formation of a H74-H74rsquo; π-stacking intermediate. Pathway 3 extends from the rear of the inducer-binding pocket, across a hydrogen-bond network at the bottom of the pocket, and transverses the monomer–monomer interface via changes in H74 and H74rsquo;. In general, intermediates detected in this study are not apparent in the crystal structures. Observations from the simulations are in good agreement with biochemical data and provide a spatial and sequential framework for interpreting existing genetic data.
Keywords: Gene regulation, allosteric mechanism, structural flexibility, conformational transition pathway, targeted molecular dynamics
Since its discovery in 1961, the lactose (lac) operon has been the prototypic model for studying genetic and allosteric regulation (Jacob and Monod 1961; Monod et al. 1965; Matthews and Nichols 1998; Matthews et al. 2000). The central component of this regulatory system—lactose repressor protein (LacI)—binds to three operator sites within the lac operon to repress gene transcription (Matthews 1992). Inducer sugars, such as allolactose (Jobe and Bourgeois 1972) or isopropyl-β-d-thiogalactoside (IPTG; Riggs et al. 1970), bind to LacI and lower the affinity of the repressor for the operator DNA through allosteric changes. This alteration in turn allows RNA polymerase access to the operon, thereby inducing the synthesis of lac mRNA (Straney and Crothers 1987; Lee and Goldfarb 1991; Schlax et al. 1995). Recently, crystal structures have been determined for the two conformations that correspond with LacI functional modes of repression and induction (Friedman et al. 1995; Lewis et al. 1996; Bell and Lewis 2000). However, these structures provide only "snapshots" of the endpoints; they do not elucidate the atomic-level mechanism by which the protein switches between conformations. Computational analysis can provide the necessary motional framework for a more complete understanding of the allosteric mechanism.
In both states, the architecture of LacI can be divided into five structural units (Friedman et al. 1995; Lewis et al. 1996; Bell and Lewis 2000). First, the N-terminal DNA-binding domain (amino acids 1–50) comprises a helix-turn-helix motif that interacts with the major groove of the DNA. Two N termini of a dimer are required for a complete DNA-binding site. Next in the primary sequence is the hinge helix (amino acids 51–61), which binds to the center of the operator within the minor groove. The hinge region appears to be disordered in the absence of DNA (Ha et al. 1989; Lewis et al. 1996; Spronk et al. 1996, 1999a,b; Kalodimos et al. 2001, 2002a,b). The hinge helix connects the DNA-binding domain to the core domain (amino acids 62–330), which comprises the interconnected N- and C-subdomains (Fig. 1A ▶). The inducer-binding pocket is located between these subdomains. Finally, the C-terminal regions (amino acids 331–360) associate into a four-helical bundle to facilitate tetramerization of LacI (not shown in Fig. 1A ▶).
Figure 1.
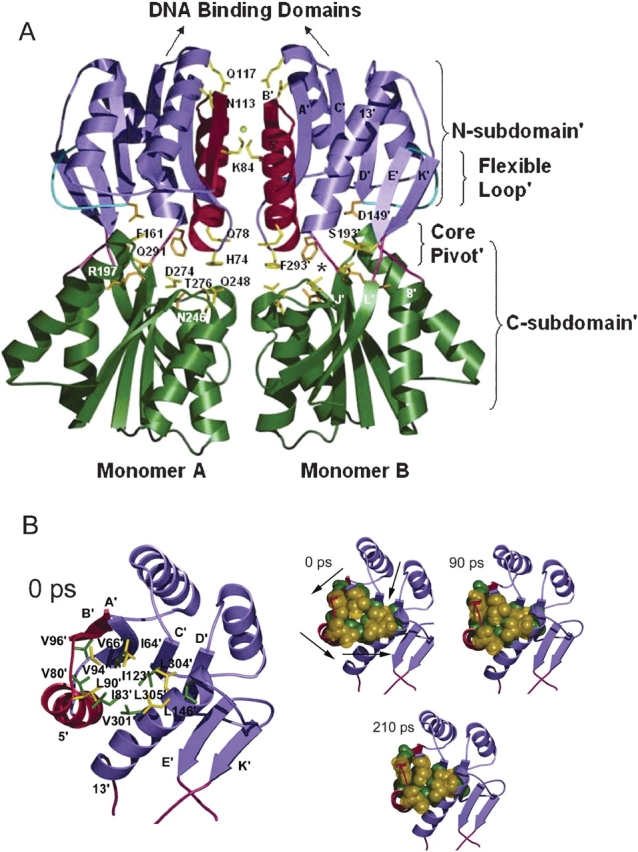
(A) Backbone representation of the core domains of LacI dimer in the repressed form (PDB code 1EFA; Bell and Lewis 2000). N-subdomains are in purple; C-subdomains, in green; N-subdomain monomer–monomer interface, in red; flexible loops, in cyan; and interconnecting strands of the core pivot, in pink. Side chains of key residues in the simulation are highlighted in yellow; those that contact inducer are in orange. The chloride anion is represented by a yellow ball. The asterisk locates one of the inducer-binding pockets. The figure was made by using Molscript (Kraulis 1991) and rendered by Povray (www.povray.org). (B) Top views of trigger monomer, illustrating the N-subdomain’ hydrophobic group changes. Time intervals are listed on the top left corner of each structure. In larger representation on the left, residues involved are labeled and indicated with a stick representation. The three smaller figures on the right show the same residues in space-filling representation as they change positions over the course of the simulation. Arrows illustrate direction of movement of each region during the simulation. Counterclockwise rotation about the N-subdomain’ hydrophobic group causes contraction in the center of this region.
Analysis of the crystal structures that represent the two states (repressed, LacI • OsymDNA bound to anti-inducer σ-nitrophenyl-β, D-fucoside [ONPF] induced, LacI•IPTG inducer) led Lewis and colleagues (Lewis et al. 1996; Bell and Lewis 2000) to postulate that the allosteric transition in LacI occurs when inducer binding shifts the N-subdomains, both with respect to each other and to the C-subdomains. As a result of this structural shift, the N-subdomain monomer–monomer interface is significantly altered. These motions in turn displace the hinge helices from the minor groove of the operator, disrupting specific protein-DNA contacts and increasing the dynamic mobility of the N-terminal DNA-binding domains (Lewis et al. 1996; Spronk et al. 1996, 1999a,b; Swint-Kruse et al. 1998; Kalodimos et al. 2001, 2002a,b). In this study, the time course and detailed interactions along the allosteric pathway in the LacI core are simulated—from the DNA/anti-inducer–bound structure (Protein Data Bank [PDB] code 1EFA; Bell and Lewis 2000) to the inducer-bound structure (PDB code 1LBH; Lewis et al. 1996)—by using the method of targeted molecular dynamics (TMD; Schlitter et al. 1993; Ma and Karplus 1997; Ma et al. 2000, 2002). The results of this study culminate in the construction of an integrated framework for the allosteric transition pathway of LacI that is consistent with the biochemical, genetic, and structural data accumulated over the past four decades.
Results and Discussion
The allosteric transition pathway
A protein conformational change is requisite for the function of LacI transcription regulation. Recently, X-ray crystallographic structures have been determined for both states: (1) repressed (Bell and Lewis 2000), which has high affinity for specific operator DNA; and (2) induced (Lewis et al. 1996), which has low DNA-binding affinity. The binding of a small molecule inducer (IPTG) facilitates the switch to the latter state. For the allosteric conformational shift to occur, the effects of inducer binding must be propagated from the inducer-binding pocket, located between the C- and N-subdomains, to the DNA-binding domains, which lie above the N-subdomains (Fig. 1A ▶). This conformational change is accomplished by an inward clamping and rotation of the N-subdomains. The two C-subdomains remain stationary during the transition and anchor the mobile N-subdomains (Fig. 2B ▶; Lewis et al. 1996; Bell and Lewis 2000).
Figure 2.
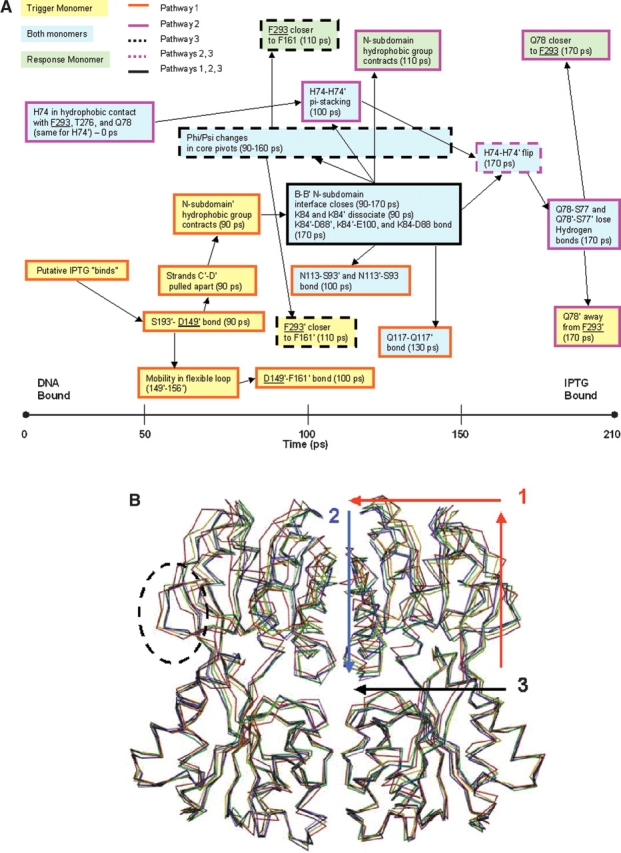
(A) Timeline of the allosteric transition pathway as simulated by TMD. Residues that contact inducer are underlined. Trigger monomer interactions are highlighted in yellow boxes; response monomer interactions, in green; and interactions involving both monomers, in blue. Boxes with dark orange borders are used to delineate pathway 1; pink borders, pathway 2; borders with black dotted lines, pathway 3; pink dotted lines, pathway 2/3; and a black border, involvement in all three pathways. (B) The structures of five representative time points along the simulated trajectory were aligned using the Cα residues of the stationary C-subdomains. The structures are colored as follows: red (0 psec), yellow (60 psec), green (110 psec), blue (160 psec), and purple (210 psec). The flexible loop (residues 149–156) is enclosed by a dotted circle. The directionality of pathway 1 is illustrated by the red arrows; pathway 2, by the blue arrow; and pathway 3, by the black arrow.
Crystal structures do not provide direct information about the mechanism by which proteins change conformation. However, they can be subjected to molecular dynamics simulations to interpolate the relevant motions. Here, the transition pathway from the DNA/anti-inducer–bound form of LacI to the inducer-bound form was simulated by the method of TMD (Schlitter et al. 1993; Ma and Karplus 1997; Ma et al. 2000, 2002), providing the opportunity to detect intermediate states. The advantage of using TMD is that it forces the starting structure to the target structure over a period of several hundred picoseconds, compared with conventional molecular dynamic simulations that would require microseconds. Thus, larger biological systems can be explored.
TMD does require that the starting and ending structures have identical sets of atoms. Therefore, the present simulations include the only domain in common between the two structures—the LacI core domains of a dimer. Neither the DNA-binding domains nor the operator DNA is present in the induced structure. The small molecule ligands differ in the two structures (IPTG and ONPF) and the DNA-bound structure was generated from LacI protein that lacked the C-terminal tetramerization domain. Thus, these features were not included in the simulation. Analysis of the CHARMM trajectories (Brooks et al. 1983) was performed by aligning the stationary C-subdomains (residues 162–290 and 321–329) followed by (1) inspecting the trajectory file computationally and visually with Quanta (www.msi.com), and (2) using the Contacts of Structural Units (CSU) software program (Sobolev et al. 1999) to compare equilibrated structures and those from representative time points during the trajectory (Fig. 2B ▶).
When LacI binds inducer, the induction signal might be expected to propagate through the structure in a spatially and temporally ordered manner. For these TMD simulations, which cannot force inducer into the binding pocket, space/time ordering may or may not be the same as for the actual protein (Fig. 2 ▶). However, the changes observed for LacI in the present simulations occur in a logical sequence. During the trajectory, a striking observation is that the conformational changes undergone by each monomer are not identical. This phenomenon is intriguing. Experimentally, the LacI dimer•operator DNA complex binds two molecules of inducer cooperatively, and the LacI allostery may be described equally well by the Monod-Wyman-Changeaux (MWC) and Koshland models (O’Gorman et al. 1980; Daly and Matthews 1986). Further, the LacI K84L mutant binds IPTG with slowed and biphasic kinetics (Chang et al. 1993); this mutation may have slowed the conformational transition so that the asymmetry is experimentally observable (see Interpretation of Experimental Data).
We have actively explored the origin of the asymmetric conformational change. An early feature of the trajectory is an asymmetric change in hydrogen-bond formation between the side chains of D149 and S193 (see Pathway 1, below). Residues that directly contact IPTG are underlined in the text, tables, and figures (Table 1). The region around D149 is essentially identical for all three monomers of the crystallographic asymmetric cell of the DNA-bound structure (Bell and Lewis 2000). However, D149 interactions in the structures of intact inducer-bound LacI and the proteolytic fragment of inducer-bound core demonstrate some differences in distances and interaction partners (Friedman et al. 1995; Lewis et al. 1996). During equilibration at the beginning of the simulation, all three structures undergo asymmetric changes between two monomers of a dimer around position 149. Multiple equilibrations demonstrate a wide variety of configurations adopted by D149 and its partners, especially S193. Both the asymmetry and the conformational changes observed during the simulation are repeatable over seven TMD trajectories (repeatability is illustrated in Fig. 3 ▶). In the simulations, the conformational change originates on a "trigger monomer," then propagates through various noncovalent interactions to the second "response," monomer (Fig. 2 ▶). In this article, we discriminate between the two monomers by adding a prime (’) symbol to residues and regions on the trigger monomer.
Table 1.
Residues contacting inducer IPTG in crystal structure 1LBHa or 1TLFb
| Residue | Nature of Contacta | Changes in TMD |
| S69 | SC Hydrogen bond | N |
| L73 | Hydrophobic | Y |
| A75 | Hydrophobic | N |
| P76 | Hydrophobic | N |
| I79 | Hydrophobic | N |
| R101 | SC Hydrogen bond | N |
| D149 | SC Hydrogen bond | Y |
| R197 | SC Hydrogen bond | Y |
| W220 | Hydrophobic/vdW | N |
| N246 | SC Hydrogen bond | Y |
| D274 | SC Hydrogen bond | Y |
| F293 | Hydrophobic | Y |
| L296 | Hydrophobic | Y |
Figure 3.

Reproducibility of seven TMD trajectories. The distance changes in the monomer–monomer interface for residues of pathway 2 provide a good example for the repeatability of changes during the simulation. Over the course of the transition, the distance between the side-chains of K84 and K84’ increases (A), whereas the distance between the Cαs of V96 and V96’ (B), and hence β-strands B and B’, decreases. (C) Notice that the squeezing together of the β-strands (V96) coincides with the departure of the lysine pair (K84).
To facilitate discussion of the allosteric transition pathway, we chose to decompose the overall pathway into three smaller pathways. Pathway 1 begins in the inducer-binding site of the trigger monomer and travels through the central β-sheet of the core N-subdomain’ to the N-subdomain monomer–monomer interface, terminating in changes at position 84. The second pathway comprises interactions that occur across the N-subdomain monomer–monomer interface after the disruption of the K84–K84’ interaction: specifically the loss of Q78 and Q78’ hydrogen bonds and stable π-stacking between H74 and H74’. The third pathway extends from F161’, at the rear of the inducer-binding pocket on the trigger monomer, through the bottom of this pocket, and across the monomer–monomer interface to the response monomer via the H74–H74’ π-stacking. The resulting conformational changes reposition the N-subdomains so that they can no longer form the contacts to the DNA-binding domain presumed requisite for high-affinity binding (Bell and Lewis 2000; Swint-Kruse et al. 2002).
Origin of asymmetry: Repeatability of equilibration and TMD trajectory
We find the asymmetry observed in the pathways to be a compelling result of the simulation. The simulated asymmetry has a number of possible origins, ranging from experimentally relevant to random occurrence during TMD.
We first examined the crystal structures to determine whether these starting structures gave rise to the observed behavior. The structure of the DNA-bound LacI (1EFA) is symmetric for all three monomers present in the crystallographic unit cell (Bell and Lewis 2000). However, all eight available monomers in the inducer-bound state—four each from 1LBH and 1TLF—have subtle differences in the region around D149 (Friedman et al. 1995; Lewis et al. 1996). A potential cause for the observed asymmetry is different occupancies of the inducer-binding sites, but this source is unlikely at the high IPTG concentrations used (Friedman et al. 1995; Lewis et al. 1996). Finally, the fact that both 1TLF and 1LBH are variable around amino acid 149 indicates that crystal packing artifacts are an unlikely source of the observed asymmetry.
To ascertain whether asymmetry was a random occurrence during the simulation, the TMD trajectory was carried out five times by using the same equilibrated start and target structures but varying the random number seed in the simulation. In each trajectory, the same monomer always emerged as the trigger monomer. When a different equilibrated target structure was used to generate two additional trajectories, the opposite monomer functioned as the trigger in both cases. Therefore, although some asymmetry is present in the crystal structures, asymmetry of the simulated conformational change appears to be dictated during equilibration.
Energy equilibration/minimization algorithms are used before virtually every molecular dynamics simulation to relieve stresses inherent to the experimental structure or those that result from modeling (e.g., the addition of solvent molecules or point charges). Equilibration usually does not result in "meaningful" conformational changes, but the structures of the current simulation are subjected to experimentally relevant forces. Prior to equilibration, the DNA-bound N termini, which make cross-domain interactions with the core N-subdomain, were removed from the starting structure; anti-inducer ONPF was also deleted from this structure; and the inducer IPTG was removed from the target structure. Therefore, both structures were in the unliganded state for 10 psec of computer time while they were equilibrated. Experimentally, the allosteric constant for unbound protein is ∼1 when LacI dimer function can be described by a MWC model, which indicates that unliganded LacI is equally likely to adopt either the repressed or induced conformation (O’Gorman et al. 1980; Daly et al. 1986). Equilibration provides opportunity for the now unliganded structures to initiate relevant changes toward the alternate conformation.
Indeed, several residues that are asymmetrically involved in the subsequent trajectory of the LacI conformational change are affected during equilibration. The first step in the trajectory involves changes at residue 149 of the trigger monomer (see Fig. 2 ▶, pathway 1). Interestingly, the region around D149—especially S193 and R197—is affected during equilibration of both the DNA- and inducer-bound structures. Note that two of these residues contact the inducer/anti-inducer ligand (Table 1), and thus, changes in this region during equilibration are consistent with the structural manipulations required to execute TMD. Although 1TLF could not be used as a target structure, we also subjected this structure to ligand removal, solvation, and equilibration and saw changes similar to those in 1LBH (Table 2).
Table 2.
Structural effects of equilibrationa
| Contact surface (Å2) | Bond length (Å) | |||
| Monomer A D149:S193 | Monomer B D149:S193 | Monomer A D149O:L128N | Monomer B D149 O:L128N | |
| Operatorb | ||||
| AB dimer | 28.3 | 26.7 | 3.1 | 3.2 |
| Monomer C | 27.0 | 3.2 | ||
| Equil. 1c | 34.9 | 48.0 | 2.8 | 2.8 |
| Equil. 2 | 30.3 | 39.3 | 3.5 | 3.1 |
| Equil. 3 | 49.8 | 30.3 | 2.9 | 3.4 |
| Equil. 4 | 18.3 | 44.0 | 3.3 | 2.9 |
| Inducerd | ||||
| AB dimer | 30.5 | 40.4 | 3.2 | 3.5 |
| CED dimer | 37.0 | 36.5 | 3.4 | 4.0 |
| Equil. 1e | 16.5 | 49.0 | 3.1 | >6 |
| Equil. 2 | 44.4 | 22.7 | 3.0 | >6 |
| Equil. 3 | 39.7 | 35.6 | 3.6 | >6 |
| Equil. 4 | 44.1 | 35.6 | >6 | 3.4 |
| Equil. 5 | 17.7 | 36.4 | 3.3 | >6 |
| Equil. 6 | 41.2 | 25.2 | 3.7 | 3.2 |
| Equil. 7 | 43.3 | 24.2 | 2.9 | 4.6 |
| Equil. 8f | 24.1 | 23.4 | 4.5 | 3.7 |
| Equil. 9 | 19.4 | 46.0 | 4.2 | 3.4 |
| Inducer (proteolytic core)g | ||||
| AB dimer | 31.3 | 34.5 | 3.4 | 3.1 |
| CD dimer | 25.2 | 27.9 | 3.2 | 3.2 |
| Equil. 1 | 32.5 | 42.7 | >6 | 3.4 |
| Equil. 2 | 30.3 | 23.2 | 3.3 | 3.9 |
| Equil. 3 | 41.7 | 27.4 | >6 | 4.2 |
| Equil. 4 | 33.6 | 26.9 | 3.1 | 4.4 |
| Equil. 5 | 34.4 | 20.0 | 2.9 | >6 |
a Values determined by CSU analysis (Sobolev et al. 1999).
b Operator-bound crystal structure (PDB code 1EFA; Bell and Lewis 2000), which has three LacI monomers in the crystallographic unit cell.
c Equilibrated starting structure used for all seven TMD trajectories, generated from AB dimer.
d Inducer-bound crystal structure (PDB code 1LBH; Lewis et al. 1996; ).
e Equilibrated target structure used for TMD trajectories 1–5, generated from AB dimer. Bold values indicate which monomer functioned as the trigger monomer.
f Equilibrated target structure used for TMD trajectories 6 and 7, generated from AB dimer. Bold values indicate which monomer functioned as the trigger monomer.
g Crystal structure of inducer-bound tryptic core (PDB code 1TLF; Friedman et al. 1995; ).
More specifically, the structure of DNA-bound LacI (1EFA) evinces a hydrogen bond between the side chain of R197 and the side chain of S193. During equilibration repetitions, this bond breaks on seven of the eight monomers and one to two new hydrogen bonds form between the R197 backbone and S193 side chain or backbone. This side chain/side chain bond between 197 and 193 is not present in either of the inducer-bound structures. The monomers of 1LBH have one to two hydrogen bonds between the R197 backbone and S193 backbone or side chain whereas those of 1TLF have one bond between the backbone atoms of the two residues. Equilibration of the inducer-bound structures affects little change for these bonds, and the 193/197 side chain/side chain bond found in 1EFA only forms on one of 28 equilibrated monomers from the inducer-bound structures. Thus, we surmise that this bond is important to the DNA/anti-inducer structure.
Freeing the S193 side chain of the DNA-bound structure allows this residue to form stronger interactions with D149 as the simulation proceeds toward the inducer-bound conformation. In 1EFA, these two residues form only one bond between the backbone N of 193 and the side chain of 149. Upon ligand removal and equilibration, these residues form any of several combinations of six different hydrogen bonds between backbone/side chain of 193 and the side chain of 149, with one to four bonds per monomer. In 1LBH, the S193 side chain forms two bonds to the D149 side chain per monomer. The structure of the proteolytic fragment, 1TLF, has one of these bonds. During equilibration of either inducer-bound structure, these residues form combinations of zero to three hydrogen bonds in variety of side chain/side chain, side chain/backbone, and backbone/side chain combinations.
The D149-S193 hydrogen bonds exhibit no obvious patterns that correlate with the asymmetry of the TMD. However, the total surface area in contact between these two residues should correlate with the total strength of the interactions. These values are presented in Table 2 and provide insight into the origin of TMD asymmetry: When a trajectory used equilibration 1 for the DNA-bound conformation and equilibration 1 for the inducer-bound target, monomer B served as the trigger monomer. Consistent with this, monomer B of inducer-bound equilibration 1 has much larger contact surface between D149 and S193 than does monomer A. Further, when equilibration 8 was substituted for the inducer-bound target, monomer A emerged as the trigger monomer. In this case, the monomers of the target had similar contact surface (Table 2), and thus, the moderate difference in start structures appears to dominate: Monomer A of the starting structure had less contact surface—and thus may be easier to interrupt—than is monomer B. This correlation between contact surface of D149/S193 and trigger monomer will be tested in future studies.
Finally, a backbone hydrogen bond is frequently lost between β-strands C and D (D149-O:L128-NH; Table 2) during equilibration of the target monomer. Intriguingly, this change can occur on either monomer, but the distance is always longer on one monomer within a dimer. This pattern persisted during equilibration of the proteolytic fragment 1TLF. Further, the monomer with the broken bond functioned as the trigger monomer in the TMD trajectories. Because the bonds are formed in both the DNA-bound and inducer-bound crystal structures, this feature of the equilibrated structures may approximate a feature of apo-LacI that allows the protein to switch between conformations.
The TMD trajectory was repeated seven times (illustrated in Fig. 3 ▶), and the features of all trajectories were highly similar. For the purpose of clarity, the main body of this article uses the time points of the first trajectory and is summarized in Figure 2 ▶. The reverse trajectory—from the inducer- to DNA-bound conformation—was also simulated. Although a detailed examination is beyond the scope of this article, the key features of the process are near-mirror images of the data presented for the forward transition.
Pathway 1
In pathway 1, changes in the inducer-binding pocket of the trigger monomer propagate through β-strands C’ and D’ to a hydrophobic cluster in the N-subdomain’, then to the monomer–monomer interface, and finally result in significant changes at K84’ (Fig. 2B ▶). Pathway 1 only occurs on the trigger monomer, and we assume that this simulates the changes that occur when the trigger monomer binds the first inducer molecule. Toward the end of pathway 1, the response monomer undergoes changes so that the two monomers adopt similar configurations in the final structure.
As mentioned above, changes in D149’ appear critical to propagating the message from the inducer-binding pocket to the N-subdomain’. D149’ lies at a key junction: It contacts IPTG and is located between the base of β-strand D’ and at the start of a flexible loop (residues 149’-156’;Fig. 1A ▶). Flexibility in this loop allows the side chain of D149’ to move ∼4 Å, thus forming the hydrogen bond with S193′ (at 90 psec; Fig. 4A ▶, arrow 1). Note that the distance between D149 and S193 of the response monomer remains constant throughout the simulation (Fig. 4B ▶). Next, at 100 psec, the side chain of D149’ forms another hydrogen bond with the backbone -NH of F161’ (Fig. 4C ▶, arrow 3). Finally, the backbone oxygen of D149’ forms a hydrogen bond to the side chain of S193′ 15 psec later (Fig. 4A ▶, arrow 2). Note that most of the motions in the flexible loop stabilize after 110 psec (Fig. 4E,G) ▶, most likely due to the two D149’ hydrogen bonds, which essentially lock the flexible loop in place.
Figure 4.

Interatomic distance versus time plots of hydrogen bonds formed between main chain of D149’ and side chain of S193′ on trigger monomer (A), main chain of D149 and side chain of S193 on response monomer (B), side chain of D149’ and main chain of F161’ on trigger monomer (C), and side chain of D149 and main chain of F161 on response monomer (D). Arrow 1 in A notes time when hydrogen bond forms between side chains of D149’ and S193′ (D149’-OD1 and S193′-HG). Arrow 2 in A illustrates hydrogen bonding between main chain of D149’ and the side chain of S193′ (D149’-O and S193′-HG). Arrow 3 in C marks the beginning of hydrogen bonding between the side chain of D149’ and the main chain of F161’. (E–H) Mobility of the flexible loop (residues 149–156) versus time for both monomers. Distances to the relevant Cα in the flexible loop were measured from a stationary anchor point in the C-subdomains (P284’-Cα and P284-Cα). The plots stabilize near 110 psec, after the N-subdomain interface is closed. (I, J) Hydrogen bond formation between atoms in β-strands C and D versus time. Filled circles indicate the distance between L128-NH and D149-O; open diamonds, Y126-O to D149-NH; filled triangles, I124-O to L148-NH; and plus symbols, I124-NH to L146-O. Notice the break in hydrogen bonding between L128’–D149’ and Y126’–D149’ at 90 psec between β-strands C’ and D’ (arrow 4). The asymmetry at the two endpoints of the graphs is not present in the inducer-bound crystal structure but occurs during the equilibration.
The hydrogen bonds formed by D149’ concurrently pull the lower portion of strand D’ away from strand C’. Strand D’ is in the center of the six-stranded parallel β-sheet, which makes up the nucleus of each of the N-subdomains and contributes to the monomer–monomer interface (Fig. 1A ▶). In the starting conformation, D’ forms six backbone hydrogen bonds with C’. The distances of four of these bonds are plotted in Figure 4 ▶, I and J. At 90 psec (Fig. 4I ▶–arrow 4) two bonds are broken and do not re-form (L128’-D149’ and Y126’-D149’), effectively shortening the lower portion of the β-sheet. In contrast, β-strands C and D of the response monomer are never separated (Fig. 4J ▶). Thus, the β-sheet of the 90-psec structure now resembles that of the asymmetric target structure formed during equilibration.
The changing hydrogen-bond distances between strands C’ and D’ endow greater mobility to the region of the N-subdomain’ that is closest to the monomer–monomer interface. A cluster of hydrophobic residues in this region contract at ∼90 psec (Fig. 1B ▶, arrows). This apparently leads to a similar change in the response monomer ∼110 psec (Fig. 2 ▶), resulting in a more closely packed monomer–monomer interface.
Interface rearrangement: The intersection of pathways 1 and 2
When pathway 1 changes are completed on the trigger monomer structure, the LacI dimer is poised to make changes that affect both the cross-subunit (trigger monomer to response monomer) and cross-domain (core to DNA binding) interfaces. This arrangement provides opportunity to transmit the allosteric message from the inducer-binding site of one monomer (presumably the trigger monomer) to (1) the inducer-binding site of the second monomer, so that the second inducer molecule binds with higher affinity than the first, and (2) the DNA-binding site, so that it adopts the conformation with low affinity for the operator sequence.
As stated above, changing hydrophobic interactions in N-subdomain’ alter the interactions across the monomer–monomer N-subdomain interface (Fig. 1A,2B). This entire region plays a key role in determining the conformational state of LacI (Lewis et al. 1996; Bell and Lewis 2000; Bell et al. 2001), but the electrostatic interactions of the lysine residues (K84 and K84’) are particularly important. In the initial (repressed) state, the side chains of residue 84 are located in the middle of this interface, in plane with but interrupting many polar groups capable of forming hydrogen bonds (Fig. 5A ▶). The side chains of K84 and K84’ point toward each other, and their conformations appear to be anchored by an intervening anion (Bell and Lewis 2000). For the purposes of this study, we have assumed this to be a chloride anion (see Materials and Methods). Above and below the lysine plane are two layers of hydrophobic side chains, including members of the N-subdomain hydrophobic groups mentioned above. In addition, an extensive network of polar and ionic interactions forms the monomer–monomer interface of the DNA-bound state (Fig. 5A ▶).
Figure 5.

Schematic representation of N-subdomain monomer–monomer interface in the repressed (A) and the induced (B) conformations. The view is from above the N-subdomains looking down into the interface. Filled circles indicate the side chain is above the plane of the β-sheets; open circles, the side chain lies below the plane. Note that residue 84 is in the plane of the β-sheets, even though its side chain is white. The repressed interface (A) is dominated by hydrogen bond and electrostatic interactions (dotted lines) that stabilize the K84 pair and chloride anion in the space located between the two β-strands. In the induced conformation (B), the K84 side chains are outside the interface, allowing the backbones of V96 and V96’ to form hydrogen bonds, creating an antiparallel β-sheet across the interface. V94 and V96 are also members of the N-subdomain hydrophobic group (see Fig. 1B ▶).
In the inducer-bound conformation, this network changes dramatically (Fig. 5B ▶). The side chains of K84 and K84’ lie outside of the N-subdomain interface and engage in ionic interactions with D88 of the same subdomain and E100’ of the opposite subdomain. Consequently, the B and B’ β-strands of the two N-subdomains are close enough to make main-chain contacts and form a continuous 12-stranded β-sheet across the dimer interface. The chloride anion is no longer present in the inducer-bound structure.
The shift in the N-subdomain interface is more or less concerted. The interatomic distance between the backbones of V96 and V96’ decreases nearly linearly throughout the transition, whereas the distance between the side chains of K84 and K84’ increases (Fig. 3C ▶). The simulation indicates the following pathway: After contraction of the N-subdomain’ hydrophobic cluster on the trigger monomer, the -NH3+ groups of the side chains of both K84s slowly dissociate from a sandwiched position between the main-chain carbonyl groups of V94’ and V96 (90 psec). These moieties then move to a transient position between the main-chain carbonyl of V94’ and the side chain of S97. The displacement of the lysine side chains allows secondary structural contacts to form across the interface between the main-chain amino and carbonyl groups of V96 and V96’. Finally, the second N-subdomain hydrophobic cluster on the response monomer contracts. The side chain of K84’ soon detaches from V94’ and S97, and moves outward to briefly contact the main-chain carbonyl of M98 before engaging the two acidic residues (170 psec), D88’ and E100, in a charge–charge interaction. Interestingly, during the transition, the hydrocarbon stem of the K84 side chain maintains a close hydrophobic contact with the side chain of M98’, even though the ionic moiety of K84 flips in the opposite direction (same for K84’). The K84 motions are essentially symmetric. The only difference in the two monomers is that K84 and E100’ do not interact in the equilibrated target structure.
Water molecules also participate in the N-subdomain transition. In the DNA-bound state, the side chains of K84 and K84’ and the chloride anion are partially exposed to the solvent, as proposed by Bell et al. (2001). However, the water molecules are squeezed out of the interface completely upon the formation of the inducer-bound conformation due to the extensive hydrophobic interactions. The chloride anion escapes to the solvent before the final transformation to the inducer-bound state. The complete departure of water molecules and the chloride ion from the interface occurs approximately in the middle of the transition.
As the N-subdomains move closer to form a continuous 12-stranded β-sheet, N113 and Q117 also undergo dramatic changes. Both N113 and Q117 are located on the top surface of the N-subdomain and interact with the DNA-binding domain in the repressed structure (Fig. 1A ▶). In the DNA-bound form, the side chains of Q117 and Q117’ do not contact each other, but in the induced state, their side chains form a hydrogen bond. In the simulation, this event occurs at 130 psec. The side chain of N113 lies ∼5 Å from the side chain of S93′ (same for N113′ and S93) in the repressed conformation. During the simulation, the distances between these side chains decrease to 3 Å to form new hydrogen bonds (beginning at 100 psec), further tightening the monomer–monomer interface. Thus, these changes may interrupt the cross-domain interface interactions requisite for high-affinity DNA-binding (Bell and Lewis 2000; Swint-Kruse et al. 2002).
Pathway 2
The second pathway involves interactions across the N-subdomain monomer–monomer interface after changes in the K84 pair. Three significant pairs of residues change: K84–K84’, H74–H74’, and Q78–Q78’. The K84 interactions link pathway 1 to pathways 2 and 3, and the H74 interactions connect pathway 2 to 3 (see pathway 3; Fig. 2B ▶).
H74 and H74’ are located at the bottom of helices 5 and 5′ in the N-subdomains, very close to the inducer-binding pockets (Fig. 1A ▶), and undergo extensive changes during the course of the transition. Initially, H74 forms intra-subunit hydrophobic contacts with F293, T276, and the hydrophobic region of Q78, whereas H74’ forms similar contacts within the partner monomer. Because of the loss of the K84–K84’ interaction (90 psec), the distance between the core N-subdomains also decreases. This distance eventually decreases enough (110 psec) to allow H74 and H74’ to engage in stable parallel π-stacking across the interface (Fig. 6A,B ▶, arrow 1). Histidine rings must be within 4.5 Å of each other and have similar dihedral angles for parallel π-stacking to occur (McGaughey et al. 1998). This π-stacking was not present in either crystal structure and was only detected by the TMD simulation. At 170 psec, the histidines simultaneously rotate 180°, then reform their π-stacking interaction (Fig. 6B ▶, arrow 2). The π-stacking ceases at 200 psec (Fig. 6B ▶, arrow 3) and adopts the final equilibrated inducer-bound structure. These π-stacking interactions appear to be important for transmitting signals across the N-subdomain interface. This π-stacking and the concomitant flip were present in every TMD trajectory and are shown in Figure 6 ▶ for trajectories 1, 5, and 7.
Figure 6.

Interatomic distance (A, C, E) and dihedral angle (B, D, F) versus time plots for H74 and H74’ for representative trajectories (1, 5, and 7). (B, D, F) The H74 dihedral angle of the trigger monomer is represented by circles; that for the response monomer, by the solid line. In order for stable parallel π-stacking to occur, the histidine side chains must have a similar dihedral angle (indicating that the rings are oriented in parallel planes), and the rings must be ∼4.5 Å apart (shown as dashed line in A,C, E). These conditions first exist concurrently in trajectory 1 at 110 psec (arrow 1), in trajectory 5 at 95 psec (arrow 4), and in trajectory 7 at 95 psec (arrow 7). The two histidines undergo simultaneous 180° rotations in trajectory 1 at 170 psec (arrow 2), in trajectory 5 at 140 psec (arrow 5), and in trajectory 7 at 140 psec (arrow 8), which allows them to reform π-stacking until they adopt their final conformations. The noise following arrows 3 and 6 is due to His74’ adopting the equilibrated inducer-bound target structure in the simulation, in which π-stacking does not exist.
The third interaction across the N-subdomain monomer–monomer interface involves Q78 and Q78’. The side chains of these glutamines lie in a plane directly above H74 and H74’ (Fig. 1A ▶). The -NH2 side chain of Q78 forms hydrogen bonds to the backbone oxygen of L71’ and the side chain oxygen of S77, whereas Q78’ forms an identical set of bonds with corresponding residues. As the simulation progresses (110 psec), Q78 forms an additional bond with the backbone oxygen of H74; Q78’ follows (data not shown). However, these new bonds are disrupted when H74 and H74’ flip at 170 psec. The side chain oxygen atoms of both Q78s then form a hydrogen bond with the backbones of their respective E277 -NH. This alteration moves Q78 closer to F293, but Q78’ moves further from F293’. This asymmetric interaction also exists in the inducer-bound crystal structure. F293 and F293’ both contact IPTG in the induced state (Table 1) and are part of the core pivot hydrophobic groups (see Core Pivot).
In summary, three monomer–monomer interface interactions of the DNA-bound state are disrupted (K84–K84’, Q78–L71’, and Q78’–L71), but a stable π-stacking interaction is transiently gained as the simulation progresses from the DNA-bound to the inducer-bound structure.
Core pivot
The gross clamping motion of the N-subdomains relative to the fixed C-subdomains must logically involve the three sets of residues that covalently interconnect the N- and C-subdomains: (1) amino acids 161–164, connecting strand E of the N-subdomain and helix 8 of the C-subdomain; (2) residues 290–293, joining helix 13 of the N-subdomain and strand J of the C-subdomain; and (3) positions 318–322, linking strand K of the N-subdomain to L of the C-subdomain (Fig. 1A ▶). We denote these sets of connecting residues at the N- and C-core junction as the core pivot (Figs. 1A, 7A ▶ ▶). Similar regions in other proteins are often referred to as hinge, but we have chosen this alternate description to avoid confusion with the LacI hinge helix at residues 51–61.
Figure 7.
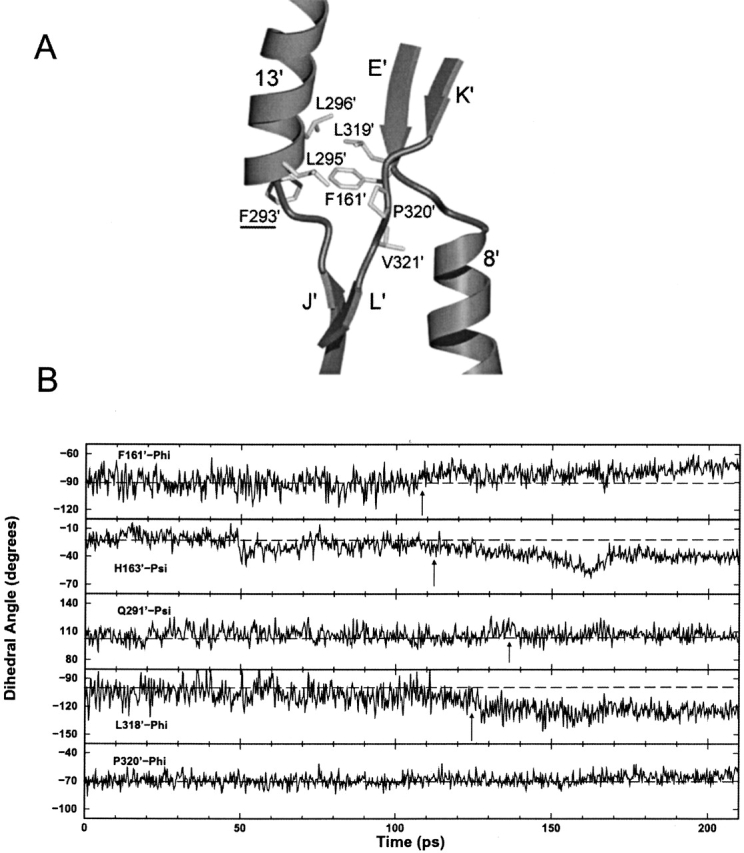
(A) Detailed view of the core pivot hydrophobic group in the trigger monomer. Interactions between residues are centered on F161’. (B) Backbone φ and ξ plots versus time for core pivot residues in trigger monomer. All residues plotted have standard deviations >9°, with the exception of P320’-Phi, which is displayed for comparison. Dotted lines indicate the approximate average angles at the start of the simulation. Arrows call attention to significant changes in φ/ξ angle.
To analyze the TMD data in this region, we found it useful to follow the procedure of Magnusson et al. (2002). These investigators examined the φ and ξ angles of the backbone of three conformations of d-allose-binding protein (ALBP; a structural homolog of the LacI core domain) to determine the mechanism by which coupled torsional changes of the backbone produced large conformational changes in the region analogous to the core pivot of LacI. Plots of the φ and ξ angles for the residues of the core pivot were constructed over the entire 210-psec trajectory (Fig. 7B ▶). The standard deviation from the mean φ/ξ angle for each data set was also evaluated for the complete 210-psec simulation (Supplemental Material). Examination of φ/ξ versus time identified those data sets with a standard deviation >9° that adopted at least two distinct conformations at some point in the trajectory. The ξ angles of P320’, P320, V321’, and V321 were exceptions; each had a standard deviation >9° due to a flip of the pucker in the proline ring. The data illustrate that the highest degree of mobility resides in the backbones of residues F161’, H163′, E164’, Q291’, L318’, L319’, V321’, and S322’ in the trigger monomer, and F161, E164, K290, Q291, D292, L318, L319, V321, and S322 in the response monomer (Supplemental Material). The majority of backbone rearrangements occur after 90 psec and contribute to pathway 3.
In addition, many side chains in the core pivot form a hydrophobic cluster located at the back of the inducer-binding pocket. On both monomers, most of these hydrophobic side chains make van der Waals contact with F161 (Fig. 7A ▶). In the equivalent region of ALBP, Magnusson et al. (2002) theorized that water molecules acted as structural "ball bearings" to facilitate the subdomain conformational change. Similarly, Mowbray and Bjorkman (1999) studied conformational changes in ribose-binding protein (RBP) and discovered that water molecules play an important role in its hinge region (equivalent to the core pivot in LacI), but they noted that LacI likely cannot use a similar mechanism due to hydrogen-bonding differences involving residues F161 and S162. We propose that LacI may use the inherent hydrophobicity of the core pivot, especially F161, to facilitate the conformational change—literally "greasing" the transition machinery.
Pathway 3
The third pathway extends from the back of the inducer-binding pocket across the monomer–monomer interface via the H74–H74’ π-stacking (Fig. 1A ▶). This pathway appears to propagate the additional changes needed for cooperative inducer-binding on the second monomer. After the loss of the K84–K84’ interaction (90 psec), both N-subdomains clamp inward, thereby exerting bending forces at their respective core pivots. Accordingly, substantial backbone rearrangements take place in the core pivots to accommodate the conformational changes at the interface. As a possible consequence of these core hinge backbone motions, the side chain of F293 moves ∼2 Å closer to the side chain of F161 at 110 psec; F293’ and F161’ do the same (Fig. 8 ▶). The φ angle of F161’ also has a noticeable change at 110 psec (Fig. 7B ▶).
Figure 8.

Interatomic distance versus time plots between F293 and F161, two residues in the core pivot hydrophobic cluster. The dotted line represents the approximate average distance at the start of the simulation and aids visual inspection. F293’ moves toward F161’ at 110 psec, whereas F293 moves closer to F161 at the same time. The asymmetry of the two endpoints is the result of asymmetry from the original crystal structure.
The F161 ring makes van der Waals contact with the aliphatic carbons of Q291 in the DNA-bound state, and F161’ and Q291’ are symmetric. These glutamines (291 and 291’) provide two ends of hydrogen-bond networks that extend from the back of the inducer-binding pockets, through the base of the pockets, and out to the N-subdomain interface separating the monomers (Fig. 1A ▶). These hydrogen-bond networks undergo substantial changes throughout the simulation. Other residues involved are R197, N246, Q248, D274, and L73. Even though each monomer uses the same network, changes in the trigger and response monomers are asymmetric at intermediate time points, as well as in the equilibrated target structure.
The other termini of the hydrogen-bond networks are positioned near the entrance to the inducer-binding pockets at the N-subdomain interface (Q248 and Q248’; Fig.1A ▶). This region of the protein is extensively solvated in both conformations and has significant hydrogen bonding. As the simulation progresses (155 psec), the side chain of Q248’ flips away from the inducer-binding pocket and interacts with the Nɛ2 atom of H74, momentarily destabilizing the π-stacking interaction across the interface between H74 and H74’. In addition, a single water molecule forms new hydrogen bonds with the side chain oxygen of N246’ and the backbone oxygen of L73’. Consequently, this Q248’ flip may be due to a combination of three factors: (1) stabilization of the monomer–monomer interface after the onset of the H74-H74’ π-stacking (110 psec), (2) closure of the N-subdomain interface at β-strands B and B’ (140–170 psec), and (3) the presence of a lone water molecule, which preserves the continuity of the hydrogen bonding between N246’ and L73’, allowing Q248’ to break its contacts with these two residues. The transient hydrogen bonds mentioned above are only detected via the use of TMD.
The disruption created by the Q248’ flip allows the side chains of H74 and H74’ to change conformation, rotating 180° (170 psec), after which they regain their π-stacking interaction (Fig. 6B ▶, arrow 2). In turn, this rotation causes the backbones of H74 and H74’ to flip, momentarily disrupting the hydrogen bonding between the side chain oxygen of Q248 and the backbone -NH of H74. Finally, the side chain of Q248 rotates 180° (185 psec), forming a hydrogen bond with both the backbone oxygen of L73 and the side chain oxygen of N246. The side chain-NH of N246 is hydrogen-bonded to one of the side chain oxygens of D274, thereby continuing the hydrogen-bond network across the interface. Hence, movement generated on one monomer (the flip by Q248’) passes across the interface between the two monomers (by means of the histidine π-stacking), and is felt by the second monomer (rotation of Q248). Communication between the monomers across this interface at this position could contribute to cooperativity of inducer binding.
However, the Q248’ flip is the only feature that is not consistently present in repeated trajectories. Thus, changes at Q248 cannot be always responsible for the change in conformation of the H74–H74’ π-stacking. In the remaining six trajectories, the π-stack flip appears to be mediated by water molecules present in the interface. The flip occurs only after the N-subdomain interface completely closes (denoted by the stabilization of the interatomic distance between the backbones of V96’ and V96; see Fig. 3B ▶).
Interpretation of experimental data
Both phenotypic and biochemical studies demonstrate that a number of residues implicated by the TMD simulation are important to LacI function. The simulated allosteric transition pathway provides a spatial and temporal framework for interpreting some of these data.
Mutations of nearly every residue of lac repressor have been studied phenotypically to detect whether a mutant affects loss of either repression or induction (Suckow et al. 1996). Lack of repression could be due to improper folding, assembly defects, or changes in DNA-binding affinity, whereas loss of induction could result from changes in either inducer binding or propagation of the allosteric signal to the DNA-binding domain. In the intact protein (with residues 2–329 individually mutated), mutations at ∼56% of the sites alter function (Suckow et al. 1996). Here, ∼75% of residues that participate in the simulated pathways are affected by mutation, a result indicating that TMD identified functionally relevant positions. In addition, residues identified in this study comprise 60% of all positions that generate inducer insensitivity (is) positions. Because another 20% of is residues are located in the immobile C-subdomain, TMD identified the majority of residues with this phenotype (Suckow et al. 1996).
Of necessity, biochemical analysis of purified repressor protein is limited to a smaller number of mutant proteins. However, these studies provide a more detailed picture of mutational effects on function. Several interesting residues are summarized below, whereas other residues that participate in the conformational change are summarized in Table 3.
Table 3.
Mutational data for pathways 1 through 3
| Residuea | Mutantsb | Locationc | Residuea | Mutantsb | Locationc |
| Pathway 1 | Pathway 2 | ||||
| D149d | is, i− | BP | H74d,j | is | π-stacking |
| Y126 | BP | Q78 | is | π-stacking | |
| L128 | BP | 276d | is | π-stacking | |
| S193 | is | BP | F293 | is | HP |
| R197e | is | BP | L71 | is, i− | π-stacking |
| I64 | i− | NSH | S77k | is, i− | π-stacking |
| V66 | is, i− | NSH | Pathway 3 | ||
| V80 | is | NSH | F161 | is, i− | CP, HP |
| I83 | is, i− | NSH | S162 | CP | |
| L90 | NSH | H163 | CP | ||
| V94 | is, i− | NSH | E164 | CP | |
| V96 | is | NSH | K290 | CP | |
| I123 | i− | NSH | Q291 | is | CP, HB |
| L146 | i− | NSH | D292 | is, i− | CP |
| V301 | is, i− | NSH | L295 | CP, HP | |
| L304 | is, i− | NSH | L296d | is, i− | CP, HP |
| L305 | is | NSH | L318 | CP | |
| K84f,g,h | is | MM | L319 | is, i− | CP, HP |
| V95 | is | MM | P320 | CP, HP | |
| S97 | is | MM | V321d | is, i− | CP, HP |
| M98d | is, i− | MM | S322 | CP | |
| D88i | is | MM | L73 | is | HB |
| E100 | MM | N246d | is, i− | HB | |
| S93 | DNA | Q248d | is | HB | |
| N113 | is, i− | DNA | D274d,l | is, i− | HB |
| Q117 | DNA |
a Underlined residues contact inducer.
bSuckow et al. 1996. Phenotypic analysis of mutations. i− means no repression, whereas is signifies no induction.
c BP indicates inducer binding pocket; NSH, N-subdomain hydrophobic group; MM, N-subdomain monomer–monomer interface; DNA, top of N-subdomain, beneath DNA binding domains; π-stacking, near H74-H74’; HP, core pivot hydrophobic group; CP, core pivot; and HB, hydrogen bond network that extends from back of inducer binding pocket to monomer–monomer interface.
dSwint-Kruse et al. 2001; . Mutation restores repressor function to Y282D monomer mutant, presumably through restoring dimerization.
eSwint-Kruse et al. 1991; . Mutation of R197 (L, G, and K) decreases affinity for inducer.
fBell et al. (2001). K84L mutant has reduced affinity for operator and decreased inducer-binding rate-constants.
gChang et al. 1993. K84 mutants (A and L) coupled to Y282D monomer mutant result in 100- to 200-fold decrease in association and dissociation inducer-binding rate constants.
hMatthews and Nichols 1997;. Triple mutants composed of K84L, deletion of 11 C-terminal residues, and Y282D demonstrated that apolar mutation at residue 84 stabilizes the monomer–monomer interface.
iChang et al. 1994. D88 mutants (A, E, R, and N) decrease inducer-binding affinity by increasing inducer dissociation rate constants.
jBarry and Matthews (1999). H74A and H74L decreased operator binding, whereas H74W and H74F increased affinity for operator.
k Chou and Matthews 1989. S77L binds DNA nonspecifically.
l Chang and Matthews 1995. D274N and D274E lower inducer-binding affinity.
Mutations in the region of the inducer-binding pocket dramatically affect induction. Most substitutions for R197, S193, D149, F293, D274, N246, and Q248 yield a phenotype to that is unable to respond to inducer (Suckow et al. 1996). Although several of these sites contact inducer, TMD illustrates additional roles that S193 and D149 may play in propagating the allosteric signal. One subset of the simulations also provides a possible mechanism through which Q248 relays the effects of inducer binding both within and between monomers. Hence, altering Q248 could have a twofold effect on allostery—blocking propagation of the inducer-binding signal and disrupting binding pocket cooperativity.
Mutations of the N-subdomain interface also affect allostery. Apolar substitutions at K84 yield a phenotype that is unresponsive to inducer (Suckow et al. 1996), although K84L can still bind IPTG with wild-type affinity, albeit with significantly slower and biphasic kinetic rate constants (Chang et al. 1993). In wild-type repressor, the highly charged lysine side chains may destabilize the otherwise hydrophobic monomer–monomer interface. When the lysines are mutated to hydrophobic residues, the interface is highly stabilized (Nichols and Matthews 1997), presumably by enhanced hydrophobic interactions, especially from contacts similar to those made by the hydrocarbon stem of K84 with the side chain of M98’. The enhanced stability of the interface may prevent the monomers from effectively sliding against one another during the transition and, therefore, reduce the inducibility of the protein. The recent crystal structure of the K84L mutant provides direct evidence for modified hydrophobic packing interactions within this interface (Bell et al. 2001). We speculate that stabilization of the K84L interface slows the asymmetric conformational change predicted by TMD so that it may be detected experimentally as biphasic kinetics.
Mutations of D88, which anchors K84 in the wild-type induced conformation, also decrease inducibility of the repressor (Chang et al. 1994; Suckow et al. 1996). Mutating D88 would deprive the K84 cation of a negative anchor in the induced state. In particular, the D88K substitution (Chang et al. 1994) could repel the K84 pair and disfavor the inducer-bound conformation. During equilibration of the inducer-bound structure (1LBH), K84 tends to form electrostatic interactions only with D88 and not with E100. Interestingly, mutations of E100 have no effect on inducibility (Suckow et al. 1996). Structurally, the differential importance of D88 and E100 may be due to their distinct structural environments. D88 lies on a rigid helix (helix 5), whereas E100 lies on a flexible loop (Bell et al. 2001). Greater mobility of E100 could compromise the strength of its interactions with K84. This possibility is further supported by the fact that although K84’ could form an interaction with E100 during one equilibration, K84 did not interact with E100’. This preference of K84 for D88 was also observed in most of the target structure (inducer-bound) equilibrations.
Substitutions at other positions may influence the interactions delineated above. For example, the observed decrease in inducibility for the A110K mutation (Müller-Hartmann and Müller-Hill 1996) could derive from the lysine side chain at position 110 forming an ionic interaction with D88’, thus destabilizing the interaction between K84’ and D88’.
As noted previously, H74 appears to be integral to signal transmission across the subunit interface. Early examination of the crystal structure led to the hypothesis that forming an H74–D278’ ion pair in the induced conformation is critical to the conformational change (Lewis et al. 1996). However, Barry and Matthews (1999) demonstrated that although many mutations of H74 and D278 affect the allosteric conformational change, these effects were independent of ionic charge. The observed π-stacking of H74–H74’ provides a mechanism that explains the biochemical results (Barry and Matthews 1999). In addition, H74W and H74F mutants increase apparent DNA-binding affinity by changing the allosteric constant to favor the state with high DNA-binding affinity (Barry and Matthews 1999). Replacing histidine with these large aromatic residues likely stabilizes the DNA-bound structure of the protein in which H74 and H74’ form intra-subunit hydrophobic contacts. Furthermore, these large aromatic residues are not likely to engage in π-stacking that appears to facilitate induction.
Finally, F161, located in the core pivot region, is in a highly hydrophobic environment and may be key to the mobility of the N-subdomain. Consistent with this observation, F161 mutants demonstrate that changing this residue can affect either repression or induction (Suckow et al. 1996). Mutations at this position may "lock" the protein in one conformation or the other. All members of the core pivot hydrophobic group, except L295 and P320, are phenotypically affected by mutations (Table 3; Suckow et al. 1996). In contrast, of the remaining polar core pivot residues, mutation only affected Q291 and D292 (Table 3; Suckow et al. 1996). These differential effects may be the result of side chain versus backbone involvement for each of the core pivot residues in the allosteric transition. As pointed out earlier, the side chains of the core pivot hydrophobic group cluster together around F161 (Fig. 7B ▶). In contrast, the side chains of S162, H163, E164, K290, L318, and S322 all point outward and away from the protein interior and interact extensively with solvent during the simulation, indicating a possible explanation for minimal effects of mutations at these residues.
Additional structural insight may be obtained by comparing structures of homologous proteins. As noted earlier, the hydrophobic region of the core pivot may function like the water molecule "ball-bearings" of ALBP and RBP (Mowbray and Bjorkman 1999; Magnusson et al. 2002). Conformational changes are only known for one other related repressor, Escherichia coli purine repressor (PurR). In PurR, the "allosteric switch loop" is analogous to the LacI flexible loop, and PurR mutations of W147 (homologous to LacI V150) show that this residue plays an intimate part in the allosteric response of the protein (Huffman et al. 2002). The side chain of PurR W147 undergoes significant conformational changes in the transition between the two regulatory states, apparently stabilizing two distinct conformations of the allosteric switch loop (Huffman et al. 2002). The TMD results for LacI reveal a different mechanical role: Changes in side chains are more subtle (e.g., hydrogen bonding at D149), whereas mobility in this loop propagates the inducer-binding signal from the binding pocket to the monomer–monomer interface that lies below the DNA-binding site.
Finally, a new computational technique that uses orthologous and paralogous proteins to identify specificity-determining residues in proteins has been applied to the LacI/PurR family (Mirny and Gelfand 2002). By using this method, a dozen specificity-determining residues were identified (note that the bold residues below are implicated by TMD in the allosteric transition). Four of these residues are in the DNA-binding domain (Y17, Q18, V52, and A57), six are in the inducer-binding pocket (N125, D149, V150, F161, W220, and Q248), and two "false positives" lie further from the DNA and ligand binding sites (R101 and Q117). Interestingly, TMD indicates that the specificity false positive (Q117) plays a role in transmitting the allosteric signal.
Conclusion
This article reports the allosteric conformational pathway of LacI predicted by targeted molecular dynamics simulations. In this method, the effect of the ligand binding was achieved by using the inducer-bound protein conformation as the target of the simulated conformational transition from the DNA/anti-inducer-bound form. The C-subdomains do not undergo significant motions during the transition and anchor the N-subdomains. Therefore, our discussion focuses primarily on the changes of the extensive interactions both within and across the two N-subdomains.
The simulated trajectories indicate that the allosteric signal originates asymmetrically in the inducer-binding site of one (trigger) monomer and propagates to the other (response) monomer through various noncovalent interactions of three interconnected pathways. Asymmetry originates in the interactions involved with D149 during equilibration of the target structure. We believe this reflects experimentally relevant changes between the bound crystal structure and the unliganded equilibration structure. Neither D149 nor S193 has been targeted for biochemical studies of substitutions at these sites. Therefore, this work predicts results for new projects in vitro (biochemical characterization of mutants at 149 and 193) and in silico (determining whether the contact surface between the two residues dictates the trigger monomer).
Both the asymmetry and conformational changes observed in the trajectory are repeatable, and either monomer may function as the trigger. The K84–K84’ interaction connects pathway 1 to pathways 2 and 3, whereas the H74–H74’ π-stacking links pathways 2 and 3. The conformational changes observed reposition the N-subdomains so that they no longer make the contacts with the DNA-binding domain that appear to be requisite for high affinity binding. The detection of the intermediate features of the allosteric transition pathway, such as the H74–H74’ π-stacking interaction, demonstrates the power of molecular dynamics simulations. Such intermediates are not apparent in the available crystal structures. Overall, the results from the simulated trajectories are in agreement with a wide range of experimental biochemical and genetic data, and provide a spatial and temporal framework for interpreting these existing data and for designing new experiments. Targeted molecular dynamics provides an extremely useful tool to assess intermediate states along allosteric or conformational transition pathways. The asymmetric interactions and the three pathways delineated by this approach offer new insights into the atomic-level functional mechanism of LacI.
Materials and methods
The targeted molecular dynamics method (Schlitter et al. 1993), interfaced to the CHARMM program (version 28; Brooks et al. 1983), was used to simulate the conformational transition in LacI. This method uses a standard molecular dynamics potential with a time-dependent constraining force that is directly correlated to the difference between the conformation of the moving structure and the target structure at each time step. Hence, the transition is achieved by slowly "pulling" the initial structure toward the target by minimizing the RMS difference between the two structures over time.
The coordinates of a certain conformation j are given by vector xj = (x1j,x2j,x3j…x3Nj), here N is the number of atoms, where x1, x2, and x3 represent the components of a Cartesian coordinate system for the first atom, and so forth. The distance, γ, between two conformations j and k can be calculated as 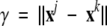 . γ is then used in the time-dependent constraint as a directing control parameter:
. γ is then used in the time-dependent constraint as a directing control parameter:
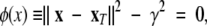 |
where x represents the current conformation in the simulation, and xT the target conformation (Schlitter et al. 1993). Consequently, decreasing γ forces the system to undertake the conformational transition toward the target. γ is decreased after each time step (Δt) by the following equation:
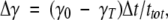 |
here 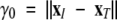 , where xI and xT are the initial and target conformations in TMD, respectively; γT is the final distance allowed between the structures at the end of the simulation (≤0.3 Å); and ttot is the total time of the simulation predefined by the user. Holding γ constant at each time step limits only one of the total 3N degrees of freedom. Hence, the system still has the ability to adequately sample the conformational space (Schlitter et al. 1994).
, where xI and xT are the initial and target conformations in TMD, respectively; γT is the final distance allowed between the structures at the end of the simulation (≤0.3 Å); and ttot is the total time of the simulation predefined by the user. Holding γ constant at each time step limits only one of the total 3N degrees of freedom. Hence, the system still has the ability to adequately sample the conformational space (Schlitter et al. 1994).
The initial (ONPF-DNA-bound; Bell and Lewis 2000) and target (IPTG-bound; Lewis et al. 1996) crystal structures were obtained from the PDB (PDB codes 1EFA and 1LBH, respectively). The structure of the core tetrameric proteolytic fragment, 1TLF, was also obtained from the PDB (Friedman et al. 1995). LacI naturally occurs as a homo-tetramer but can be considered as a dimer of dimers, because each dimer is a functional repressor (Chakerian et al. 1991). Residues 1–62 and 330–360 were omitted from the structures used in the simulation. Residues 1–61 contain the DNA-binding domain and are not resolved in the IPTG-bound crystal structure. Residues 330–360 contain the tetramerization domains and were deleted because 1EFA is a truncated dimer mutant.
A chloride anion was chosen for use as the anion present in the 1EFA structure (Bell and Lewis 2000). Larger polyatomic anions, such as phosphate, were found to exceed size limitations imposed by the surrounding groups (data not shown). The ligands, inducer (IPTG) and anti-inducer (ONPF), were also not included in the simulation because TMD requires identical sets of atoms in the initial and target conformations. A further limitation of the X-ray studies is that these ligands could be fit to the density in two different orientations (Lewis et al. 1996; Bell and Lewis 2000). A shell of 925 water molecules was placed around each protein (1EFA and ILBH), yielding a system with 7677 atoms. A modified TIP3P potential was used for the water molecules (Jorgensen 1981; Neria et al. 1996). 1TLF was treated similarly. To compensate for the limited number of water molecules, the atomic charges on the ionic side chains of arginine, lysine, glutamate, and aspartate were scaled by a factor of 0.25 (Ma et al. 2002), and a distance-dependent dielectric constant was used (Brooks et al. 1988). The atomic charge of the chloride ion was scaled by a factor of 0.7, which was chosen iteratively to match the scaling of side chains.
Because water shells were added to the initial (1EFA) and the target (1LBH) structures, equilibrations were performed on both the initial (1EFA, water, and chloride) and target (1LBH, water, and chloride) systems. The water shells were first equilibrated for 500 steps of steepest descent, whereas the two proteins were fixed. Both systems (initial and target) were then minimized by 500 steps of steepest descent, followed by 1000 steps of adopted basis Newton-Raphson, and finally 10 psec of molecular dynamics, whereas a small harmonic constraining force was imposed on the main chain and side chains of the proteins. 1TLF was treated similarly.
To explore asymmetric changes that originate during this procedure, the equilibration was carried out four times for the operator-bound starting structure, nine times for the inducer-bound target structure, and five times for the core proteolytic fragment (Table 2). The crystal structure of 1EFA is symmetric, and repeated equilibration of the modified starting structure elicited similar structural changes. The most significant result involves the loss of the side chain hydrogen bonds between R197 and S193 on both monomers of the starting structure. These side chains adopt distances characteristic of the inducer-bound crystal structure, which may be the result of the removing ONPF from the binding pocket. In addition, the side chains of Q248, Q248’, Q291, and Q291’ change orientation. These changes are likely because glutamine can easily form hydrogen bonds in varied conformations.
In contrast, the inducer-bound crystal structure, 1LBH, itself has some asymmetry in the regions around F161 and D149. In addition, Q248 and Q248’ have different side chain conformations (180° flip at Cγ). During equilibration of the modified induced structure, both inducer-bound structures were subjected to changes in the region around D149 and S193. In all cases, the chloride anion of the target structure equilibrated to the highly solvated environment outside of the N-subdomain monomer–monomer interface. The crystal and equilibrated structures for 1TLF showed similar patterns. The 210-psec TMD trajectory was performed a total of seven times to analyze the repeatability of the pathway (see Table 2). The first 10 psec of each trajectory is essentially a "final equilibration" to ensure that no angular momentum is generated as the initial structure is pulled toward the target structure and was included in the plots/pathway for completeness.
No other constraints were imposed during the actual simulation other than the standard TMD constraint, φ(x) (Schlitter et al. 1993); that is, no portions of the molecule were artificially anchored in space. A dynamic time step of 2 fsec was used with the leapfrog integration scheme (Allen and Tildesley 1980), and the TMD transition was achieved by 210-psec simulation for the results described in detail. The CHARMM polar hydrogen potential function (PARAM19) was used (Neria et al. 1996). The length of all bonds involving hydrogen atoms was kept constant by the SHAKE algorithm (Ryckaert et al. 1977). The system was coupled to a 300 K heat bath to keep the temperature relatively constant (Berendsen et al. 1984).
The simulation was performed on an SGI Indigo. The approach implemented here is based on previous studies of the oncogenic protein ras p21 (Ma and Karplus 1997), the molecular chaperonin GroEL (Ma et al. 2000), and the rotary motor F1-ATPase (Ma et al. 2002), each of which undergoes significant conformational changes upon ligand binding.
For data analysis, alignments of the C-subdomains were performed by using CHARMM (Brooks et al. 1983), and structures were visualized with Quanta (Accelrys). Contacts in specific regions were analyzed with CSU (Sobolev et al. 1999).
Acknowledgments
We would like to thank Michelle Calabretta and Hongli Zhan for helpful discussions. This research was supported in part by grants to J.M. from the American Heart Association (AHA-TX0160107Y), the Robert A. Welch Foundation (Q-1512), the NIH (R01-GM067801), and a National Science Foundation Career Award (MCB-0237796). J.M. is a recipient of the Award for Distinguished Young Scholars Abroad from National Natural Science Foundation of China. This work was supported by grants to K.S.M. from the National Institutes of Health (GM22441) and the Robert A. Welch Foundation (C-576). L.S.K. was supported in part by a training grant from the National Library of Medicine to the W.M. Keck Center for Computational and Structural Biology.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03188303.
Footnotes
The DNA-binding domain was intact on the protein used to create crystals for 1LBH; however, this region did not exhibit an electron density, presumably due to high mobility in the absence of DNA (Lewis et al. 1996).
The crystal structure with PDB code 1TLF was generated from LacI protein that was proteolytically treated to remove the DNA-binding domain (Friedman et al. 1995). Even though the 1LBH structure does not have any electron density for this domain (Lewis et al. 1996), its physical presence might affect small but important changes in the structure that could influence the TMD of the core domain. Further, the protein used to create 1TLF differs at position 109 (alanine) from repressor used to create 1LBH and 1EFA (threonine 109). Thus, the TMD requirement for identical atoms precludes the use of 1TLF without further modeling, which would introduce additional computational error into the trajectory.
References
- Allen, M.P. and Tildesley, D.J. 1980. Computer simulation of liquids, pp. 78–82. Clarendon Press, Oxford.
- Barry, J.K. and Matthews, K.S. 1999. Substitutions at histidine 74 and aspartate 278 alter ligand binding and allostery in lactose repressor protein. Biochemistry 38 3579–3590. [DOI] [PubMed] [Google Scholar]
- Bell, C.E. and Lewis, M. 2000. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 7 209–214. [DOI] [PubMed] [Google Scholar]
- Bell, C.E., Barry, J., Matthews, K.S., and Lewis, M. 2001. Structure of a variant of lac repressor with increased thermostability and decreased affinity for operator. J. Mol. Biol. 313 99–109. [DOI] [PubMed] [Google Scholar]
- Berendsen, H.J.C., Postma, J.P.M., von Gunsteren, W.F., DiNola, A., and Haak, J.R. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 3684–3690. [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Brooks III, C.L., Karplus, M., and Pettitt, B.M. 1988. Proteins: A theoretical perspective of dynamics, structure, and thermodynamics. Adv. Chem. Phys. 71 1–249. [Google Scholar]
- Chakerian, A.E., Tesmer, V.M., Manly, S.P., Brackett, J.K., Lynch, M.J., Hoh, J.T., and Matthews, K.S. 1991. Evidence for leucine zipper motif in lactose repressor protein. J. Biol. Chem. 266 1371–1374. [PubMed] [Google Scholar]
- Chang, W.I. and Matthews, K.S. 1995. Role of Asp274 in lac repressor: Diminished sugar binding and altered conformational effects in mutants. Biochemistry 34 9227–9234. [DOI] [PubMed] [Google Scholar]
- Chang, W.I., Olson, J.S., and Matthews, K.S. 1993. Lysine 84 is at the subunit interface of lac repressor protein. J. Biol. Chem. 268 17613–17622. [PubMed] [Google Scholar]
- Chang, W.-I., Barrera, P., and Matthews, K.S. 1994. Identification and characterization of aspartate residues that play key roles in the allosteric regulation of a transcription factor: Aspartate 274 is essential for inducer binding in lac repressor. Biochemistry 33 3607–3616. [DOI] [PubMed] [Google Scholar]
- Chou, W.-Y. and Matthews, K.S. 1989. Mutation in hinge region of lactose repressor protein alters physical and functional properties. J. Biol. Chem. 264 6171–6176. [PubMed] [Google Scholar]
- Daly, T.J. and Matthews, K.S. 1986. Allosteric regulation of inducer and operator binding to the lactose repressor. Biochemistry 25 5479–5484. [DOI] [PubMed] [Google Scholar]
- Friedman, A.M., Fischmann, T.O., and Steitz, T.A. 1995. Crystal structure of lac repressor core tetramer and its implications for DNA looping. Science 268 1721–1727. [DOI] [PubMed] [Google Scholar]
- Ha, J., Spolar, R.S., and Record Jr., M.T. 1989. Role of hydrophobic effect in stability of site-specific protein-DNA complexes. J. Mol. Biol. 209 801–816. [DOI] [PubMed] [Google Scholar]
- Huffman, J.L., Lu, F., Zalkin, H., and Brennan, R.G. 2002. Role of residue 147 in the gene regulatory function of the Escherichia coli purine repressor. Biochemistry 41 511–520. [DOI] [PubMed] [Google Scholar]
- Jacob, F. and Monod, J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3 318–356. [DOI] [PubMed] [Google Scholar]
- Jobe, A. and Bourgeois, S. 1972. lac repressor-operator interaction, VI: The natural inducer of the lac operon. J. Mol. Biol. 69 397–408. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L. 1981. Transferable intermolecular potential functions for water, alcohols, and ethers: Application to liquid water. J. Am. Chem. Soc. 103 335–340. [Google Scholar]
- Kalodimos, C.G., Folkers, G.E., Boelens, R., and Kaptein, R. 2001. Strong DNA binding by covalently linked dimeric lac headpiece: Evidence for the crucial role of the hinge helices. Proc. Natl. Acad. Sci. 98 6039–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalodimos, C.G., Boelens, R., and Kaptein, R. 2002a. A residue-specific view of the association and dissociation pathway in protein–DNA recognition. Nat. Struct. Biol. 9 193–197. [DOI] [PubMed] [Google Scholar]
- Kalodimos, C.G., Bonvin, A.M., Salinas, R.K., Wechselberger, R., Boelens, R., and Kaptein, R. 2002b. Plasticity in protein–DNA recognition: lac repressor interacts with its natural operator O1 through alternative conformations of its DNA-binding domain. EMBO J. 21 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lee, J. and Goldfarb, A. 1991. lac repressor acts by modifying the initial transcribing complex so that it cannot leave the promoter. Cell 66 793– 798. [DOI] [PubMed] [Google Scholar]
- Lewis, M., Chang, G., Horton, N.C., Kercher, M.A., Pace, H.C., Schumacher, M.A., Brennan, R.G., and Lu, P. 1996. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 271 1247–1254. [DOI] [PubMed] [Google Scholar]
- Ma, J. and Karplus, M. 1997. Molecular switch in signal transduction: Reaction paths of the conformational changes in ras p21. Proc. Natl. Acad. Sci. 94 11905–11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J., Sigler, P.B., Xu, Z., and Karplus, M. 2000. A dynamic model for the allosteric mechanism of GroEL. J. Mol. Biol. 302 303–313. [DOI] [PubMed] [Google Scholar]
- Ma, J., Flynn, T.C., Cui, Q., Leslie, A.G., Walker, J.E., and Karplus, M. 2002. A dynamic analysis of the rotation mechanism for conformational change in F1-ATPase. Structure 10 921–931. [DOI] [PubMed] [Google Scholar]
- Magnusson, U., Chaudhuri, B.N., Ko, J., Park, C., Jones, T.A., and Mowbray, S.L. 2002. Hinge-bending motion of d-allose-binding protein from Escherichia coli: Three open conformations. J. Biol. Chem. 277 14077–14084. [DOI] [PubMed] [Google Scholar]
- Matthews, K.S. 1992. DNA looping. Microbiol. Rev. 56 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, K.S. and Nichols, J.C. 1998. Lactose repressor protein: Functional properties and structure. Prog. Nucl. Acid Res. Mol. Biol. 58 127–164. [DOI] [PubMed] [Google Scholar]
- Matthews, K.S., Falcon, C.M., and Swint-Kruse, L. 2000. Relieving repression. Nat. Struct. Biol. 7 184–187. [DOI] [PubMed] [Google Scholar]
- McGaughey, G.B., Gagne, M., and Rappe, A.K. 1998. π-Stacking interactions: Alive and well in proteins. J. Biol. Chem. 273 15458–15463. [DOI] [PubMed] [Google Scholar]
- Mirny, L.A. and Gelfand, M.S. 2002. Using orthologous and paralogous proteins to identify specificity-determining residues in bacterial transcription factors. J. Mol. Biol. 321 7–20. [DOI] [PubMed] [Google Scholar]
- Monod, J., Wyman, J., and Changeux, J.-P. 1965. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12 88–118. [DOI] [PubMed] [Google Scholar]
- Mowbray, S.L. and Bjorkman, A.J. 1999. Conformational changes of ribose-binding protein and two related repressors are tailored to fit the functional need. J. Mol. Biol. 294 487–499. [DOI] [PubMed] [Google Scholar]
- Müller-Hartmann, H. and Müller-Hill, B. 1996. The side chain of the amino acid residue in position 110 of the Lac repressor influences its allosteric equilibrium. J. Mol. Biol. 257 473–478. [DOI] [PubMed] [Google Scholar]
- Neria, E., Fischer, S., and Karplus, M. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105 1902–1921. [Google Scholar]
- Nichols, J.C. and Matthews, K.S. 1997. Combinatorial mutations of lac repressor. J. Biol. Chem. 272 18550–18557. [DOI] [PubMed] [Google Scholar]
- O’Gorman, R.B., Rosenberg, J.M., Kallai, O.B., Dickerson, R.E., Itakura, K., Riggs, A.D., and Matthews, K.S. 1980. Equilibrium binding of inducer to lac repressor/operator DNA complex. J. Biol. Chem. 255 10107–10114. [PubMed] [Google Scholar]
- Riggs, A.D., Newby, R.F., and Bourgeois, S. 1970. lac Repressor–operator interaction. J. Mol. Biol. 51 303–314. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J.P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23 327–341. [Google Scholar]
- Schlax, P.J., Capp, M.W., and Record Jr., M.T. 1995. Inhibition of transcription initiation by lac repressor. J. Mol. Biol. 245 331–350. [DOI] [PubMed] [Google Scholar]
- Schlitter, J., Engels, M., Kruger, P., Jacoby, E., and Wollmer, A. 1993. Targeted molecular dynamics simulation of conformational change: Application to the T-R transition in insulin. Mol. Sim. 10 291–308. [Google Scholar]
- Schlitter, J., Engels, M., and Kruger, P. 1994. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 12 84–89. [DOI] [PubMed] [Google Scholar]
- Sobolev, V., Sorokine, A., Prilusky, J., Abola, E.E., and Edelman, M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15 327–332. [DOI] [PubMed] [Google Scholar]
- Spotts, R.O., Chakerian, A.E., and Matthews, K.S. 1991. Arginine 197 of lac repressor contributes significant energy to inducer binding: Confirmation of homology to periplasmic sugar binding proteins. J. Biol. Chem. 266 22998–23002. [PubMed] [Google Scholar]
- Spronk, C.A., Slijper, M., van Boom, J.H., Kaptein, R., and Boelens, R. 1996. Formation of the hinge helix in the lac repressor is induced upon binding to the lac operator. Nat. Struct. Biol. 3 916–919. [DOI] [PubMed] [Google Scholar]
- Spronk, C.A., Bonvin, A.M., Radha, P.K., Melacini, G., Boelens, R., and Kaptein, R. 1999a. The solution structure of lac repressor headpiece 62 complexed to a symmetrical lac operator. Structure 7 1483–1492. [DOI] [PubMed] [Google Scholar]
- Spronk, C.A., Folkers, G.E., Noordman, A.M., Wechselberger, R., van den Brink, N., Boelens, R., and Kaptein, R. 1999b. Hinge-helix formation and DNA bending in various lac repressor-operator complexes. EMBO J. 18 6472–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straney, S.B. and Crothers, D.M. 1987. Lac repressor is a transient gene-activating protein. Cell 51 699–707. [DOI] [PubMed] [Google Scholar]
- Suckow, J., Markiewicz, P., Kleina, L.G., Miller, J., Kisters-Woike, B., and Müller-Hill, B. 1996. Genetic studies of the lac repressor XV†: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J. Mol. Biol. 261 509–523. [DOI] [PubMed] [Google Scholar]
- Swint-Kruse, L., Matthews, K.S., Smith, P.E., and Pettitt, B.M. 1998. Comparison of simulated and experimentally determined dynamics for a variant of the Lacl DNA-binding domain, Nlac-P. Biophys. J. 74 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swint-Kruse, L., Elam, C.R., Lin, J.W., Wycuff, D.R., and Matthews, K.S. 2001. Plasticity of quaternary structure: 22 ways to form a LacI dimer. Protein Sci. 10 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swint-Kruse, L., Larson, C., Pettitt, B.M., and Matthews, K.S. 2002. Fine-tuning function: Correlation of hinge domain interactions with functional distinctions between LacI and PurR. Protein Sci. 11 778–794. [DOI] [PMC free article] [PubMed] [Google Scholar]