

Abstract
Human respiratory syncytial virus (RSV) encodes a small hydrophobic (SH) protein, whose function in the life cycle of the virus is unknown. Recent channel activity measurements of the protein suggest that like other viroporins, SH may assemble into a homo-oligomeric ion channel. To further our understanding of this potentially important protein, a new strategy was implemented in order to model the transmembrane oligomeric bundle of the protein. Global searching molecular dynamic simulations of SH proteins from eight different viral strains, each at different oligomeric states, as well as different lengths of the putative transmembrane domain, were undertaken. Taken together, a total of 45 different global molecular dynamic simulations pointed to a single pentameric structure for the protein that was found in all of the different variants. The model of the structure obtained is a channel-like homopentamer whose minimal transmembrane pore diameter is 1.46 Å.
Keywords: HRSV, membrane protein, molecular dynamics, SH protein, protein structure, viroporin
Human respiratory syncytial virus (HRSV; genus Pneumovirus, subfamily Pneumovirinae, family Paramyxoviridae) is the most important cause of lower respiratory tract infections in infants and young children (Collins et al. 1996). In adults, studies indicate that HRSV is also an important cause of pneumonia (Dowell et al. 1996). RSV is an enveloped, single-stranded, negative-sense RNA virus. The RSV envelope contains three viral encoded proteins, that is, the G (attachment) and F (fusion) glycoproteins that are the major determinants of the virus (Collins et al. 1996), and SH (small hydrophobic), whose function is as yet unknown.
The SH protein consists of 64 amino acids (antigenic subgroup A) or 65 amino acids (antigenic subgroup B). One hypothesis with regard to the function of SH is that it forms ion channels (Collins and Mottet 1993), as suggested by permeability changes of Escherichia coli membranes induced by the expression of HRSV SH protein-subgroup B (Perez et al. 1997). Other studies point toward different or additional functions; SH may be required for stabilization of the viral envelope in nature, or SH may represent a viral encoded virulence factor (Bukreyev et al. 1997; Whitehead et al. 1999; Chen et al. 2000). A recent functional analysis of recombinant mutants suggests that SH is not involved in binding or infectivity, and that it inhibits virus fusion and spreading in cell culture, at least in the absence of the G protein (Techaarpornkul et al. 2001). Insight into the three-dimensional structure of the SH protein may enable a better understanding of SH’s role in the life cycle of the virus, and potentially facilitate focused screening of candidate drugs.
Prediction of a transmembrane domain structure is facilitated by its tendency to adopt (in most cases) an α-helical fold, limiting the number of possible conformations. Brünger and coworkers (Treutlein et al. 1992; Adams et al. 1995) have developed a procedure to explore transmembrane helix interactions on the basis of global searching molecular dynamics simulations. In this method, multiple symmetric bundles of helices are constructed, each differing from the other by the rotation of the helices about their axes. These are then used as starting positions for molecular dynamics and energy minimization protocols. The output structures from these simulations are compared and grouped into clusters that contain similar structures. An average of the structures forming a cluster represents a model with characteristic interhelical interactions and helix tilt.
Until recently, the correct model was selected among the several different clusters, on the basis of existing experimental data, either from mutagenesis (Lemmon et al. 1992a,b; Arkin et al. 1994) or orientational data from site-specific infrared dichroism used as spatial restraints (Kukol et al. 1999; Torres et al. 2000). Recently, an improvement to this method was suggested, in which simulations are performed on close sequence variants that are likely to share the same structure (Briggs et al. 2001; Kukol et al. 2002; Torres et al. 2002).
Here, we present a model for the structure of the transmembrane domain of SH protein of HRSV, on the basis of global molecular dynamics, using silent amino acid substitution modeling to discriminate between the different candidate structures obtained. Ambiguities with regard to the oligomeric state of the protein, and the span of the transmembrane segment of the protein, were overcome through simulation of oligomers of different sizes and sequences of different lengths. The results of the simulation incorporating eight different variants point to a model in which SH assembles into homopentameric helical bundle.
Materials and methods
SH sequences
Simulations of SH were performed using the transmembrane sequence from residues 14–41 (long TM segment), or 23–41 (short TM segment). SH variants (Fig. 1 ▶) with at least 75% identity to one another were obtained by searching the NCBI database (Altschul et al. 1997). The results consist of seven subgroup A variants (NCBI accession nos. AAG28084, AAG28086, AAG28107, AAG28111, AAG28127, AAG28139 [Chen et al. 2000], and NP-044594 [Tolley et al. 1996]), and one subgroup B variant (VSH-HRSV1 [Collins et al. 1990]). The amino acid sequence of the short TM segment is identical for the variants AAG28107 and AAG28127, and therefore, the simulations were performed for seven variants only when using the short TM segments.
Figure 1.

Sequences of the transmembrane segment of SH (TM-SH) used in the simulations. The numbers indicate the starting points for the long TM segment (14) and the short TM segment (23).
GSMDS protocol
Calculations were performed using either CNS or PCNS, the parallel-processing version of the Crystallography and NMR System (CNS version 0.3, Brunger et al. 1998), with the OPLS parameter set and united atom topology (Jorgensen and Tirado-Rives 1988), explicitly describing only polar and aromatic hydrogens. A global search was carried out in vacuo as described elsewhere in detail (Adams et al. 1995), using CHI (CNS Helical Interactions), assuming a symmetrical interaction between the helices in the homo-oligomer. Briefly, trials were carried out starting from either left or right crossing angles (i.e., ±25°, respectively). For each of these cases, the helices were rotated a total of 350° about their helical axes in 10° increments (Adams et al. 1995), so that all possible interhelical interactions were explored. Four trials were carried out from each starting configuration by use of different initial random velocities, making a total of 36 × 2 × 4 = 288 trials, each producing a final structure. Clusters of output structures were identified, defined as those that contain a minimum number of structures (typically six). Any structure belonging to a particular cluster was typically within 1.4 Å α-carbon RMSD from any other structure within that cluster. Therefore, some clusters overlap, and output structures may be members of more than one cluster. The structures belonging to each cluster were averaged and subjected to a further simulated annealing protocol. This final structure was taken as the representative of the cluster.
Analysis of the simulations
The results from the global searching molecular dynamics simulations were represented graphically by plotting each cluster representative as a function of two parameters, the helix rotation angle φ, and the crossing angle Ω, as described previously (Briggs et al. 2001). φ is the helix rotational angle about the long axis of the helices relative to some arbitrary starting position. The helical axis is a vector with starting and end points above and below a defined residue, in which the points correspond to the geometric mean of the coordinates of the five α carbons amino-terminal and the five α carbons carboxy-terminal to the defined residue.
Precise comparisons between similar clusters obtained from different variants were made by calculating the RMSD between their α carbon backbones. In the simulations, the handedness of the bundle is indicated by the helix tilt sign, positive or negative, which corresponds to left- and right-handed bundles, respectively.
Results
Analysis of the SH protein by sedimentation on sucrose gradients indicated that it assembles into a homo-oligomeric form (Collins and Mottet 1993). Chemical cross-linking generated species that appeared to represent dimers, trimers, tetramers, and pentamers (Collins and Mottet 1993). In this study, we tested the hypotheses that it assembles as a homotetramer or homopentamer. The reasoning behind this approach is that for a helical bundle to form a channel, a minimum of four helices are most likely needed. For the sake of completeness, however, homotrimers were simulated as well (see below). Furthermore, the length of the single hydrophobic region spanning the membrane is unclear as well. Two studies suggested that amino acids 14–41 (Olmsted et al. 1989; Perez et al. 1997) span the membrane, whereas another study suggested that the spanning segment consists of amino acids 23–41 (Collins and Mottet 1993).
Figure 2 ▶ shows the results of a hydrophobicity analysis of SH using the GES scale (Engelman et al. 1986), with a window size of 15 residues and a hydrophobicity threshold of ΔGwater→oil = −25 kcal/mole (Stevens and Arkin 2000). Despite the fact that the value obtained for amino acid 23 is higher than that obtained for amino acid 14, we simulated SH using both sequences, that is, 14–41 and 23–41. Thus, six different combinations were simulated for each of the eight different variants (seven different variants for the short TM segment) as follows: (1) a trimeric short bundle, (2) a trimeric long bundle, (3) a tetrameric short bundle, (4) a tetrameric long bundle, (5) a pentameric short bundle, and (6) a pentameric long bundle. The total number of global molecular dynamics searches performed was therefore 45, analyzing a total of 12,960 structures.
Figure 2.

Hydrophobicity analysis plot of the putative TM domains of SH protein, using GES scale (Engelman et al. 1986). The parameters used are a window size of 15 and the hydrophobicity threshold of ΔGwater→oil = −25 kcal/mole (Stevens and Arkin 2000), shown as a dotted line.
Tetramer simulations
The results of the global search molecular dynamics protocol of amino acids 14–41 (long TM segment) for eight variants of SH (HRSV), assuming tetrameric oligomerization, are shown in Figure 3 ▶. As in the results from glycophorin A and CD3ζ (Briggs et al. 2001), multiple clusters are obtained. However, only one conformation located at φ ~122° and Ω ~14°, persists in all sequences. No structure within this complete set differs from any other in the set by more than 0.82 Å Cα RMSD. Note that the actual calculation of the rotation pitch angle φ is not as accurate a representation of the similarity between two structures as is the Cα RMSD. As such, structures with slightly differing rotational pitch angles might still be relatively similar and vice versa.
Figure 3.
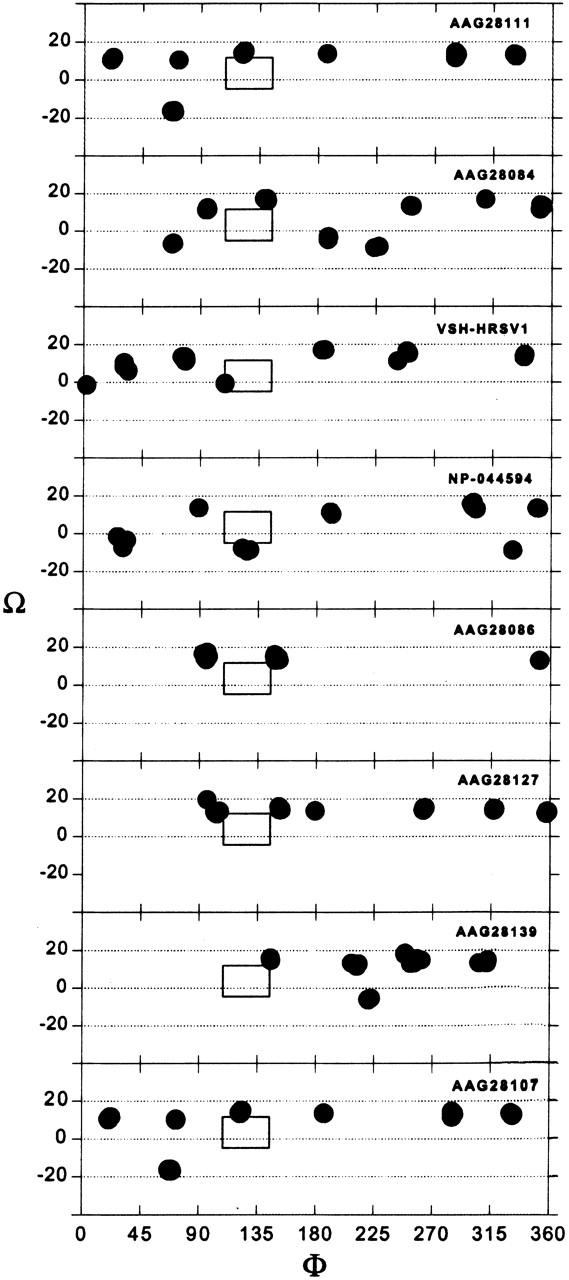
Results of the global search molecular dynamics protocol for the long TM segment (14–41) of eight different variants of SH HRSV protein simulated as a tetramer. The clusters obtained are indicated in terms of their helix rotation angles φ and crossing angles Ω. The box marks the position of structures that persisted in all of the simulations.
When simulating seven SH variants in tetrameric configuration, using amino acids 23–41 (short TM segment), a similar picture is obtained (Fig. 4 ▶). A complete set is found at φ ~176°, Ω ~11°, with <0.81 Å Cα RMSD between the different variants.
Figure 4.
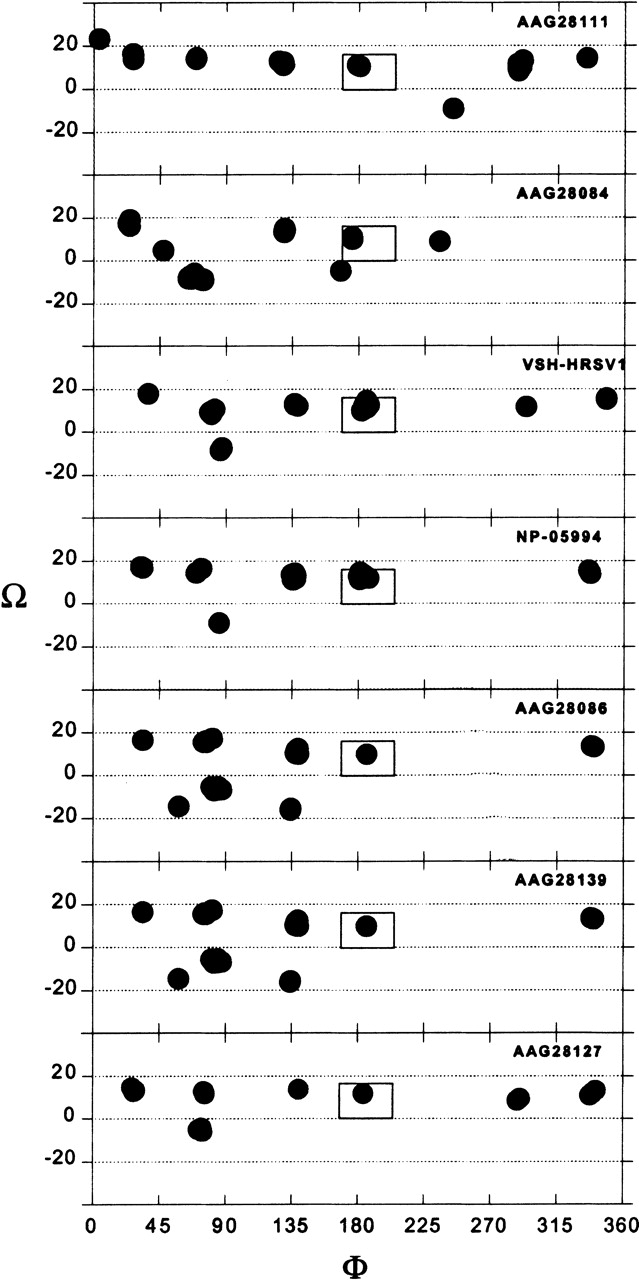
Results of the global search molecular dynamics protocol for the short TM segment (23–41) of seven different variants of SH HRSV protein simulated as a tetramer. The clusters obtained are indicated in terms of their helix rotation angles φ and crossing angles Ω. The box marks the position of structures that persisted in all of the simulations.
Thus, a single consensus structure was found when simulating the short TM segment from different SH variants as a homotetramer. Similarly, a single consensus structure was obtained, simulating the long TM segment. Superimposition was used in order to determine the similarity between the two resulting structures. The results shown in Figure 5 ▶ revealed a Cα RMSD of 4.58 Å and a shift of the φ angles of 90° between the two structures.
Figure 5.

Superimposition of the short TM segment structure (dark gray) onto the long TM segment (light gray), each obtained from the global searching molecular dynamics simulations of multiple variants of SH as a homo-tetramer. Residue 29 (serine) of each helix is shown in CPK representation. Figure generated by MOLSCRIPT (Kraulis 1991).
Pentamer simulations
Figure 6 ▶ depicts the results of the simulation of the long TM segment for the above HRSV variants, assuming pentameric oligomerization. The results point to a single structure that persists in all instances (φ ~263°, Ω ~13°). The complete set found here can be defined at <0.99 Å Cα RMSD.
Figure 6.
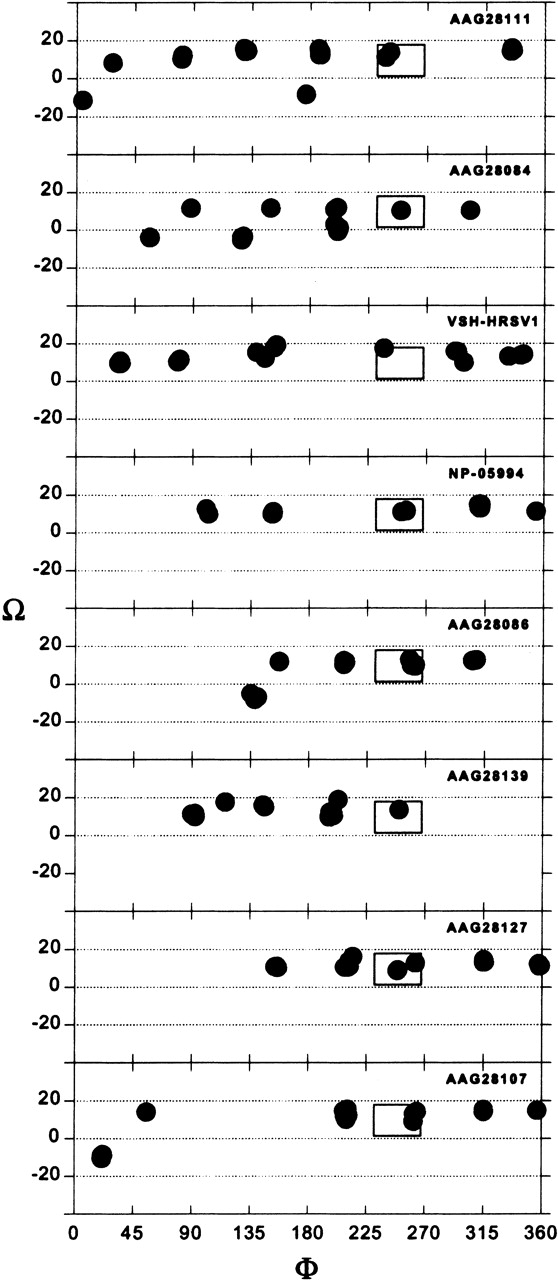
Results of the global search molecular dynamics protocol for the long TM segment (amino acids 14–41) of variants of SH HRSV protein as a homopentamer. The clusters obtained are indicated in terms of their helix rotation angles φ and crossing angles Ω. The box marks the position of structures that persisted in all of the simulations.
Similar results are obtained when using the short TM segment (Fig. 1 ▶). Here, the complete set can be defined at <0.86 Å Cα RMSD. The conformation that persists in all variants is at position φ ~25°, Ω ~15° (Fig. 7 ▶).
Figure 7.
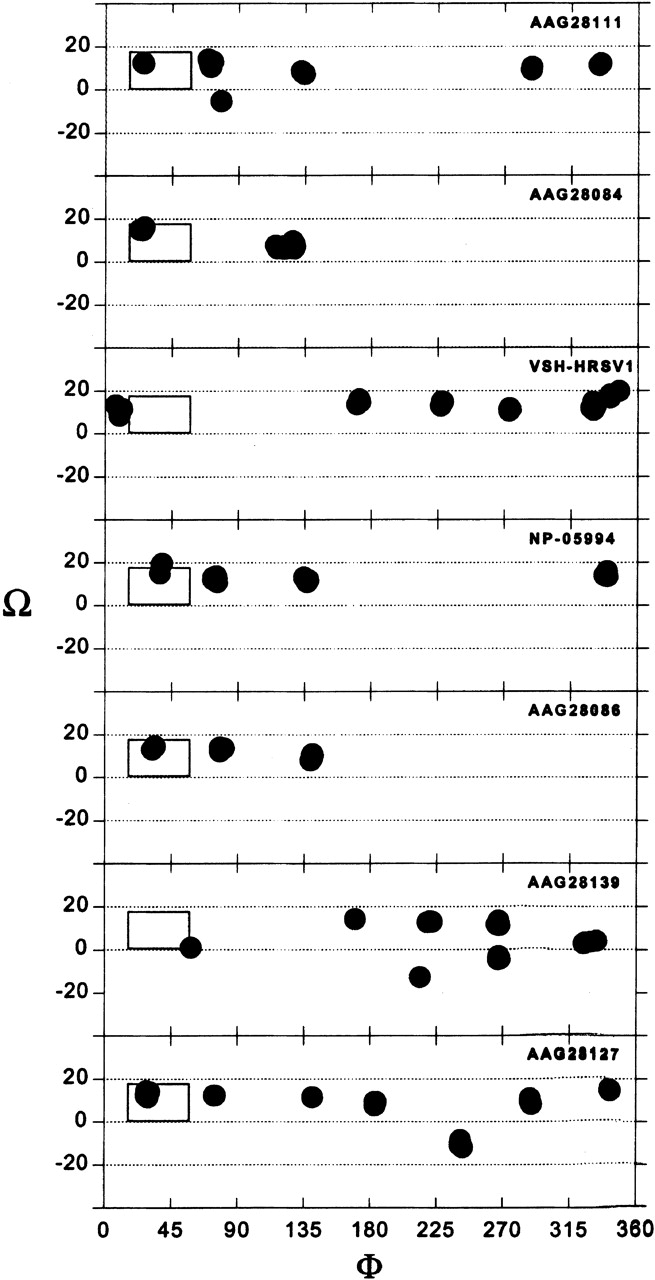
Results of the global search molecular dynamics protocol for the short TM segment (amino acids 23–41) of variants of SH HRSV protein as a homopentamer. The clusters obtained are indicated in terms of their helix rotation angles φ and crossing angles Ω. The box marks the position of structures that persisted in all of the simulations.
Superimposition of the structure obtained from searching the short TM segment onto that obtained from the long TM segment (Fig. 8 ▶) revealed a Cα RMSD of 1.9 Å, and a shift of the φ angles of 30°.
Figure 8.

Superimposition of the short TM segment structure (dark gray) onto the long TM segment (light gray) each obtained from the global searching molecular dynamics simulations of multiple variants of SH as a homopentamer. Residue 29 (serine) of each helix is shown in CPK representation. Figure generated by MOLSCRIPT (Kraulis 1991).
Trimer simulations
Finally, SH trimeric structures were simulated using global molecular dynamics searches (data not shown). These simulations performed on the short TM segment revealed that a complete set was only found when raising the Cα RMSD threshold to 1.85 Å. Similarly, simulations of the long TM segment as a trimer could not find any complete set, unless the Cα RMSD threshold was raised to 1.45 Å. Superimposing the complete sets from the long and short TM segments resulted in a Cα RMSD of 2.22 Å between the two structures.
Discussion
Implementation of the new Silent-Substitution method (Briggs et al. 2001) for selecting the correct model after performing global molecular dynamic simulations for Human RSV SH protein revealed complete sets for homotrimeric, homotetrameric, and homopentameric transmembranal structures. It seems unlikely, however, that the SH protein oligomers into a trimer considering the high Cα RMSD required in order to form a complete set, 1.85 Å and 1.45 Å when using the long TM segment or the short TM segment, respectively. Furthermore, it is difficult to imagine a trimeric helical bundle functioning as an ion channel, on the basis of structural considerations.
Determining whether the correct oligomerizing form of SH is tetrameric or pentameric was found to be harder, as the complete set for both of oligomeric forms could be formed using a Cα RMSD cutoff <1.0 Å. The Cα RMSD between all the structures in the tetrameric complete set is less than that obtained for the pentameric complete set (0.82 Å and 0.99 Å, respectively).
Nevertheless, further evidence points to the fact that the SH protein does not form a tetrameric structure: (1) The Cα RMSD obtained by superimposing amino acids 23–41 of the tetrameric long TM segment structure upon those of the tetrameric short TM segment structure, is 4.58 Å, and (2) a shift of the φ angles of 90°. Clearly, the results obtained when simulating a homotetrameric assembly depend, therefore, on the construct simulated. This would indicate that the hypothesized tetramer structure is unstable.
On the other hand, superimposing the results of the short TM segment and long TM segment obtained when simulating SH as a homopentamer revealed a much higher degree of similarity: Cα RMSD of 1.9 Å and a Δφ = 30°, pointing toward a stable conformation. Therefore, the results point to the fact that the SH protein oligomers into a homopentameric, channel-like structure (Fig. 9 ▶), whose minimal pore diameter is 1.46 Å.
Figure 9.
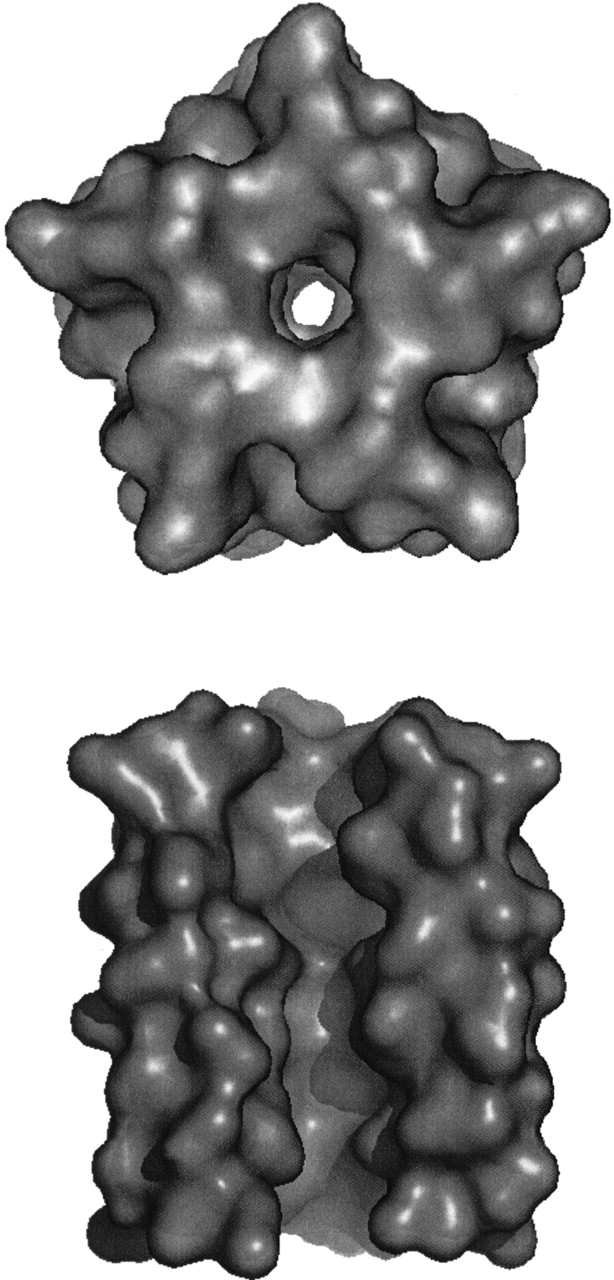
(Top) Surface model of the structure of HRSV SH protein, looking along the putative pore from the carboxyl terminus. (Bottom) Surface model of structure of HRSV SH protein, rotated by 90°. One helix has been removed in order to show the putative inner channel. Figure generated by PYMOL (Delano).
Although the exact function of SH protein of HRSV is as yet unknown, membrane permeability changes to low molecular-weight compounds induced by its expression in E. coli (Perez et al. 1997) are suggestive of SH being a viroporin. Our model of a homopentameric structure supports this hypothesis (Collins and Hay 1989), in that a pore is revealed in the pentameric helical bundle. However, further experimental data (FTIR, NMR, etc.) are required in order to strongly establish this conclusion.
Acknowledgments
This work was supported in part by a grant from The Israel Science Foundation (grant no. 784/01-16.1).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RMSD, root mean square deviation
CNS, crystallography and NMR system
CHI, CNS searching of helix interactions
HRSV, human respiratory syncytial virus
SH, small hydrophobic
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03151103.
References
- Adams, P.D., Arkin, I.T., Engelman, D.M., and Brunger, A.T. 1995. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 2 154–162. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin, I.T., Adams, P.D., MacKenzie, K.R., Lemmon, M.A., Brunger, A.T., and Engelman, D.M. 1994. Structural organization of the pentameric transmembrane α-helices of phospholamban, a cardiac ion channel. EMBO J. 13 4757–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs, J.A., Torres, J., and Arkin, I.T. 2001. A new method to model membrane protein structure based on silent amino acid substitutions. Proteins 44 370–375. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Bukreyev, A., Whitehead, S.S., Murphy, B.R., and Collins, P.L. 1997. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J. Virol. 71 8973–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.D., Vazquez, M., Buonocore, L., and Kahn, J.S. 2000. Conservation of the respiratory syncytial virus SH gene. J. Infect. Dis. 182 1228–1233. [DOI] [PubMed] [Google Scholar]
- Collins, P.L. and Mottet, G. 1993. Membrane orientation and oligomerization of the small hydrophobic protein of human respiratory syncytial virus. J. Gen. Virol. 74 1445–1450. [DOI] [PubMed] [Google Scholar]
- Collins, P.L., Olmsted, R.A., and Johnson, P.R. 1990. The small hydrophobic protein of human respiratory syncytial virus: Comparison between antigenic subgroups A and B. J. Gen. Virol. 71 1571–1576. [DOI] [PubMed] [Google Scholar]
- Collins, P.L., McIntosh, K., and Chanock, R.M. 1996. Respiratory syncytial virus, 3rd ed. Lippincott-Wilkins, Philadelphia.
- Delano, W.L. The PYMOL molecular graphics system. Delano Scientific LLC, San Carlos, CA. http://www.pymol.org.
- Dowell, S.F., Anderson, L.J., Gary Jr., H.E., Erdman, D.D., Plouffe, J.F., File Jr., T.M., Marston, B.J., and Breiman, R.F. 1996. Respiratory syncytial virus is an important cause of community-acquired lower respiratory infection among hospitalized adults. J. Infect. Dis. 174 456–462. [DOI] [PubMed] [Google Scholar]
- Engelman, D.M., Steitz, T.A., and Goldman, A. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15 321–353. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L. and Tirado-Rives, J. 1988. The OPLS potential function for proteins, energy minimization for crystals of cyclic peptides and crambin. J. Amer. Chem. Soc. 110 1657–1666. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Kukol, A., Adams, P.D., Rice, L.M., Brunger, A.T., and Arkin, T.I. 1999. Experimentally based orientational refinement of membrane protein models: A structure for the Influenza A M2 H+ channel. J. Mol. Biol. 286 951–962. [DOI] [PubMed] [Google Scholar]
- Kukol, A., Torres, J., and Arkin, I.T. 2002. A structure for the trimeric MHC class II-associated invariant chain transmembrane domain. J. Mol. Biol. 320 1109–1117. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A., Flanagan, J.M., Hunt, J.F., Adair, B.D., Bormann, B.J., Dempsey, C.E., and Engelman, D.M. 1992a. Glycophorin A dimerization is driven by specific interactions between transmembrane α-helices. J. Biol. Chem. 267 7683–7689. [PubMed] [Google Scholar]
- Lemmon, M.A., Flanagan, J.M., Treutlein, H.R., Zhang, J., and Engelman, D.M. 1992b. Sequence specificity in the dimerization of transmembrane α-helices. Biochemistry 31 12719–12725. [DOI] [PubMed] [Google Scholar]
- Olmsted, R.A., Murphy, B.R., Lawrence, L.A., Elango, N., Moss, B., and Collins, P.L. 1989. Processing, surface expression, and immunogenicity of carboxy-terminally truncated mutants of G protein of human respiratory syncytial virus. J. Virol. 63 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, M., Garcia-Barreno, B., Melero, J.A., Carrasco, L., and Guinea, R. 1997. Membrane permeability changes induced in Escherichia coli by the SH protein of human respiratory syncytial virus. Virology 235 342–351. [DOI] [PubMed] [Google Scholar]
- Stevens, T.J. and Arkin, I.T. 2000. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins 39 417–420. [DOI] [PubMed] [Google Scholar]
- Techaarpornkul, S., Barretto, N., and Peeples, M.E. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 75 6825–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolley, K.P., Marriott, A.C., Simpson, A., Plows, D.J., Matthews, D.A., Longhurst, S.J., Evans, J.E., Johnson, J.L., Cane, P.A., Randolph, V.B., et al. 1996. Identification of mutations contributing to the reduced virulence of a modified strain of respiratory syncytial virus. Vaccine 14 1637–1646. [DOI] [PubMed] [Google Scholar]
- Torres, J., Adams, P.D., and Arkin, I.T. 2000. Use of a new label, 13C=18O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol. 300 677–685. [DOI] [PubMed] [Google Scholar]
- Torres, J., Briggs, J.A., and Arkin, I.T. 2002. Convergence of experimental, computational and evolutionary approaches predicts the presence of a tetrameric form for CD3-ζ. J. Mol. Biol. 316 375–384. [DOI] [PubMed] [Google Scholar]
- Treutlein, H.R., Lemmon, M.A., Engelman, D.M., and Brunger, A.T. 1992. The glycophorin A transmembrane domain dimer: Sequence-specific propensity for a right-handed supercoil of helices. Biochemistry 31 12726–12732. [DOI] [PubMed] [Google Scholar]
- Whitehead, S.S., Hill, M.G., Firestone, C.Y., St. Claire, M., Elkins, W.R., Murphy, B.R., and Collins, P.L. 1999. Replacement of the F and G proteins of respiratory syncytial virus (RSV) subgroup A with those of subgroup B generates chimeric live attenuated RSV subgroup B vaccine candidates. J. Virol. 73 9773–9780. [DOI] [PMC free article] [PubMed] [Google Scholar]