

Abstract
A combined molecular dynamics simulation and multiple ligand docking approach is applied to study the roles of the anionic subsite residues (W86, E202, Y337) in the binding of acetylcholine (ACh) to acetylcholinesterase (AChE). We find that E202 stabilizes docking of ACh via electrostatic interactions. However, we find no significant electrostatic contribution from the aromatic residues. Docking energies of ACh to mutant AChE show a more pronounced effect because of size/shape complementarity. Mutating to smaller residues results in poorer binding, both in terms of docking energy and statistical docking probability. Besides separating out electrostatics by turning off the partial charges from each residue and comparing it with the native, the mutations in this study are W86F, W86A, E202D, E202Q, E202A, Y337F, and Y337A. We also find that all perturbations result in a significant reduction in binding of extended ACh in the catalytically productive orientation. This effect is primarily caused by a small shift in preferred position of the quaternary tail.
Keywords: Acetylcholine, acetylcholinesterase, binding, electrostatic interactions, ligand docking, molecular dynamics, size shape complementarity
The catalytic power of an enzyme stems from its ability to facilitate a low barrier reaction. An important capability of the enzyme is to position the substrate in the desired productive orientation. The relationship between substrate binding specificity and enzyme catalysis features from the interconversion of binding and chemical reaction activation energies (Fersht 1987). To further understand the structural origin of enzyme activity, we apply a computational approach, the combination of molecular dynamics simulation and multiple ligand docking, to study the roles of specific protein residues involved in binding. In particular, we are interested in quantifying energetic contributions of electrostatic versus size/shape complementarity of these residues, and investigating how these energetic components affect substrate binding orientation.
The enzyme under study is acetylcholinesterase (AChE, EC 3.1.1.7), a serine hydrolase responsible for the termination of impulse signaling at cholinergic synapses. It catalyzes the hydrolysis of the neurotransmitter acetylcholine (ACh) with a remarkably high catalytic efficiency, and is also a promising drug-design target for the treatment of Alzheimer’s disease (Quinn 1987; Giacobini 2000). In fact, it is the only target that has provided the few palliative drugs presently marketed for the treatment of Alzheimer’s.
The active site of AChE is located at the base of a long and narrow 20 Å gorge (Sussman et al. 1991). It consists of two subsites: an “esteratic” subsite containing the catalytic machinery, and an “anionic” subsite responsible for binding the quaternary trimethylammonium tail group of ACh (Quinn 1987). The essential catalytic functional unit of AChE is the catalytic triad consisting of S203, H447, and E334 (Quinn 1987). (Throughout this article, the sequence numbers follow the amino acid abbreviations of mouse AChE.) The oxyanion hole, formed by the peptidic NH groups of G121, G122, and A204, is another important functional unit in the esteratic subsite. The X-ray structure of a transition-state analog complex with Torpedo californica AChE (Harel et al. 1996) revealed that the acetyl head group of ACh, directly involved in making and breaking bonds, is held in place by the oxyanion hole. When the acetyl group is held in place, nucleophilic attack from the side-chain oxygen of S203 to the acetyl carbonyl carbon constitutes the first acylation step (see Fig. 1 ▶). More details of the acylation step can be found in our recent combined quantum mechanical and molecular mechanical study of this reaction (Zhang et al. 2002) and references therein.
Figure 1.

The acylation step in catalysis of ACh.
In the anionic subsite, the X-ray structure indicates that W86, E202, and Y337 form a concave binding site providing a close fit with the tail group (Harel et al. 1996). Site-directed mutagenesis also indicates that W86 (Ordentlich et al. 1993; Quinn et al. 2000), E202 (Radic et al. 1992), and Y337 (Radic et al. 1993) play an important role in binding the quaternary trimethylammonium tail group. These studies found that the mutations E202D and Y337F of these residues hardly change KM; for W86F, E202Q, and Y337A, there was a small increase in KM (within an order of magnitude). The only exception was W86A, in which KM was reduced by three orders of magnitude (Ordentlich et al. 1993; Shafferman et al. 1998). W86 and Y337 are generally thought to bind to the cationic moiety mainly through cation–π interactions, whereas the interaction between E202 and the cationic moiety is often attributed to be electrostatic. Recent more detailed mutation studies (Quinn et al. 2000) indicate that the interaction between Trp86 and the quaternary trimethylammonium moiety is approximately evenly split between cation–π and dispersion/hydrophobic interactions.
Our study focuses on the relative energetic contributions of electrostatics versus shape/size complementarity in the binding of the quarternary tail of ACh to the pocket formed by W86, E202, and Y337. To take into account the important dynamic behavior of the protein, we use a method that combines the conventional methods of molecular dynamics simulation and molecular docking. This method allows us to make perturbations and mutations to the residues of interest, quantify the energetic stabilization due to binding from a docking energy scoring function, and observe changes in orientation of the ligand. The structure of the enzyme is allowed to evolve dynamically in time, and ACh is docked to equally spaced snapshots of the enzyme. The statistical average values of docking energies and best docked orientations over many snapshots allow us to incorporate some of the complexity in the dynamical nature of ligand–receptor binding. For recent reviews of studies that incorporate protein flexibility in docking, see Ma et al. (2002) and Carlson and McCammon (2000) and references therein.
We have successfully applied this method to study ACh, choline, and closely related analogs binding to native AChE (Kua et al. 2002). The present application is to study the natural substrate, ACh, binding to W86, E202, and Y337 mutants of AChE. To study the electrostatic contribution, we first docked ACh to molecular dynamics trajectories of native AChE with the partial charges of each of these residues set to zero. This artificial method of separating electrostatics from shape/size fit was complemented with docking ACh to E202Q and Y337F, which gives rise to perturbations in electrostatics while retaining roughly similar size. We then dock ACh to W86F, W86A, Y337A, E202D, and E202A to study the effect of increasing the size of the tail-binding cavity. Calculated average docking energies and relative binding modes can then be compared with the native. All perturbations and mutations are confined to single residues. Although no double, or triple, mutations were performed in this study, our results indicate complex interplay among these three residues, and also between the anionic and esteratic subsites.
Materials and methods
The initial structure was chosen from a snapshot of a 10-nsec molecular dynamics simulation of the apo-mAChE with explicit water molecules (Tai et al. 2001). The ring of His 447 was first rotated into its productive orientation. The apo-enzyme system was then constructed by retaining the whole protein, the sodium cation in the active site and water molecules within a 24 Å radius of the S203 side chain oxygen (S203-O). The active site was finally equilibrated by a series of minimizations interspersed with short 20-psec molecular dynamics simulations. This equilibrated structure was the starting structure used for the 1-nsec native AChE simulation in the apo form (Kua et al. 2002). From this structure, the mutant AChE simulations were set up by first replacing the appropriate side chain of residue 86, 202, or 337, by its mutant amino acid. The mutants simulated were W86A, W86F, E202D, E202Q, E202A, Y337A, and Y337F. In cases in which the newly replaced side chain was smaller in size compared with its original, the optimum number of water molecules was added to fill the space. This optimum number was decided by multiple trials of adding between one and five water molecules in random configurations (drawn from an equilibrated water box) and choosing the number of waters that most successfully fit the space. We then equilibrated this structure by performing a series of minimizations interspersed by short 20-psec molecular dynamics simulations. This newly equilibrated structure was used as the starting structure for all mutant AChE simulations in the apo form.
The structure of the ACh ligand was constructed in its fully extended conformation according to early experimental and molecular modeling studies (Sussman et al. 1991; Harel et al. 1996). The ligand was then optimized using quantum mechanics at the Hartree-Fock level with the 6–31G* basis set (Hehre et al. 1986). The ligand charges were obtained from electrostatic potential (ESP) fitted charges (Besler et al. 1990) from the HF/6–31G* quantum mechanics calculation. This procedure was also used in preparing all the ligand analogs and choline for docking. A study to determine if π–cation interactions can be calculated accurately using molecular mechanical methods concluded that using quantum-mechanical methods to obtain the charges for the cationic moiety combined with an existing molecular mechanics force field gives good results (Felder et al. 2001).
The native ACh–AChE complex was constructed by docking ACh into the equilibrated apo-enzyme simulation using Autodock 3.0.4 (Morris et al. 1998) and equilibrated as described in our previous study (Kua et al. 2002). The mutant ACh–AChE simulations were set up according to the same procedure used to set up the mutant apo-AChE simulations described above.
All 1-nsec molecular dynamics simulations were carried out using the TINKER program (Ponder 1998). Because our focus is on the active site, only atoms within 20 Å of S203-O were allowed to move. A twin-range cutoff method was used to treat the nonbonded interactions (van Gunsteren et al. 1984), a long-range cutoff of 12 Å and a short-range cutoff of 8 Å. The nonbonded pair list was updated every 20 steps. The time step used was 2 fsec. Bond lengths involving hydrogen atoms were constrained using the SHAKE (Ryckaert et al. 1977) algorithm. The temperature of the simulations was maintained at 300 K using the weak coupling method with a coupling time of 0.1 psec (Berendsen et al. 1984). The molecular mechanics force field used in the present study is the AMBER95 all-atom force field for the protein (Cornell et al. 1995; Fox et al. 1997) and the TIP3P model for water (Jorgensen et al. 1983).
The ACh ligand was docked to 999 snapshots, each a picosecond apart, of the 1-nsec simulations. Water and sodium ions were removed prior to docking. The Autodock 3.0.4 (Morris et al. 1998) program was used for all docking studies. The search method used was the Lamarckian Genetic Algorithm (LGA) with 12 LGA runs. The number of individuals in each population was 50. The maximum number of energy evaluations was set at 250,000, and the maximum number of generations was set at 27,000. The number of top individuals that automatically survive was two. The rates of gene mutation and crossover were set at 0.02 and 0.80, respectively. We found these settings to give consistent results in the distribution of the final top 12 structures reported. The ligands were kept rigid both for computational speed and also because in the 1-nsec ACh–AChE simulation, ACh remained stable in the extended conformation. We also checked the stability of ACh in the ACh–mutAChE simulations, and they also remained stable in the extended conformation, although less so than in the native case, depending on the mutant. For each of the tail-binding site residues (86, 202, 337), we also docked ACh to the native 1-nsec simulations. In this docking step (but not in the MD simulation), the partial charges of the residue are set to zero to extract the electrostatic contribution of each residue. We found no significant difference between setting the partial charges of the entire residue to zero versus just zeroing the side-chain atoms. Hence, the numbers reported are for zero charges on the entire residue.
In our previous study on the native system (Kua et al. 2002), we docked ACh and its analogs to both the apo-AChE and ACh–AChE trajectories to quantify the induced fit effect. Although the results differed in magnitude with respect to ligand size versus docking energy, the qualitative results and trends were similar for both trajectories. Therefore, we chose to limit the results presented in our present study to just docking ACh to the apo-mutAChE trajectories.
The docking free energy scoring function used by Autodock is given by:
 |
(1) |
Each of the terms is defined as follows:
 |
(2) |
 |
 |
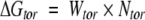 |
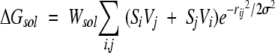 |
The hydrogen-bond term has an angle-dependent directional weight, E(t), based on the angle, t, between the probe and the target atom. Ehbond is the empirically estimated average energy of the hydrogen bonding of water with a polar atom. The electrostatic term uses a distance-dependent dielectric function to model solvent screening based on the work by Mehler and Solmajer (1991). The torsional term is proportional to Ntor, the number of sp3 bonds in the ligand. In the desolvation term, Si and Vi are the solvation parameter and the fragmental volume of atom i (Stouten et al. 1993), respectively. All five terms have weighting factors, W, obtained by fitting a large set of energetic analyses of ligand–receptor complexes (Morris et al. 1998).
This scheme of devising an artificial mutation or perturbation, docking a ligand, and evaluating the docking energy according to equation 1 is not equivalent to free-energy decomposition. The danger of attempting to separate the thermodynamic components of free energy are well documented (Mark and van Gunsteren 1994; Levy and Gallicchio 1998). For example, the artificial zeroing of charges gives us information about how the overall docking energy changes because of the perturbation, but should not be overinterpreted as an absolute numerical assignment to the electrostatic contribution to binding.
In our analysis of the docked structures, we chose only the best docked structure reported by Autodock. A structure is considered to be correctly docked for catalysis if the acetyl head group interacts with the oxyanion hole in the esteratic subsite and the tail region is located in the Trp 86–Glu 202–Tyr 337 binding pocket (anionic subsite). The distances and angles used to define the ligand–receptor interactions of docked structures are shown in Figures 2 ▶ and 3 ▶. We use these same distances and angles in our analysis of all the substrate–enzyme interactions in the 1-nsec ACh–mutAChE molecular dynamics simulations.
Figure 2.

The esteratic binding site for the acetyl head group.
Figure 3.
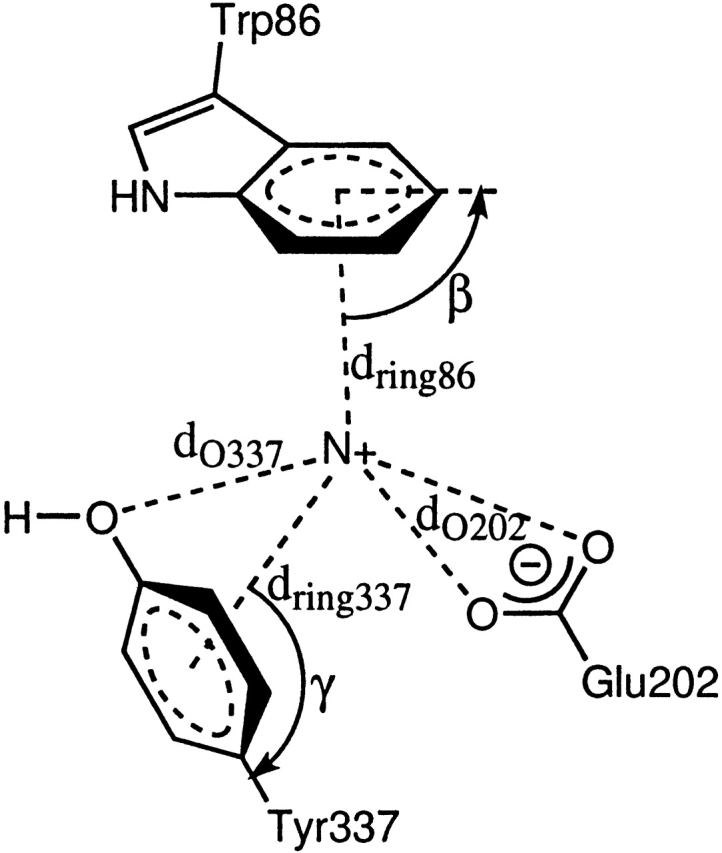
The anionic binding site for the tail group.
Results and Discussion
Electrostatic contribution to docking
To separate the electrostatic contribution to docking energy from size/shape effects, we docked ACh to the native 1-nsec trajectory where partial charges of each residue of interest (W86, E202, Y337) were set to zero in the docking step. The number of correctly docked structures and the average docking energy for each of these three perturbations are shown in Table 1. Shown in parentheses is the standard deviation calculated from the average energy of all correctly docked structures. This number indicates the spread of docking energies caused by different conformations dynamically assessed by the receptor.
Table 1.
Number of structures and docking energy (kilocalories per mole) of correctly docked ACh
| No. of structures | Docking energy | |
| Native | 958 | −7.10 (±0.30) |
| Zeroed-W86 | 771 | −7.08 (±0.41) |
| Zeroed-Y337 | 774 | −7.14 (±0.36) |
| Zeroed-E202 | 755 | −6.69 (±0.36) |
| Y337F | 674 | −6.92 (±0.35) |
| E202Q | 614 | −6.81 (±0.35) |
In terms of average docking energy, we find no significant difference when turning off the partial charges of the aromatic residues W86 and Y337. There is, however, a significant decrease in the percentage of correctly docked structures from 96% to 77%. This decrease can be attributed primarily to an increase in the number of structures that correctly dock the ACh quaternary tail but not its acetyl head, 21% for zeroed-W86 and 18% for zeroed-Y337.
Hence, correct binding of the tail group still occurs at least 95% of the time. The average distances of the quaternary N to these three residues in the correctly docked structures are shown in Table 2. The schematic for these distances is shown in Figure 3 ▶. The decrease in percentage can be illustrated by imagining ACh to be a rod with its tail acting as a pivot. A small change in orientation of the tail can lead to a large change in the position of the pivoting head. Turning off the charges leads to such a change as seen from changes in key distances of the quaternary N to the tail residues.
Table 2.
Average distances and angles of docked ACh ligand in the anionic (tail) subsite
| No. of structures | dring86 (Å) | dring337 (Å) | dO202.1 (Å) | |
| Native | 958 | 4.50 | 5.13 | 3.82 |
| Zeroed-W86 | 771 | 4.69 | 4.75 | 4.14 |
| Zeroed-Y337 | 774 | 4.67 | 4.75 | 4.13 |
| Zeroed-E202 | 755 | 4.69 | 4.57 | 4.31 |
| Y337F | 778 | 4.86 | 4.73 | 4.11 |
| E202A | 614 | 5.02 | 4.57 | 5.03 |
We do find a reduction in the average docking energy of ACh to zeroed-E202. This is not surprising because the negatively charged carboxyl is expected to provide the main electrostatic stabilization of the positively charged tail of ACh. We also see a decrease in percentage of correctly docked structures. Structures that dock the tail correctly but not the head constitute 13%, leading to a total probability of binding the tail correctly at 89%.
The balance in interactions among the three tail group residues is both complex and subtle. We first consider the orientation of the three residues when binding the quaternary tail as shown in Figure 4 ▶. E202 and Y337 are opposite to each other in space, sandwiching the quaternary tail of ACh between them. W86 is on the side, almost opposite to the acetyl head group of ACh. In space (not so obvious from the figure), W86 is closer to E202 than to Y337. When charges on E202 are zeroed, we observe an expected shorter dring337 on average compared with the native. However, when charges on Y337 are zeroed, we still have shorter dring337. In fact, these distances are similar when zeroing W86 instead of Y337. We have observed the same trend when docking the neutral analog of ACh (where the N[Me]3 tail is replaced by tBu) to native AChE, where the average distances were 4.68, 4.69, and 4.20 Å for dring86, dring337, and dO202,1, respectively (Kua et al. 2002). It therefore turns out that neutralizing charge, whether it be the ligand tail or the protein tail-binding site residues, changes the binding orientation in the same way.
Figure 4.
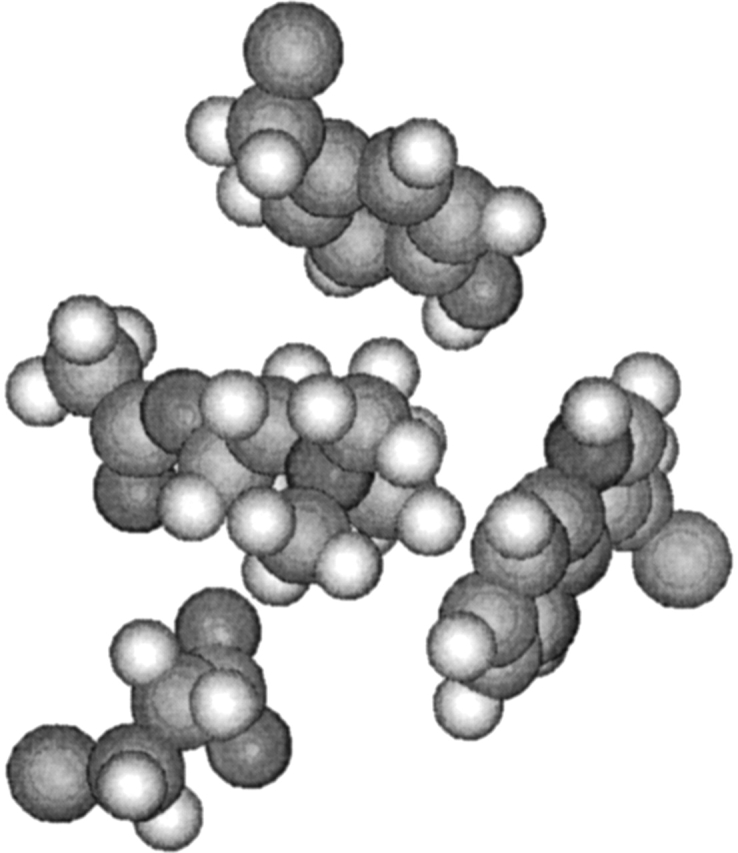
Orientation of W86, E202, and Y337 around ACh.
We also examined the effect of these perturbations on key distances of the acetyl head group in the esteratic subsite (see Table 3). The decrease in percentage of correctly docked structures for these three zero-charge mutants is accompanied by shorter distances for formation of the first bond in catalysis, that is, between S203-O and the carbonyl carbon of ACh. When the charges on any of the three residues are zero, the average distance is ~3.60 Å instead of 3.78 Å in the native. The angle of attack, α, to the normal for nucleophilic addition in the acylation step is farther away from the ideal of 0° and averages 33°. These results are consistent with our previous study docking the neutral tail analog of ACh to native AChE (Kua et al. 2002).
Table 3.
Average distances and angles of docked ACh ligand in the esteratic (head) subsite
| No. of structures | acetyl-C Ser 203-O (Å) | acetyl-O Gly 121-N (Å) | acetyl-O Gly 122-N (Å) | acetyl-O Ala 204-N (Å) | Angle α (degrees) | |
| Native | 958 | 3.78 | 2.92 | 3.00 | 3.66 | 18.67 |
| Zeroed-W86 | 771 | 3.57 | 3.05 | 2.87 | 3.59 | 32.46 |
| Zeroed-Y337 | 774 | 3.57 | 3.04 | 2.88 | 3.61 | 32.20 |
| Zeroed-E202 | 755 | 3.62 | 3.01 | 2.94 | 3.79 | 33.89 |
| Y337F | 778 | 3.75 | 3.08 | 2.87 | 3.77 | 33.13 |
| E202Q | 614 | 3.17 | 3.38 | 3.41 | 4.33 | 30.21 |
We performed two further mutations, E202Q and Y337F, to further assess the electrostatic contribution of these two residues. We chose these mutations because they retain a roughly similar size to the native but perturb the electrostatic environment. Although this test does not cleanly partition electrostatics from shape/size effects, it provides a more realistic environment that can be further tested by experiment. Average docking energies and key distances are shown in Tables 1–3. As expected, Y337F shows no significant difference in average docking energy from the native. The percentage of correctly docked structures is 76%. The percentage that dock the quaternary tail but not the acetyl head is 18%. The geometry is quite similar to zeroed-337 except for a slightly longer dring86 in the tail region and carbonyl carbon to Ser203-O in the head region.
E202Q, on the other hand, has an average docking energy between the native and zeroed-E202. Although mutating Glu to Gln reduces the overall net negative charge, the side-chain carbonyl oxygen of Gln still provides some electrostatic stabilization of the ACh quaternary tail. The percentage of correctly docked structures falls to 61% for the same reasons as zeroed-E202. In this case, the percentage of structures that correctly dock the quaternary tail but not the acetyl head is 35%. In terms of binding geometry, Y337 is now closest in distance to the tail. Conversely, the distances to W86 and E202 show noticeable increase. In particular, the distance to E202Q is larger than even zeroed-E202 because of repulsion of the tail with the positively charged hydrogens on the side-chain NH2. Although E202Q has a reduced docking energy and the percentage of correctly docked structures decreases, for the structures that do bind correctly, the acetyl head is better set up for the first catalytic acylation step. The S203-O to carbonyl carbon distance is much shorter at 3.17 Å, although α is now 30°. Note, however, that the hydrogen bonds in the oxyanion hole have increased in distance and hence decreased in strength.
Our results suggest that electrostatic perturbations to the residues of the tail-binding site have a significant effect on the orientation and position of the quaternary tail of ACh. This, in turn, affects the probability of having the acetyl head of ACh bind correctly in the oxyanion hole. The average docking energy of correctly docked structures set up for catalysis remains unchanged for perturbation to the aromatic residues W86 and Y337. This should not be interpreted as the absence of π–cation stabilization because the change in orientation of the ligand may compensate potential loss in energetic stabilization. The average docking energy is reduced as the negative charge on the carboxyl oxygen of E202 is reduced. In this case, changes in docking orientation cannot sufficiently compensate energetic destabilization.
It may be that the docking function is not sensitive enough to properly capture the π–cation interaction. The function does not contain an explicit term representing this type of interaction. A recent study (Felder et al. 2001) suggests that our general approach of using quantum-mechanical charges for the cation moiety is reasonable; however, we did not use the same parameters that they did. It is also possible that although significant energy stabilization can be expected for gas-phase π–cation interactions, the protein environment reduces the energetic contribution via other compensating effects.
The preliminary picture that emerges indicates that the native enzyme, in its dynamic state, is electrostatically “set up” more loosely in the optimum sense of having the highest probability for binding both head and tail groups of extended ACh in the productive orientation. Perturbing this electrostatic environment narrows the probability of productive binding. In terms of energetic stabilization of the quaternary tail via electrostatics, only E202 shows significant sensitivity to perturbation. In the present study, we have not made perturbations to further increase the negative charges of the tail residues; however, we expect that this will increase the energetic stabilization toward binding ACh. Adding negatively charged residues to stabilize docking, however, may be unfavorable toward catalysis because the acylation step involves electron flow to the ligand during nucleophilic attack.
Size/shape complementarity contribution to docking
To examine the effect of size/shape complementarity, we docked ACh to the 1-nsec trajectories of W86F, W86A, E202A, E202D, and Y337A mutant AChE. The number of correctly docked structures and the average docking energy (and spread) for each of these three perturbations are shown in Table 4.
Table 4.
Number of structures and docking energy (kilocalories per mole) of correctly docked ACh
| No. of structures | Docking energy | |
| Native | 958 | −7.10 (±0.30) |
| W86F | 659 | −6.70 (±0.35) |
| W86A | 655 | −6.46 (±0.35) |
| Y337A | 148 | −6.87 (±0.36) |
| E202A | 330 | −6.38 (±0.32) |
| E202D | 554 | −7.18 (±0.37) |
As the size of W86 is decreased to W86F and W86A, respectively, the average docking energy is reduced. We also observe this when Y337 is mutated to Y337A, and when E202 is mutated to E202A, the latter being caused by both electrostatic and size/shape fit. Shortening of the E202 side chain by a methylene in the E202D mutation shows no decrease in average docking energy. In fact the average docking energy is slightly (but not significantly) stronger, but this is because of the lower probability and hence clustering of a smaller number of stronger binding structures. W86A and E202A have the lowest average docking energies among all perturbations. These are caused by reducing the size of a much larger residue in W86A, and a combination of both size reduction and loss of electrostatic stabilization in E202A.
The number of structures in all mutants is significantly lower than the native. The differences are all attributable to having a large percentage of structures that bind the tail correctly but not the head. These percentages are 28%, 32%, 81%, 48%, and 44% for W86F, W86A, Y337A, E202A, and E202D, respectively. Hence, all mutants have the tail docked correctly >95% of the time, with the exception of E202A with 81%. The number of correctly docked mutants with size/shape perturbations is lower than with electrostatic perturbations. We also find that this number is surprisingly low for Y337A.
From Table 5, we see that size perturbations to W86 follow the same trend as electrostatic perturbations, i.e., the average value of dring337 decreases significantly accompanied by a smaller increase in dO202,1. Note that for mutations to Ala, distances (to the β-carbon) are all much larger and cannot be compared directly with key distances to the native; they are hence omitted (labeled “NA”). We have also included dring86 in W86F for reference, although this value is much larger because the position of the six-membered ring in the side chains is different in Trp and Phe. Y337A is the only mutation that has a decrease in dO202,1. Because there is very little size/shape complementarity with residue 337, the tail is now dominated by the electrostatic attraction from E202. We do not see this with W86A because W86 and E202 are close in proximity, in contrast to Y337 and E202 on opposite sides to each other. Y337 is important in how it affects catalysis because with E202 pulling the tail group toward it, the probability of the head group binding correctly decreases dramatically to 15%. For E202A, the tail now moves toward Y337, and hence farther away from W86. The probability of the head group binding is also quite low at 33%. Note that these low percentages for Y337A and E202A, and to a lesser extent all the other mutations, are for catalytically productive binding orientation of the extended form of ACh. Although we find that ACh remains stable in the extended form in AChE, we do observe some decrease in this stability for the mutants, through rotation of the more conformationally flexible dihedral ω3 (see Fig. 5 ▶; Kua et al. 2002). We have not included these results at present because they cannot be directly compared with those obtained from the rigid ligand docking because of the additional entropic terms used by AutoDock for flexible ligand docking.
Table 5.
Average distances and angles of docked ACh ligand in the anionic subsite
| No. of structures | dring86 (Å) | dring337 (Å) | dO202.1 (Å) | |
| Native | 958 | 4.50 | 5.13 | 3.82 |
| W86F | 659 | 5.58 | 4.70 | 3.93 |
| W86A | 655 | NA | 4.59 | 4.06 |
| Y337A | 148 | 5.00 | NA | 3.61 |
| E202A | 330 | 5.09 | 4.68 | NA |
| E202D | 554 | 5.19 | 5.41 | 3.96 |
Figure 5.

Schematic of dihedral angles along backbone of ACh.
The mutant E202D is an interesting case. Although dO202,1 is expected to be slightly longer because the side chain of residue 202 has been shortened by a methylene group, it is not much longer (3.96 Å) than the native. The negatively charged Asp still pulls the ACh tail toward it, and simultaneously much farther away from Y337. This results in an effect similar to, although less dramatic than, Y337A; that is, the probability of binding the head group correctly drops to 55%.
The way in which these mutations affect the productive binding of the acetyl head group is similar (see Table 6). In all five cases, the distance from the acetyl carbon to S203-O decreases. We also observed this when we docked neutral analogs of ACh with reduced tail sizes to native AChE (Kua et al. 2002). The angle of attack to the normal, α, also increases, although to a value not as large as observed from the electrostatic perturbations. Mutations to E202 have a smaller α value, closer to the native, but show the hydrogen bond in the oxyanion hole to G121 weakening. Y337A has a weaker hydrogen bond to G122 instead of G121.
Table 6.
Average distances and angles of correctly docked analogs in the esteratic subsite
| No. of structures | acetyl-C Ser 203-O (Å) | acetyl-O Gly 121-N (Å) | acetyl-O Gly 122-N (Å) | acetyl-O Ala 204-N (Å) | Angle α (degrees) | |
| Native | 958 | 3.78 | 2.92 | 3.00 | 3.66 | 18.67 |
| W86F | 659 | 3.31 | 3.04 | 3.02 | 3.91 | 26.59 |
| W86A | 655 | 3.16 | 3.08 | 2.93 | 3.81 | 26.19 |
| Y337A | 148 | 3.03 | 3.07 | 3.18 | 3.51 | 25.63 |
| E202A | 330 | 3.10 | 3.36 | 3.09 | 3.67 | 22.04 |
| E202D | 554 | 3.10 | 3.23 | 3.02 | 3.47 | 20.46 |
Our results imply that shape/size perturbations have a larger effect than electrostatic perturbations both in terms of docking energies and in terms of probability of productive binding orientation. We do find that as the size of the cavity is increased by mutating to smaller residues, the docking energy is reduced because of lessened stabilization via van der Waals forces. The effect of a mutation repositioning the quaternary tail and thus changing the binding in the head region is most significant for perturbations to the opposing residues Y337and E202.
Our calculated average docking energies are in good qualitative agreement with experimental KM values (see Fig. 6 ▶; we converted KM to RT × ln[KM] in kilocalories per mole). The experimental KM values (Radic et al. 1992, 1993; Shafferman et al. 1998) for W86A, W86F, E202Q, E202D, Y337F, and Y337A are shown in Table 7. Our calculated average docking energies are shown in Tables 1 and 4. We compared our results to KM because the ligand is reactive and therefore Ki values are not available. The value of KM is complex and depends on the mechanism and the relative rates of binding, catalysis, and intermediate steps. Under some conditions, KM may be equal or proportional to the substrate dissociation constant (Fersht 1987).
Figure 6.

Comparison of calculated average docking energies with experimental RT × ln(KM) values in kilocalories per mole.
Table 7.
Experimental KMvalues of AChE mutants
| KM (μM) | |
| Native | 46 |
| W86F | 276 |
| W86A | 30,636 |
| E202Q | 660 |
| E202D | 43 |
| Y337F | 53 |
| Y337A | 110 |
Setup of catalytic triad from MD simulation
The setup of the catalytic triad formed by S203–H447–E334 is crucial for catalytic activity; however, we were also interested to examine if its setup was important for binding. Our previous study on native AChE (Kua et al. 2002) indicated that in the absence of the ligand ACh, the hydrogen bond between S203-OH and His 447-N is only set up about half the time, whereas the hydrogen bond between His 447-NH and Glu 334-O is stable. This first metastable hydrogen bond, formed part of the time, is stable in blocks of 100–200 psec (see Fig. 7 ▶). The catalytic triad is set up when both hydrogen bonds are formed as shown in Figure 8 ▶.
Figure 7.

The setup of the catalytic triad along the 1-nsec apo-AChE trajectory. The + symbols refer to the Ser 203–His 447 hydrogen bond, and the × symbols refer to the His 447–Glu 334 hydrogen bond.
Figure 8.

Hydrogen bonds in the setup of the catalytic triad.
Using an arbitrary cutoff of 2.5 Å to define if the S203–His 447 hydrogen bond is present, the percentage of structures with this distance <2.5 Å is 45%. Preliminary analysis of the native trajectory indicates that in the majority of cases when this hydrogen bond is broken, it is because S203-OH forms a hydrogen bond with the backbone carbonyl of nearby Ser 229 instead. Initial analyses indicate that E202 is primarily responsible for ordering the arrangement of water molecules and influencing water traffic in the vicinity of S203 and H447, and that it stabilizes water close to H447 that periodically disrupts the S203–H447 hydrogen bond. Further study is required, however, before proposing a more detailed mechanism.
Results for the S203–H447 hydrogen-bond stability of the mutants are shown in Table 8. In all cases, the H447–E334 hydrogen bond remains stable throughout the 1-nsec MD simulation. Mutations of E202 all result in higher stability of the S203–H447 hydrogen bond, ranging from 85% to 98%. These can be explained by our preliminary suggestion of the role of E202 above. E202Q and E202A both reduce the ordering ability of residue 202, resulting in the absence or transience of a disruptive water in the vicinity of S203–H447. Shortening the side chain in E202D orders the water, but now the disruptive water is no longer in the vicinity of S203–H447. We also find that W86 mutations also increase S203–H447 hydrogen-bond stability, but Y337 mutations decrease it. In both cases, mutations to Phe result in lower stability; however, it is not clear why this is so.
Table 8.
Number of structures with Ser 203–His 447 hydrogen-bond setup
| No. of structures | |
| Native | 452 |
| W86F | 850 |
| W86A | 981 |
| Y337F | 300 |
| Y337A | 374 |
| E202Q | 985 |
| E202D | 936 |
| E202A | 848 |
Comparing the number of structures with catalytic triad setup and number of correctly docked structures, there seems to be no correlation between the two factors; that is, lack of setup of the catalytic triad does not seem to preclude correct docking. However, once a ligand is bound productively in the active site, it favors setup of the catalytic triad, if not already in position. This is expected because the ligand displaces the potentially disruptive water near S203–H447. If the ligand is included in the MD simulation both in the native and mutant AChE, the catalytic triad remains stable throughout the simulation.
Conclusion
We find that any perturbation to W86, E202, or Y337, results in a significant reduction in binding of extended ACh in the catalytically productive orientation. The reduction is primarily due to a small shift in preferred position of the quaternary tail. The percentage of structures that dock the tail correctly still remains high, above 85% in all cases. This shift in position is due to the interplay of effects among all three residues.
From an energetic point of view, our results indicate that size/shape complementarity has a larger effect than electrostatics, although the latter contributes significantly when the charges on E202 are reduced or zeroed. E202A and W86A show the largest reduction in docking energy among the mutants. As expected, we do find that as the size of the cavity is increased by mutating to smaller residues, the docking energy is reduced owing to lessened stabilization via van der Waals forces. The magnitude in docking energy reduction is small for any single mutation, and it is possible that the docking function used underestimates the electrostatic contribution. We find good qualitative agreement between our calculated docking energies and experimental KM values. We also find that setup of the catalytic triad is not important to induce favorable docking; however, a bound ligand does stabilize having the catalytic triad setup.
The present study has not directly addressed the connection between binding energy and the activation energy of the acylation reaction, the first step in catalysis. Combined quantum-mechanical and molecular mechanical investigation indicate a qualitative connection between specificity (or “tightness”) of binding correlating with lowering of the reaction barrier (Zhang et al. 2003). In this case, the sum of combined distances of the acetyl head to esteratic subsite residues and combined distances of the quaternary tail to anionic subsite residues was used to define the tightness of binding. Because our mutations all result in “looser” binding from a geometric point of view compared with the native, it would indicate that W86A would have the lowest catalytic efficiency, in good agreement with experimental results.
Acknowledgments
We are grateful for helpful discussions with Palmer Taylor. This work has been supported in part by grants from the NSF and the NIH. Additional support has been provided by the San Diego Supercomputer Center (SDSC), the National Biomedical Computing Resource (NBCR), and the W.M. Keck Foundation. A.C.E. was funded in part by an institutional award from the American Society for Pharmacology and Experimental Therapeutics.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03318603.
References
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., DiNola, A., and Haak, J.R. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 3684–3690. [Google Scholar]
- Besler, B.H., Merz Jr., K.M., and Kollman, P.A. 1990. Atomic charges derived from semiempirical methods. J. Comp. Chem. 11 431–439. [Google Scholar]
- Carlson, H.A. and McCammon, J.A. 2000. Accommodating protein flexibility in computational drug design. Mol. Pharmacol. 57 213–218. [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Felder, C., Jiang, H., Zhu, W., Chen, K., Silman, I., Botti, S.A., and Sussman, J.L. 2001. Quantum/classical mechanical comparison of cation–π interactions between tetramethylammonium and benzene. J. Phys. Chem. A 105 1326–1333. [Google Scholar]
- Fersht, A. 1987. Enzyme structure and mechanism, 2nd ed. W.H. Freeman, New York.
- Fox, T., Scanlan, T.S., and Kollman, P.A. 1997. Ligand binding in the catalytic antibody 17e8. A free energy perturbation study. J. Am. Chem. Soc. 119 11571–11577. [Google Scholar]
- Giacobini, E. 2000. Cholinesterases and cholinesterase inhibitors. Martin Dunitz Ltd., London.
- Harel, M., Quinn, D., Nair, H.K., Silman, I., and Sussman, J.L. 1996. The x-ray structure of a transition state analog complex reveals the molecular origins of the catalytic power and substrate specificity of acetylcholinesterase. J. Am. Chem. Soc. 118 2340–2346. [Google Scholar]
- Hehre, W., Radom, L., Schleyer, P., and Pople, J. 1986. Ab initio molecular orbital theory. John Wiley & Sons, New York.
- Jorgensen, W.L., Chandrasekhar, J., Madura, J., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–933. [Google Scholar]
- Kua, J., Zhang, Y., and McCammon, J.A. 2002. Studying enzyme binding specificity in acetylcholinesterase using a combined molecular dynamics and multiple docking approach. J. Am. Chem. Soc. 124 8260–8267. [DOI] [PubMed] [Google Scholar]
- Levy, R.M. and Gallicchio, E. 1998. Computer simulations with explicit solvent: Recent progress in the thermodynamic decomposition of free energies and in modeling electrostatic effects. Annu. Rev. Phys. Chem. 49 531–567. [DOI] [PubMed] [Google Scholar]
- Ma, B.Y., Shatsky, M., Wolfson, H.J., and Nussinov, R. 2002. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 11 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark, A.E. and van Gunsteren, W.F. 1994. Decomposition of the free energy of a system in terms of specific interactions: Implications for theoretical and experimental studies. J. Mol. Biol. 240 167–176. [DOI] [PubMed] [Google Scholar]
- Mehler, E.L. and Solmajer, T. 1991. Electrostatic effects in proteins: Comparison of dielectric and charged models. Prot. Eng. 4 903–910. [DOI] [PubMed] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19 1639–1662. [Google Scholar]
- Ordentlich, A., Barak, D., Kronman, C., Flashner, Y., Leitner, M., Segall, Y., Ariel, N., Cohen, S., Velan, B., and Shafferman, A. 1993. Dissection of the human acetylcholinesterase active-center determinants of substrate-specificity—Identification of residues constituting the anionic site, the hydrophobic site, and the acyl pocket. J. Biol. Chem. 268 17083–17095. [PubMed] [Google Scholar]
- Ponder, J.W. 1998. TINKER, software tools for molecular design, version 3.6. http://dasher.wustl.edu/tinker. [DOI] [PMC free article] [PubMed]
- Quinn, D.M. 1987. Acetylcholinesterase—Enzyme structure, reaction dynamics, and virtual transition-states. Chem. Rev. 87 955–979. [Google Scholar]
- Quinn, D.M., Feaster, S.R., Nair, H.K., Baker, N.A., Radic, Z., and Taylor, P. 2000. Delineation and decomposition of energies involved in quaternary ammonium binding in the active site of acetylcholinesterase. J. Am. Chem. Soc. 122 2975–2980. [Google Scholar]
- Radic, Z., Gibney, G., Kawamoto, S., MacPhee-Quigley, K., Bongiorno, C., and Taylor, P. 1992. Expression of recombinant acetylcholinesterase in a baculovirus system—Kinetic-properties of glutamate 199 mutants. Biochemistry 31 9760–9767. [DOI] [PubMed] [Google Scholar]
- Radic, Z., Pickering, N.A., Vellom, D.C., Camp, S., and Taylor, P. 1993. Three distinct domains in the cholinesterase molecule confer selectivity for acetylcholinesterase and butyrylcholinesterase inhibitors. Biochemistry 32 12074–12084. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J.-P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the Cartesian equations of a motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23 327–341. [Google Scholar]
- Shafferman, A., Ordentlich, A., Barak, D., Kronman, C., Ariel, N., and Velan, B. 1998. Contribution of the active center functional architecture to AChE reactivity toward substrates and inhibitors. In Structure and function of cholinesterases and related proteins (eds. B.P. Doctor et al.), pp. 203–209. Plenum Press, New York.
- Stouten, P.F.W., Frömmel, C., Nakamura, H., and Sander, C. 1993. Mol. Simul. 10 97–120. [Google Scholar]
- Sussman, J.L., Harel, M., Frolow, F., Oefner, C., Goldman, A., Toker, L., and Silman, I. 1991. Atomic structure of acetylcholinesterase from Torpedo californica—A prototypic acetylcholine-binding protein. Science 253 872–879. [DOI] [PubMed] [Google Scholar]
- Tai, K., Shen, T.Y., Borjesson, U., Philippopoulos, M., and McCammon, J.A. 2001. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys. J. 81 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gunsteren, W., Berendsen, H., Colonna, F., Perahia, D., Hollenberg, J., and Lellouch, D. 1984. On searching neighbours in computer simulations of macromolecular systems. J. Comput. Chem. 5 272–279. [Google Scholar]
- Zhang, Y., Kua, J., and McCammon, J.A. 2002. Role of the catalytic triad and oxyanion hole in catalysis: An ab initio study. J. Am. Chem. Soc. 124 10572–10577. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Influence of structural fluctuation on enzyme reaction barriers in combined quantum mechanical/molecular mechanical studies. J. Phys. Chem. B 107 4459–4463. [Google Scholar]