

Abstract
An alternative core packing group, involving a set of five positions, has been introduced into human acidic FGF-1. This alternative group was designed so as to constrain the primary structure within the core region to the same threefold symmetry present in the tertiary structure of the protein fold (the β-trefoil superfold). The alternative core is essentially indistinguishable from the WT core with regard to structure, stability, and folding kinetics. The results show that the β-trefoil superfold is compatible with a threefold symmetric constraint on the core region, as might be the case if the superfold arose as a result of gene duplication/fusion events. Furthermore, this new core arrangement can form the basis of a structural “building block” that can greatly simplify the de novo design of β-trefoil proteins by using symmetric structural complementarity. Remaining asymmetry within the core appears to be related to asymmetry in the tertiary structure associated with receptor and heparin binding functionality of the growth factor.
Keywords: Fibroblast growth factor, de novo design, superfold, protein evolution, β-trefoil
The protein-folding problem is one of the most important unsolved problems in modern biophysics. The ability to accurately predict tertiary structure from primary structure will permit a tremendous advance in deciphering the information generated by the sequencing of the human genome. One of the most exciting applications resulting from such knowledge would be the de novo design of proteins to develop entirely new functionality. Intertwined with these challenges is the question of how the complex tertiary structure of proteins evolved, and what the structural, kinetic, and thermodynamic restrictions are on such evolution.
Structural studies of globular proteins have demonstrated that despite the tens of thousands of uniquely different proteins within living organisms, almost all tertiary structures can be categorized into 1 of 10 fundamental protein folds (Orengo et al. 1994; Thornton et al. 1999). With these 10 fundamental protein folds, nature has identified the appropriate structural, kinetic, and thermodynamic solutions that result in a “foldable” polypeptide. The majority of the 10 fundamental superfolds (including the αβ plait, TIM barrel, β-trefoil, “jelly roll,” IG-like, and “up-down” superfolds) exhibit symmetric tertiary structures. These symmetric superfolds have been postulated to have evolved via gene duplication and fusion events (Tang et al. 1978; Holm et al. 1984; Volbeda and Hol 1989; Bergdoll et al. 1998; Lang et al. 2000; Mukhopadhyay 2000; Ponting and Russell 2000). However, when comparing the symmetry-related domains within such symmetric superfolds, evidence of such symmetry at the level of the primary structure is often largely absent. If such proteins evolved via gene duplication and fusion events, then there appears to have been differential selective pressure regarding symmetry within the primary and tertiary structures.
The fact that the majority of known foldable polypeptides exhibit a symmetric tertiary structure indicates a strategy for de novo protein design; a complex molecular architecture might be constructed through the use of an appropriate polypeptide “building block” that is capable of kinetically and thermodynamically favorable symmetric self assembly. However, for the six superfolds that are symmetric, there is essentially no information regarding the degree to which the primary structure can be constrained to conform to the tertiary structure symmetry and still produce a foldable polypeptide. In the case of the hydrophobic core region of such folds, there may be a limited number of solutions to the packing problem using the common hydrophobic amino acids and the symmetric tertiary structure constraint. Such solutions, if possible, will identify an appropriate building block useful in de novo design.
FGF-1 is a member of the β-trefoil protein superfold (Orengo et al. 1994; Thornton et al. 1999). A diverse group of proteins shares the β-trefoil fold, including interleukin-1α and β (Priestle et al. 1989), the actin binding protein hisactophilin (Habazettl et al. 1992), plant and bacterial toxins (Rutenber et al. 1991; Tahirov et al. 1995; Lacy et al. 1998; Emsley et al. 2000), mannose receptor (Liu et al. 2000), amylase (Vallee et al. 1998), xylanase (Kaneko et al. 1999), and Kunitz soybean trypsin inhibitors (Sweet et al. 1974). This fold was first described by McLachlan (1979) and subsequently analyzed in detail by Chothia (Murzin et al. 1992). The tertiary structure consists of a six-stranded β-barrel closed off at one end by a β-hairpin triplet. This architecture results in a pseudo threefold axis of symmetry along the β-barrel axis, and the β-trefoil is the only member of the 10 fundamental superfolds to exhibit threefold structural symmetry. The repeating structural subunit consists of ~40 amino acids that comprise a pair of antiparallel β-sheets (Fig. 1 ▶).
Figure 1.

(Top panel) Relaxed stereo ribbon diagram (side and top views) detailing the β-trefoil tertiary structure of FGF-1 and the associated threefold symmetry. The green color identifies the repeated structural domain. The cyan color within this domain identifies structurally conserved elements. (Middle panel) Relaxed stereo diagram showing an overlay of the three structural domains within FGF-1 (regions 11–52, 53–93, and 94–137). The overlay was performed using the structurally conserved regions (cyan color) in the top panel. (Bottom panel) Alignment of the 140-amino-acid primary structure of FGF-1 according to the threefold tertiary structure symmetry present in the β-trefoil superfold (Murzin et al. 1992). Light gray boxes indicate residue positions with two residues in common. The dark gray box indicates the single position where all three residues are in common. The open boxes indicate the residues comprising the core packing group.
In the present study, we use directed mutagenesis to introduce a primary structure symmetry within FGF-1 that reflects the threefold tertiary symmetry present within the β-trefoil superfold. The goal of the study is to identify an appropriate solution to such a symmetric constraint within the hydrophobic core. In starting with a native protein, we begin with a foldable polypeptide and can, therefore, unambiguously identify mutation sites that negatively impact the thermodynamics or kinetics of folding. This approach is in contrast to attempting an initial de novo design of the entire structure, because the specific reasons for any failure to achieve a foldable polypeptide would be extremely difficult to elucidate. FGF-1 has a greater degree of tertiary symmetry than other members of the β-trefoil superfold, and is among the best characterized with regard to structure, thermodynamics, and folding kinetics, and is therefore an excellent choice for this study (Copeland et al. 1991; Blaber et al. 1996, 1999; Chi et al. 2001; Samuel et al. 2001; Kim et al. 2003).
The directed mutagenesis makes use of sequence analysis between structurally related subdomains within FGF-1, as well as between homologous positions for other members of the β-trefoil superfold. The hydrophobic core of FGF-1 comprises 15 amino acids (with each of the three symmetry-related subdomains contributing five residues). In the present report, we describe a combination mutant of FGF-1, involving five positions within the core, that substantially increases the threefold symmetric constraint of the primary structure. In targeting the core region, we are focusing on a key structural domain that is an important contributor to stability and folding, and we are leaving intact residues known to be associated with receptor binding or heparin binding functionality (Zhu et al. 1993; Springer et al. 1994; Seddon et al. 1995; Blaber et al. 1996). Although each of the five point mutations within the core affects the structure, thermodynamics, and folding kinetics of FGF-1 to varying degrees, the combination of all five mutations exhibits structural, thermodynamic, and kinetic properties essentially indistinguishable from the WT protein. Although further work remains to constrain all 15 core residues to threefold primary structure symmetry (potentially requiring deletion mutations in the tertiary structure), the present work identifies a solution to the symmetric core packing problem for the β-trefoil superfold.
Results
Choice of core mutations, expression, and purification
Residue positions Val 31, Leu 73, and Cys 117 comprise a group of core positions related by the threefold symmetry of the β-trefoil tertiary architecture (Fig. 1 ▶). Val occurs at a consistently high frequency at each of these positions for a sequence comparison of the entire β-trefoil family of proteins, and we have previously described a Val mutation at position 73 (Brych et al. 2001). The substitution of an additional Val residue at position 117 (thus constraining symmetry-related positions 31, 73, and 117 to Val) was evaluated in the present study. An analysis of van der Waals contacts for a Val residue modeled into position 117 in both the WT and SYM3 mutant (Table 1) did not identify any steric clashes. Furthermore, the electron density of WT in the region of residue 117 (molecule B, PDB structure factor accession R1JQZSF) indicates the WT Cys Sγ can adopt more than one conformation. Thus, the sequence and structure analysis indicated that a β-branched side chain would be accommodated at position 117 within both the WT and SYM3 mutant structures. Because of the dual orientation of the WT Cys 117 side chain, the mutant electron density maps do not unambiguously indicate that the Cys side chain has been mutated to Val. FGF-1 contains three free cysteine residues and can be stoichiometrically quantitated by reaction with DTNB reagent. DTNB titration of the WT and Cys 117→Val mutant proteins confirmed the existence of the mutation because of absorbance at 412 nm of the nitrobenzoate group that was two-thirds of the WT signal (data not shown).
Table 1.
FGF-1 core mutations, abbreviations, and their symmetry relationships
| Mutation | Abbreviation | Symmetry relationships |
| Wild type | WT | (Leu 14, Ile 56, Tyr 97) |
| (Leu 23, Leu 65, Val 109) | ||
| (Ile 25, Met 67, Leu 111) | ||
| (Val 31, Leu 73, Cys 117) | ||
| (Leu 44, Phe 85, Phe 132) | ||
| Leu 44→Phe | — | (Phe 44, Phe 85, Phe 132) |
| Met 67→Ile | — | (Ile 25, Ile 67, Leu 111) |
| Leu 73→Val | — | (Val 31, Val 73, Cys 117) |
| Val 109→Leu | — | (Leu 23, Leu 65, Leu 109) |
| Leu 111→Ile | — | (Ile 25, Met 67, Ile 111) |
| Cys 117→Val | — | (Val 31, Leu 73, Val 117) |
| Leu 73→Val/Val 109→Leu | SYM2 | (Leu 23, Leu 65, Leu 109) |
| (Val 31, Val 73, Cys 117) | ||
| Leu 44→Phe/Leu 73→Val/Val 109→Leu | SYM3 | (Leu 23, Leu 65, Leu 109) |
| (Val 31, Val 73, Cys 117) | ||
| (Phe 44, Phe 85, Phe 132) | ||
| Leu 44→Phe/Leu 73→Val/Val 109→Leu/Cys 117→Val | SYM4 | (Leu 23, Leu 65, Leu 109) |
| (Val 31, Val 73, Val 117) | ||
| (Phe 44, Phe 85, Phe 132) | ||
| Leu 44→Phe/Leu 73→Val/Val 109→Leu/Leu 111→Ile/Cys 117→Val | SYM5 | (Leu 23, Leu 65, Leu 109) |
| (Ile 25, Met 67, Ile 111) | ||
| (Val 31, Val 73, Val 117) | ||
| (Phe 44, Phe 85, Phe 132) | ||
| Leu 44→Phe/Met 67→Ile/Leu 73→Val/Val 109→Leu/Leu 111→Ile/Cys 117→Val | SYM6 | (Leu 23, Leu 65, Leu 109) |
| (Ile 25, Ile 67, Ile 111) | ||
| (Val 31, Val 73, Val 117) | ||
| (Phe 44, Phe 85, Phe 132) |
Bold type indicates the mutated positions within the symmetry-related sets of core residues.
Residue positions Ile 25, Met 67, and Leu 111 comprise another group of core positions related by the threefold symmetry of the β-trefoil tertiary architecture (Fig. 1 ▶). β-Branched residues occur with a 53% frequency at position 25 for a comparison of all members of the β-trefoil family of proteins. A similar analysis of position 67 indicates β-branched residues are present with a frequency of occurrence of 32%. Likewise, β-branched residues are found with a frequency of occurrence of 42% at position 111. Thus, although no single residue type is generally conserved, there is a significant preference for β-branched residues at these symmetry-related positions. With regard to a choice between Ile or Val residues, the preexisting Ile at position 25 and a consideration of the packing volume contributed by this group indicated that Ile would be the more appropriate choice. An analysis of packing interactions indicated that an Ile substitution at position 67 (with rotamer identical to Ile 25) would not introduce any significant close contacts, but that a 24 Å3 cavity (detectable using a 1.0 Å radius probe) would be created with the loss of the Sδ and Cɛ groups of Met 67. Conversely, an Ile substitution at position 111 would not introduce any close contacts and no significant increase in microcavity space within the core. Therefore, Ile substitutions were selected at positions 67 and 111 to constrain the symmetry-related positions at 25, 67, and 111 to Ile.
The Cys 117→Val mutation was constructed in both the WT and SYM3 mutant backgrounds (resulting in the SYM4 mutant). The Leu 111→Ile mutation was constructed in both the WT background and the SYM4 mutant background (resulting in the SYM5 mutant). The Met 67→Ile mutation was constructed in both the WT background and the SYM5 mutant background (resulting in the SYM6 mutant; Table 1). Each mutation expressed at a level similar to the WT protein (~30–100 mg/L) with the exception of the Met 67→Ile substitutions. The Met 67→Ile point mutation in the WT background exhibited precipitation during purification (attributed to substantial destabilization resulting in unfolding) that precluded isolation with any significant yield (<1 mg/L of culture). The SYM6 mutant likewise precipitated; however, a few milligrams of purified protein was obtained from the combination of several liters of culture.
Structural studies
Structures for the Leu 44→Phe, Leu 73→Val, and Val 109→Leu point mutations and their combination have previously been reported (Brych et al. 2001). In the present study, high-resolution X-ray structures were determined for the Cys 117→Val and Leu 111→Ile point mutations as well as the SYM4 and SYM5 mutations (Table 2). The SYM5 mutation was observed to crystallize in a lower-symmetry spacegroup (C2, four molecules/asu) in comparison to the other structures. This crystal form has previously been observed for the Leu 44→Phe and SYM3 mutants, and appears to be related to a naturally occurring alternative side-chain conformation for residue His 93, located at a crystal contact (Brych et al. 2001).
Table 2.
Crystallographic data collection and refinement
| WTa | Cys 117→Val | Leu 111→Ile | SYM4 | SYM5 | |
| Crystal data | |||||
| Space group | C2221 | C2221 | C2221 | C2221 | C2 |
| a (Å) | 74.1 | 74.2 | 74.4 | 74.2 | 96.7 |
| b (Å) | 96.8 | 96.2 | 96.7 | 97.0 | 74.0 |
| c (Å) | 109.0 | 109.0 | 107.8 | 108.9 | 109.1 |
| β (°) | — | — | — | — | 89.98 |
| Mol/ASU | 2 | 2. | 2. | 2. | 4. |
| Matthews constant | 2.96 | 2.95 | 2.94 | 2.97 | 2.96 |
| Max resolution (Å) | 1.65 | 1.70 | 1.60 | 1.70 | 1.95 |
| Data collection and processing | |||||
| Total/unique reflections | 1,633,618/41,378 | 1,103,929/49,432 | 554,851/41,034 | 738,828/55,365 | |
| % completion (highest shell) | 98.4 (95.8) | 98.8 (88.4) | 88.3 (90.5) | 100.0 (100.0) | |
| I/σ (highest shell) | 60.1 (3.3) | 46.4 (4.3) | 32.1 (3.3) | 21.8 (2.9) | |
| Wilson B (Å2) | 21.1 | 16.7 | 20.2 | 15.4 | |
| Rmerge (%) | 6.3 | 5.0 | 4.7 | 6.0 | |
| Refinement | |||||
| Rcryst (highest shell) | 20.9 (37.0) | 21.9 (32.0) | 22.0 (34.0) | 19.4 (25.0) | |
| Rfree (highest shell) | 24.7 (37.0) | 25.1 (36.0) | 24.9 (36.0) | 25.3 (27.0) | |
| R.m.s. bond length dev (Å) | 0.007 | 0.009 | 0.008 | 0.008 | |
| R.m.s. bond angle dev (°) | 1.5 | 1.7 | 1.5 | 1.6 | |
| R.m.s. B factor dev (σ)b | 1.9 | 2.9 | 1.9 | 1.9 | |
| PDB accession | 1JY0 | 1P63 | 1M16 | 1NZK | |
Equilibrium thermodynamics
Thermodynamic values derived from isothermal equilibrium denaturation follow closely the results previously reported for DSC stability measurements for point mutations Leu 44→Phe, Leu 73→Val, and Val 109→Leu, as well as the SYM2 and SYM3 mutants (Brych et al. 2001; Kim et al. 2002). Both the Cys 117→Val and Leu 111→Ile point mutations were accommodated within the WT, SYM3, and SYM4 mutants, respectively, with minimal effects on all relevant thermodynamic parameters (Table 3). The SYM6 mutant was purified and the isothermal equilibrium data indicated partial denaturation even in 0 M denaturant and a transition midpoint of ~0.54 M GuHCl (Table 3). These results support the substantial instability postulated from the precipitation during purification for the Met 67→Ile mutation when constructed in either the WT or SYM5 backgrounds.
Table 3.
Thermodynamic parameters for WT and core packing mutations as determined by both isothermal equilibrium denaturation fluorescence and DSC
| Protein | ΔG0 (kJ/mole) | m-value (kJ/mole M) | Cm (M) | ΔΔGa (kJ/mole) | Tm (K)d | ΔH (kJ/mole)d |
| WTb | 21.1 ± 0.6 | 18.9 ± 0.6 | 1.11 ± 0.01 | — | 313.2 ± 0.7 | 261 ± 14 |
| Leu 44→Pheb | 25.1 ± 0.3 | 20.4 ± 0.2 | 1.23 ± 0.01 | −2.4 | 316.2 ± 0.1 | 320 ± 4 |
| Met 67→Ilec | ND | ND | ND | ND | ND | ND |
| Leu 73→Valb | 13.7 ± 0.6 | 19.7 ± 0.5 | 0.69 ± 0.05 | 8.1 | 304.7 ± 0.5 | 187 ± 3 |
| Val 109→Leub | 17.4 ± 0.5 | 20.2 ± 1.1 | 0.86 ± 0.02 | 4.9 | 309.9 ± 0.1 | 219 ± 1 |
| Leu 111→Ile | 21.1 ± 0.1 | 19.9 ± 0.2 | 1.06 ± 0.01 | 1.0 | 312.5 ± 0.2 | 274 ± 4 |
| Cys 117→Val | 21.0 ± 0.3 | 20.1 ± 0.1 | 1.05 ± 0.01 | 1.2 | 312.8 ± 0.2 | 257 ± 4 |
| SYM2b | 17.2 ± 0.3 | 20.6 ± 0.3 | 0.84 ± 0.01 | 5.3 | 308.6 ± 0.2 | 233 ± 3 |
| SYM3b | 17.9 ± 0.4 | 19.1 ± 0.3 | 0.94 ± 0.01 | 3.2 | 312.5 ± 0.7 | 257 ± 11 |
| SYM4 | 19.4 ± 0.2 | 19.2 ± 0.3 | 1.01 ± 0.02 | 1.9 | 312.0 ± 0.2 | 254 ± 6 |
| SYM5 | 20.8 ± 0.5 | 19.4 ± 0.1 | 1.07 ± 0.02 | 0.8 | 313.3 ± 0.3 | 273 ± 3 |
| SYM6e | 10.4 ± 0.8 | 19.3 ± 1.9 | 0.54 ± 0.01 | 10.9 | ND | ND |
a ΔΔG = (Cm WT − Cm mutant) |K3 (mWT + mmutant)/2 as described by Pace (Pace and Scholtz 1997). A negative value indicates a more stable mutation. All errors are stated as standard error from multiple data sets.
c Only partially folded in 0 M denaturant.
d Data from DSC analysis.
e No Tm in 0.7 M GuHCl.
Differential scanning calorimetry
DSC data for Leu 44→Phe, Leu 73→Val, and Val 109→Leu, as well as the SYM2 and SYM3 mutants, have previously been reported (Brych et al. 2001). DSC data were determined for all other mutations in the present study with the exception of the Met 67→Ile point mutation and the SYM6 mutant (Table 3). Although quantities of the SYM6 mutant could be isolated, the combination of substantial instability (Cm = 0.54 M GuHCl) and the presence of 0.7 M GuHCl in the DSC buffer (required for reversible thermal denaturation [Blaber et al. 1999]) resulted in there being no temperature at which the protein was at least 50% folded. Formally, therefore, there is no Tm value under these conditions for this mutant. We have previously reported that ΔΔG values determined by DSC are typically within 0.5 kJ/mole of values obtained by isothermal equilibrium denaturation data (Blaber et al. 1999; Kim et al. 2003), and this observation is consistent with the current set of mutations (data not shown).
Folding and unfolding kinetics
The point mutations that exhibit the most significant effects on folding rate include Leu 44→Phe (resulting in a 9.7-fold increase in kf) and the Leu 73→Val mutation (resulting in a 42-fold decrease in kf; Table 4, Fig. 2 ▶). In contrast, the Val 109→Leu, Leu 111→Ile, and Cys 117→Val point mutants introduced within the WT background exhibited minimal perturbations on kf. The unfolding rate constants were generally observed to fall within a more narrow range than the folding rate constants. The point mutation that exhibited the most significant effect on the unfolding rate was Val 109→Leu (resulting in a 3.9-fold increase in ku Table 4, Fig. 2 ▶). The SYM6 mutation appeared to be substantially unfolded in the absence of denaturant, and kinetic analysis was not possible.
Table 4.
Folding and unfolding kinetic parameters derived from global (chevron) fit
| Protein | kf (sec−1) | ΔΔG≠-D (kJ/mole) | mf (M−1) | ku (1 × 10−3 sec−1) | ΔΔG≠-N (kJ/mole) | mu (M−1) | βT |
| WTa | 3.74 | — | −6.61 | 0.808 | — | 0.46 | 1.07 |
| Leu 44→Phe | 36.35 | 5.6 | −7.21 | 1.677 | 1.8 | 0.56 | 1.08 |
| Met 67→Ileb | ND | ND | ND | ND | ND | ND | ND |
| Leu 73→Val | 0.09 | −9.2 | −4.95 | 0.357 | −2.0 | 0.56 | 1.13 |
| Val 109→Leu | 3.04 | −0.5 | −6.04 | 3.11 | 3.3 | 0.56 | 1.10 |
| Leu 111→Ile | 3.05 | −0.5 | −6.44 | 0.769 | −0.1 | 0.48 | 1.08 |
| Cys 117→Val | 3.28 | −0.3 | −7.01 | 0.872 | 0.2 | 0.46 | 1.07 |
| SYM2 | 0.14 | −8.1 | −5.36 | 0.273 | −2.7 | 0.53 | 1.11 |
| SYM3 | 2.09 | −1.4 | −6.99 | 0.612 | −0.7 | 0.57 | 1.09 |
| SYM4 | 3.37 | −0.3 | −7.74 | 0.381 | −1.9 | 0.54 | 1.08 |
| SYM5 | 1.91 | −1.7 | −6.96 | 0.388 | −1.8 | 0.48 | 1.07 |
| SYM6c | ND | ND | ND | ND | ND | ND | ND |
b Only partially folded at 0 M denaturant.
c Folding data limited by short chevron arm. Unfolding data unreliable because of low protein concentration.
Figure 2.

Chevron plot of the folding and unfolding kinetic data for WT (filled circles), Leu 44→Phe (filled diamonds), Leu 73→Val (filled triangles), Val 109→Leu (filled squares), and the SYM5 mutant (open circles). The Cys 117→Val and Leu 111→Val point mutations are essentially indistinguishable from WT and are omitted for clarity.
Discussion
Many de novo protein design efforts have used tertiary structure symmetry as a design strategy to simplify the construction of complex protein architectures, including α-helical bundles (Regan and DeGrado 1988; Hecht et al. 1990; Betz and DeGrado 1996; Betz et al. 1997; Walsh et al. 2001; Burkhard et al. 2002; Wei et al. 2003), various types of β secondary structure (Richardson and Richardson 1989; Quinn et al. 1994; Yan and Erickson 1994; West et al. 1999), and α/β-barrel proteins (Offredi et al. 2003). Although some notable successes in de novo design have been achieved, the typical designed protein suffers from flaws in the packing of hydrophobic core residues, resulting in “molten globules” or loosely packed cores (Bryson et al. 1995; Betz and DeGrado 1996; Wei et al. 2003).
Martial and coworkers have recently described the ambitious de novo design of a 216-residue α/β-barrel (TIM barrel) protein (Offredi et al. 2003). The design principle constrained a structural subdomain (comprising two strand/turn/helix/strand elements) to fourfold tertiary structure symmetry. A dead-end elimination algorithm was then used to identify a solution to the packing of side chains. The resulting primary structure, although exhibiting evidence of the tertiary structure symmetry, was, nonetheless, asymmetric. Unfortunately, no discussion was provided regarding whether a symmetric solution was possible given the symmetric tertiary structure constraint, or whether such a solution might yet be identified. The resulting protein exhibited a high content of secondary structure and substantial stability; however, it also exhibited structural rearrangement in low concentrations of denaturant, as well as the presence of a folding intermediate (Offredi et al. 2003). This intermediate showed enhanced binding to ANS in low concentrations of denaturant, indicating a loosely packed hydrophobic core under low denaturant conditions. The relationship between tertiary structure symmetry, primary structure symmetry, stability, foldability, and efficient hydrophobic core packing are critically important aspects of de novo protein design that remain to be fully elucidated for each of the fundamental superfolds.
Mutational effects on folding and unfolding kinetics, strain, and buried area
Seminal work by Serrano and coworkers, investigating alternative core packing arrangements within the spectrin SH3 domain, has highlighted the importance of elucidating the effects on folding and unfolding kinetics, in addition to stability effects, in order to gain a clear understanding of the properties of redesigned core regions and the TSE (Ventura et al. 2002). In particular, alternative core packing arrangements that bury additional hydrophobic area, but subsequently introduce strain, can exhibit an increased rate of folding (as a result of additional buried area) but also an increased rate of unfolding (as a result of additional strain). The kinetic profile for such mutations may be substantially altered (i.e., in the earlier example, both arms of the “chevron” plot will be shifted vertically in comparison to the WT protein), which can be independent of any observable effects on stability. Thus, based on stability arguments alone, an alternative core packing arrangement cannot be unambiguously identified as being “WT equivalent.”
The kinetic data reported here indicate that the Leu 44→Phe mutation increased the rate of folding by ~10-fold while increasing the rate of unfolding by approximately twofold (Table 4, Fig. 2 ▶). In terms of the study of Serrano and coworkers, these results indicate that this mutation buries additional area within the core without suffering substantial strain. This interpretation is in excellent agreement with the previously reported structural results that indicate a “microcavity” (identified using a 1.0 Å radius probe) is present within the WT core region adjacent to the side chain of position 44, and can readily accommodate the aromatic ring of the Phe mutation (Brych et al. 2001). The Leu 73→Val mutation results in an approximate fourfold reduction in the folding rate and a twofold reduction in the unfolding rate (Table 4, Fig. 2 ▶). As previously reported, this mutation results in a reduction in buried area and an increase in cavity space within the core (Brych et al. 2001). Again, the observed effects on the folding and unfolding kinetic data are in agreement with the results of Serrano and coworkers. The Val 109→Leu mutation has essentially no effect on the rate of folding, but increases the rate of unfolding approximately fourfold (Table 4, Fig. 2 ▶). A previously reported structural analysis of this mutation showed that a close contact occurs between positions 109 and 73, with the Leu mutation at position 109 adopting a strained conformation. Furthermore, although the introduced Leu side chain partially fills a centrally located cavity within the core, an additional adjacent microcavity is formed due to the deletion of the WT Val γ methyl group (Brych et al. 2001). The kinetic results indicate that this mutation is associated primarily with additional strain, and the additional buried area is of limited consequence (again, in agreement with the structural and stability data).
Although positions 44, 73, and 109 are members of the core packing group, positions 73 and 109 are immediate neighbors, and position 44 has limited contact with position 109 and no direct contact with position 73 (Fig. 3 ▶; Blaber et al. 1996; Brych et al. 2001). Thus, the effects on stability for the SYM2 mutant (i.e., Leu 73→Val/Val 109→Leu) are highly nonadditive (i.e., this is a compensating pair of adjacent mutations), whereas the combination of the SYM2 mutant and the Leu 44→Phe mutation (SYM3) is additive (Brych et al. 2001). In reference to the Leu 73→Val point mutation, the introduction of the Val 109→Leu mutation results in an ~1.5-fold increase in the rate of folding and also a slight decrease in the rate of unfolding (Table 4). These results indicate that the Val 109→Leu mutation in the Leu 73→Val background resulted in additional buried area, as well as a slight reduction in strain. Again, this interpretation is consistent with both the previously reported structural and stability data and the study of Serrano and coworkers (Ventura et al. 2002).
Figure 3.

Relaxed stereo diagram showing overlaid subdomains within WT FGF-1 (top panel) and SYM5 (bottom panel) for the core positions mutated in this study, and illustrating the symmetric rotamer orientations and packing environments for these positions.
The SYM3 mutant is only 3.2 kJ/mole less stable than the WT protein, and this decrease in stability is associated with an ~1.8-fold reduction in the rate of folding and an almost identical rate of unfolding in comparison to the WT protein (Table 4). Thus, the SYM3 mutant is similar to the WT protein with regard to both stability and kinetics. Although the difference in kinetic parameters is small, the data suggest that the SYM3 mutant is accommodated with a slight reduction in structural strain and a slightly reduced packing volume. The structural data for the SYM3 mutant do not identify any strained conformation; however, the data do indicate that the SYM3 mutant results in a reduced microcavity volume (Brych et al. 2001). Thus, although the interpretation of effects on strain is consistent, it is not immediately obvious why the SYM3 mutant should exhibit both a reduction in microcavity volume within the core and a reduced rate of folding. It may be that an analysis of cavity volume within the core using a smaller radius probe would identify somewhat less extensive van der Waals contacts for the SYM3 mutant. However, the resolution of the structural data makes it difficult to unambiguously assign cavity volumes for probe radii smaller than 1.0 Å. In any case, it appears that the SYM3 mutant is accommodated with both near WT stability and folding and unfolding kinetics (i.e., comparable strain and buried area in comparison to the WT protein).
The Cys 117→Val point mutation in the WT background results in a slight (1.2 kJ/mole) destabilization of the structure (Table 3). However, in the background of the SYM3 mutant, the Cys 117→Val point mutation stabilizes the structure by 1.3 kJ/mole. Similarly, the Leu 111→Ile point mutation in the WT background results in a slight (1.0 kJ/mole) destabilization; however, in the background of the SYM4 mutant, the Leu 111→Ile point mutation stabilizes the structure by 1.1 kJ/mole (Table 3). The resulting SYM5 mutant has essentially identical thermodynamic parameters with the WT protein (Table 3). A comparison of the kinetic data for the SYM5 and WT proteins indicates that this alternative core packing group is accommodated with an approximately twofold reduction in both the folding and unfolding rates (Table 4, Fig. 2 ▶). Again, these slight differences indicate that the alternative core packing group has been accommodated with minimal effects on buried area or strain.
Nonadditivity with respect to stability and kinetics, and a comparison of SYM5 to WT FGF-1
In the WT background, the Leu 44→Phe mutation is the only stabilizing mutation; all other point mutations are destabilizing. However, as noted earlier, the Cys 117→Val mutation is stabilizing when introduced into the SYM3 mutant background, and the Leu 111→Ile is stabilizing when introduced into the SYM4 mutant background. A simple sum of the stability effects of the point mutants would predict that the SYM5 mutant should be 12.8 kJ/mole destabilized in comparison with the WT protein (Table 3, Fig. 4 ▶). Because the SYM5 mutant is only 0.8 kJ/mole destabilizing (essentially within the error of the measurement), the cooperative effects of this alternative core packing group contribute 12.0 kJ/mole toward greater stability.
Figure 4.
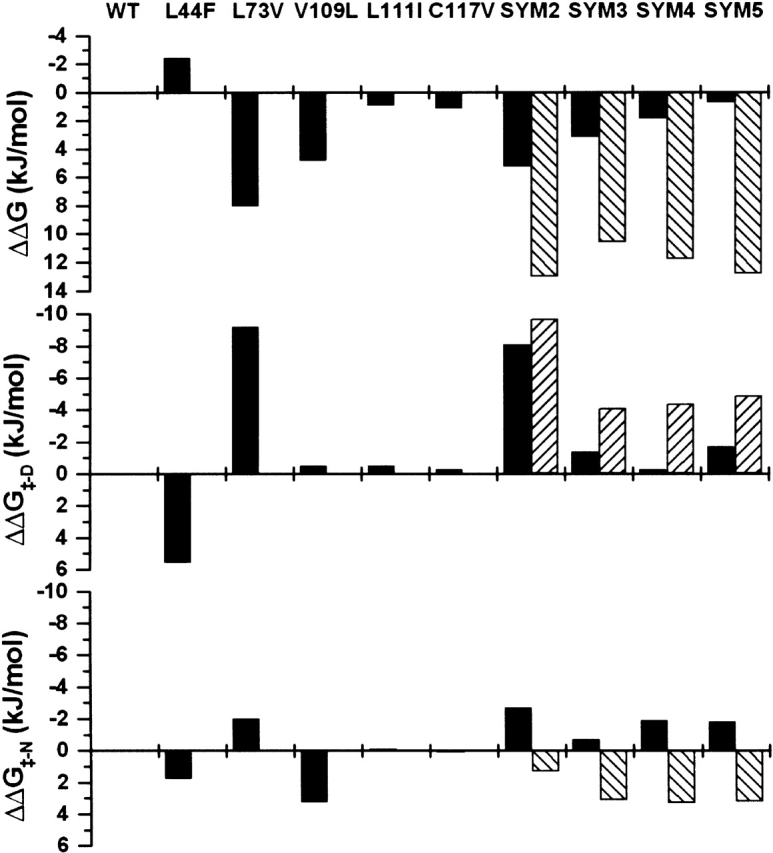
(Top panel) ΔΔG values for the effects on stability of point and combination mutations (negative values indicate a stabilizing mutation). (Middle panel) ΔΔG‡-D values for point and combination mutations derived from folding kinetic data (positive values indicate the TSE has been stabilized relative to the native state). (Bottom panel) ΔΔG‡-N values for point and combination mutations derived from unfolding kinetic data (positive values indicate the TSE has been stabilized relative to the native state). In each panel, the solid columns indicate the experimentally determined values, and the hatched columns indicate the values expected from a simple sum of the individual point mutations.
Residues 73 and 111 are core packing neighbors of position 117 (Fig. 3 ▶). The effective deletion of the Cδ1 atom of the WT Leu side chain at position 73 (by substitution to Val) provides room for a Val Cγ1 at position 117 (with a rotamer orientation consistent with symmetry-related residue Val 31). Similarly, the effective deletion of the Cδ2 atom of the WT Leu side chain at position 111 (via substitution to Ile, and adoption of a rotamer orientation consistent with symmetry-related residue Ile 25) provides room for a Val Cγ2 at position 117. X-ray structure analysis of the SYM4 mutation (containing a Cys 117→Val mutation, but with Leu at position 111) indicates that the Val side chain at position 117 adopts a χ1 rotamer angle of 71°. Only in the SYM5 mutant (with inclusion of the Leu 111→Ile mutation) does the Val side chain at position 117 adopt a trans χ1 rotamer angle (conforming to the symmetry-related χ1 rotamer angle; Fig. 3 ▶). There is, therefore, a cooperative interaction between mutations Leu 73→Val, Leu 111→Ile, and Cys 117→Val that provides a structural rationale for the cooperativity observed for the thermodynamic and kinetic data.
An analysis of the relative change in the transition state energy barrier relating to the effects on folding (ΔΔG≠-D) and unfolding (ΔΔG≠-N) kinetics also indicates a substantial nonadditive effect of the SYM5 mutant. A simple sum of ΔΔG≠-D values for the point mutations predicts a 4.9 kJ/mole increase in the TSE energy barrier, relative to the denatured state, for SYM5 (Table 4); however, the SYM5 mutant exhibits only a 1.7 kJ/mole increase. Thus, the alternative core packing group results in 3.2 kJ/mole of nonadditivity of ΔΔG≠-D values (contributing to faster-than-expected folding rates). Likewise, a simple sum of ΔΔG≠-N values for the point mutations predicts a −3.2 kJ/mole decrease in the TSE energy barrier, relative to the native state, for the SYM5 mutant; however, this mutant actually exhibits a 1.8 kJ/mole increase (Table 4). Thus, the alternative core packing group results in 5.0 kJ/mole of nonadditivity of ΔΔG≠-N values (contributing to slower-than-expected unfolding rates). Taken within the context of the work of Serrano and coworkers, these results demonstrate cooperativity in terms of both optimizing packing interactions and reduction of strain for the alternative packing arrangement in the SYM5 mutation, in addition to minimal overall energetic effects on the folding transition state ensemble. Thus, the transition state ensemble is also essentially energetically equivalent to that of WT. This conclusion is further supported by the βT values for each protein (Table 4). Each intermediate mutant exhibits a larger βT value than the WT protein, indicating that the solvent exposure of the TSE is higher than the native state. This value is lowest (identical to WT) for the C 117→Val point mutant, and highest for the Leu 73→Val mutant. These mutations correspondingly exhibit the least, and greatest, structural perturbation, respectively, on the core region. These results are consistent with a TSE that contains a native-like arrangement of the residues comprising the core region. Because of the relatively large number of mutations incorporated into the SYM5 mutant, and the very low ΔΔG value associated with these changes, application of φ value analysis to quantitate the nature of the energetic changes to the TSE is not appropriate (Fersht 1999a; Friel et al. 2003). Nonetheless, the essentially equivalent folding and unfolding kinetics, identical βT value, and ΔΔG ~ 0 are consistent with SYM5 also having a WT equivalent TSE.
Our previous study of mutations at positions 44, 73, and 109 indicated that combination mutations that increase the primary structure symmetry at these positions also result in an increasing deviation from two-state folding behavior (Brych et al. 2001). In this regard, although the WT FGF-1 protein exhibited excellent agreement between the van’t Hoff and calorimetric enthalpies of unfolding (under buffer conditions including 0.7 M added GuHCl), the SYM3 mutant exhibited a ΔHvH/ΔHcal ratio of 0.5. This result led us to postulate that properties of folding, rather than stability, may contribute to a selective pressure against symmetric primary core sequences within symmetric protein architectures (Brych et al. 2001). However, this postulate is not supported by the results of the present study involving additional sites that further increase the primary structure symmetry within the core. The SYM4 mutant exhibits a ΔHvH/ΔHcal ratio of 0.63 and the SYM5 mutant a ratio of 0.70 (and this value approaches 1.0 when the DSC data are collected under buffer conditions containing >0.70 M GuHCl). The SYM5 mutant is essentially indistinguishable from the WT protein with regard to every thermodynamic and kinetic parameter (Tables 3 and 4). Therefore, mutations contributing to a further increase in the primary structure symmetry within the core region of FGF-1 have not resulted in any identifiable deleterious effects on either the stability or the folding properties of the protein.
Symmetric nature of the structural environment around mutation sites in SYM5
The backbone atoms of each structurally conserved region of the symmetry-related subdomains (see Fig. 1 ▶) in WT FGF-1 overlay with an r.m.s. deviation of between 0.62 and 0.75 Å. These backbone atoms as a group (i.e., the entire molecule) can be rotated by the threefold symmetry and overlaid with an r.m.s. deviation of 1.06 Å. A similar analysis of the SYM5 mutant shows that the individual subdomains overlay with an r.m.s. deviation of between 0.51 and 0.70 Å, and the backbone atoms as a group overlay with an r.m.s. deviation of 1.03 Å. Thus, the combination mutations within SYM5 have not disrupted the tertiary structure (in fact, there may be a slight increase in the structural identity between subdomains). An overlay of the core positions in the WT and SYM5 mutant structures that are part of the present study are shown in Figure 3 ▶. For clarity, this figure omits main chain atoms that define additional packing interactions around these residues, but as the earlier overlay analysis confirms, these are also essentially identical for each subdomain. This overlay serves to highlight the extent of the symmetric packing relationship present in the SYM5 mutant; each symmetry-related residue is in an equivalent rotamer orientation. There are two exceptions to this symmetric packing interaction: (1) Met67 lacks the γ and δ atom interactions with Phe 85 and Val 73 (Fig. 3 ▶), and (2) the remaining residues forming the core include Leu 14, Ile 56, and Tyr 97, and these have not yet been constrained to threefold symmetry. The symmetric packing relationship between the 12 core residues in the present study, and their differences in WT versus SYM5, are illustrated in Figures 5 ▶ and 6 ▶. The juxtaposition of remaining residues 14, 56, and 97 can be appreciated by looking at the final panel in each figure; these residues essentially pack on top of residue positions 23, 65, and 109. Our approach to construct a symmetric core is to start from the “bottom” of the protein β-barrel and work upward (Figs. 5 ▶, 6 ▶). In this regard, positions 14, 56, and 97 are the topmost or last residue positions. Prior to studying these positions, the asymmetry around position 67 needs to be addressed.
Figure 5.

Relaxed stereo diagram of WT FGF-1 illustrating the core packing arrangement. Moving from top to bottom panels, the sequential packing of positions 31, 73, 117; 25, 67, 111; 23, 65, 109; and 44, 85, 132 are shown. The color of the three symmetry-related subdomains is the same as in Figure 1 ▶ (middle panel).
Figure 6.

Relaxed stereo diagram of SYM5 illustrating the core packing arrangement (details follow that of Fig. 5 ▶).
Position 67
The incorporation of point mutant Met 67→Ile into WT or the SYM5 mutant background (resulting in the SYM6 mutant) substantially destabilizes the protein (Table 3). As previously mentioned, modeling of the Met 67→Ile mutation (with a side-chain rotamer similar to symmetry-related Ile 25) in either the WT or SYM5 X-ray structure results in the formation of a 24 Å3 cavity (detected using a 1.0 Å probe radius) due to the effective deletion of the Met Sδ and Cɛ side-chain atoms (because the Met side chain adopts an alternative rotamer in comparison with Ile residues at symmetry-related positions, Fig. 3 ▶). The Met at position 67 is conserved in all known members of the FGF family of proteins. The symmetry-related positions to Met 67 are Ile 25 and Leu 111, and a comparison of the structure around these other positions provides some insight as to how a Met is accommodated at position 67 and why an Ile side chain at this position is so destabilizing. Structural overlays of position 67 with position 25 or 111 in FGF-1 indicate that the packing environment surrounding position 67 includes two regions of insertions in neighboring main chains. These insertions are defined by residue positions 104–106 and 120–122, and are apparent when comparing the primary structure between the three subdomains (Fig. 1 ▶). Conversely, the corresponding main chain regions forming the packing environment adjacent to positions 25 and 111 lack equivalent insertions. Other members of the FGF family exhibit similar insertions in their primary structure at regions predicted to pack against the Met at position 67. Other proteins belonging to the β-trefoil superfold provide compelling support for this structural relationship between Met residues at this (or a symmetry-related) position and insertions within neighboring main chain regions that pack against this side chain. Hisactophilin and barley α-amylase/subtilisin inhibitor have a β-trefoil structural fold similar to FGF-1 (Habazettl et al. 1992; Vallee et al. 1998). Hisactophilin contains an Ile residue at the equivalent position to Met 67 in FGF-1, whereas barley amylase/subtilisin inhibitor has a Met at a position equivalent to Ile 25 in FGF-1 (related by threefold symmetry to position 67). Structural analysis of hisactophilin indicates that the primary structure does not contain the characteristic insertions at positions equivalent to 104–106 and 120–122 in FGF-1 (which pack against the Ile residue at the equivalent of position 67). Conversely, analysis of the primary structure of barley amylase/subtilisin inhibitor indicates that there is a seven-residue insertion in the region that packs against the Met residue that is the structural equivalent to position 25 in FGF-1. These analyses provide strong support for the postulate that the tertiary structure of FGF-1 must be modified in order to allow a symmetric relationship of the primary structure at positions 25, 67, and 111. Specifically, residues 104–106 and 120–122 must be deleted so as to allow appropriate packing against an introduced Ile side chain at position 67. Furthermore, we note that there are important functional properties associated with regions 104–106 and 120–122. Residues 104–106 comprise the low-affinity receptor/heparin-binding site, and residues 120– 122 contribute to the heparin-binding domain (Zhu et al. 1991; Springer et al. 1994; Blaber et al. 1996; Pellegrini et al. 2000). Thus, the accommodation of these functionalities within the structure of FGF-1 appears to have disrupted the tertiary (and, conversely, primary) structural symmetry. Deletion mutations within these regions of FGF-1 are currently being investigated; however, we note that although such deletions may promote accommodation of an Ile residue at position 67, they may also abolish receptor and heparin binding. To recapitulate, some of the asymmetry within the tertiary structure appears to be associated with functionalities of receptor and heparin binding, and asymmetry within the core appears necessary to stabilize it.
Tolerance of the β-trefoil fold toward a symmetric core constraint
There have been few studies that have successfully stabilized a protein by modifying the hydrophobic core (Ishikawa et al. 1993; Ohmura et al. 2001). The vast majority of core mutations are destabilizing because of the introduction of strain, the creation of a cavity, or both (Karpusas et al. 1989; Hurley et al. 1992; Buckle et al. 1993; Eriksson et al. 1993; Itzhaki et al. 1995; Mateu and Fersht 1999; Chen and Stites 2001; De Vos et al. 2001; Holder et al. 2001). From the available thermodynamic data for core mutations involving Leu→Phe, Val→Leu, Leu→Val, and Cys→Val mutations, the vast majority of such mutations are destabilizing (by up to 28 kJ/mole). The essentially equivalent stability to WT, for the combined SYM5 mutation, highlights the cooperative native of the alternative symmetric core packing group. Furthermore, analysis of the X-ray structures indicates that these mutations are accommodated with essentially no detectable perturbation on the overall structure. Additionally, there are no significant differences in the thermal factors of the core packing group in the SYM5 mutant in comparison with the WT structure (data not shown). Thus, from a structural standpoint, the alternative core packing group appears to be WT equivalent (Figs. 5 ▶, 6 ▶). Given that a symmetric constraint severely limits the choice of an alternative packing arrangement, the cooperative interaction of the SYM5 packing group is remarkable.
Implications for protein evolution and de novo design
The symmetrically constrained alternative packing group compares favorably with the WT core arrangement by all applied metrics of structure, stability, and folding kinetics. This result indicates that symmetric design principles, as a strategy to develop complex protein architectures from simpler self-assembling structural motifs, is tractable at least for core regions within the β-trefoil superfold. The β-trefoil architecture is compatible with a symmetric core architecture; therefore, gene duplication/fusion is an entirely feasible mechanism for this superfold to have evolved. We conclude that in the case of FGF-1, if the protein architecture evolved by gene duplication/fusion events, the extant asymmetric core packing arrangement may have arisen as a result of simple drift, with the exception of position 67. Asymmetry at this position appears to be related to the unique structural requirements of functionality associated with receptor and heparin binding. However, it is interesting to speculate that incorporation of Met 67 (as part of gaining receptor and heparin binding functionality) may have initiated a series of subsequent mutational events within the core (beginning perhaps with a Val 73→Leu mutation) to optimize core packing in response to this mutation. The consequences of a symmetric constraint on other structural elements, such as turns and secondary structure, are equally important to elucidate if such protein evolution and de novo design principles are to be understood and applied.
Materials and methods
Design of core mutations
An in-depth description of the symmetric constraint mutation design process has been previously reported (Brych et al. 2001). In brief, the hydrophobic core region of FGF-1 comprises 15 residue positions: 14, 23, 25, 31, 44, 56, 65, 67, 73, 85, 97, 109, 111, 117, and 132. Each β-trefoil subunit (~40 amino acids in length) in the 140-amino-acid β-trefoil structure contributes five residues to the hydrophobic core and is related to the other β-trefoil subunits by the threefold symmetry of the tertiary architecture. Thus, residue positions 14, 56, and 97; 23, 65, and 109; 25, 67, and 111; 31, 73, and 117; and 44, 85, and 132 comprise five sets of threefold symmetry-related positions that constitute the hydrophobic core of FGF-1 (Blaber et al. 1996; Brych et al. 2001). The rational design principle for the selection of mutations with which to increase primary structure symmetry within the core region involved a combination of structure and sequence analyses. The structural analysis focused on the characterization of van der Waals contact distances and the identification of microcavities within the core region (detectable using a 1.0 Å radius probe (Brych et al. 2001). The sequence analysis included identification of any intrachain symmetry-related consensus, as well as interchain analysis of structurally related proteins within the β-trefoil family. Once a candidate residue was identified, the mutation was modeled into the 1.65 Å crystal structure of FGF-1 (1JQZ), or relevant mutation thereof. The structural analysis made note of consensus rotamer angles for symmetry-related positions, and incorporated these as a constraint in modeling of the mutant side chain. Appropriate candidate mutations were identified by acceptable van der Waals contact criteria.
Mutagenesis and expression
All studies used a synthetic gene for the 140-amino-acid form of human FGF-1 (Gimenez-Gallego et al. 1986; Linemeyer et al. 1990; Ortega et al. 1991; Blaber et al. 1996) with the addition of an amino-terminal six-residue His-tag to facilitate purification (Brych et al. 2001). The QuikChange site-directed mutagenesis protocol (Stratagene) was used to introduce the mutations Leu 44→Phe, Met 67→Ile, Leu 73→Val, Val 109→Leu, Leu 111→Ile, and Cys 117→Val (individually or in combination) using mutagenic oligonucleotides of 25 to 31 bases in length (Biomolecular Analysis Synthesis and Sequencing Laboratory, Florida State University). All FGF-1 mutants were expressed using the pET21a(+) plasmid/BL21(DE3) Escherichia coli host expression system (Invitrogen). Mutant construction, expression, and purification followed previously described procedures (Blaber et al. 1999; Culajay et al. 2000; Brych et al. 2001).
X-ray crystallography
Purified protein was equilibrated in 50 mM sodium phosphate, 100 mM NaCl, 10 mM ammonium sulfate, 2 mM DTT, and 0.5 mM EDTA (pH 7.5) and concentrated to 10 to 12 mg/mL. Crystallization was performed using hanging drop vapor diffusion as previously described (Brych et al. 2001). X-ray diffraction data were collected using a Rigaku RU-H2R rotating anode X-ray source (Rigaku MSC) equipped with Osmic Blue or Purple confocal mirrors (MarUSA) coupled with either a Rigaku R-axis IIc image plate or MarCCD 165 detector. Crystals were mounted using nylon cryo loops (Hampton Research) and the crystals were frozen in a stream of liquid nitrogen and cooled to 100K during data collection. X-ray data indexing, integration, and scaling were performed using the DENZO software package (Otwinowski 1993; Otwinowski and Minor 1997). Before structure refinement, 3%–10% of the raw data was removed into a test set to calculate Rfree (Brunger 1992). Atomic coordinate refinement was completed using the TNT least-squares refinement software package (Tronrud et al. 1987; Tronrud 1992) using knowledge-based thermal factor restraints (Tronrud 1996). Model building was accomplished using the O software program (Jones et al. 1991). All coordinate and structure factor files have been deposited in the protein data bank (Table 2).
Isothermal equilibrium denaturation
Protein samples were equilibrated overnight in 20 mM ADA, 100 mM NaCl, and 2 mM DTT (pH 6.60) at 298K in 0.1 M increments of GuHCl. All samples contained a final protein concentration of 10 μM. A Varian Eclipse fluorescence spectrophotometer (Varian) was used for all fluorescence measurements using a 5-nm slit on both excitation and emission monochrometers. The fluorescence signal arising from the single ~90% buried Trp 107 is internally quenched in the native state, and quenching is subsequently released on denaturation (Gimenez-Gallego et al. 1986; Linemeyer et al. 1987; Blaber et al. 1999). Fluorescence emission spectra were obtained by excitation of the Trp residue at 295nm. For each sample, triplicate scans were collected and averaged, and buffer traces were collected, averaged, and subtracted from the sample traces. Integration of the fluorescence scans was used to quantitate the total fluorescence per sample. The general-purpose nonlinear least-squares fitting program DataFit (Oakdale Engineering) was used to fit the total fluorescence versus GuHCl concentration data to a six-parameter, two-state model (Eftink 1994):
 |
(1) |
where [D] is the denaturant concentration; F0N and F0D are the 0M denaturant fluorescence intercepts for the native and denatured states, respectively; and SN and SD are the slopes of the native and denatured state baselines, respectively. ΔG0 and m describe the linear function of the unfolding free energy versus denaturant concentration at 298K. The effect of a given mutation on the stability of the protein (ΔΔG) was calculated by taking the difference between the Cm values for WT and mutant and multiplying by the average of the m values, as described by Pace and Scholtz (1997):
 |
(2) |
Differential scanning calorimetry
All DSC data were collected on a VP-DSC microcalorimeter (MicroCal LLC) as previously described (Blaber et al. 1999). Protein samples (0.04 mM) were equilibrated at 298K in 20 mM ADA, 100 mM NaCl, and 0.7 M GuHCl (pH 6.60). The samples were filtered (0.2 μm) and degassed for 10 min prior to loading. A scan rate of 15°C/h was used and the protein was kept at ~30 psi during the calorimetric run. All data were collected without interruption of repeated thermal cycles. An average of at least three protein traces was performed, with buffer scans subsequently subtracted from the protein and concentration normalization. The resulting DSC endotherms were analyzed using the DSCfit software package (Grek et al. 2001).
Unfolding kinetics measurements
Folding and unfolding kinetic data were collected using previously described methods (Kim et al. 2003). In brief, protein samples were dialyzed against 20 mM ADA, 100 mM NaCl, and 2 mM DTT (pH 6.60) overnight at 298K prior to data collection. Initial protein concentrations were 25 μM except for mutations Leu 73→Val and Val 109→Leu, which precipitated at this concentration. Consequently, a 10-μM concentration was used for these proteins. Unfolding was initiated by 1 : 10 dilution of the native protein into 20 mM ADA, 100 mM NaCl, and 2 mM DTT (pH 6.60), with a final GuHCl concentration in the range of 1.5 to 4.5 M in 0.5-M increments. The unfolding process was quantitated at 298K by following Trp 107 emission at 350 nm, with excitation at 295 nm, using a Varian Eclipse fluorescence spectrophotometer equipped with a temperature-controlled Peltier cell holder. Manual mixing was the method of choice for unfolding studies because of the slow unfolding kinetics of the protein. Data collection times for each protein were designed so as to quantitate the fluorescence signal over two to three half-lives, or >75% of the total expected amplitude.
Folding kinetics measurements
Although collection of unfolding kinetic data was straightforward, the collection of folding kinetic data presented several previously described technical challenges (Kim et al. 2003). In brief, the low thermal stability of FGF-1 presents a relatively short “folding arm” of the chevron plot (Fig. 2 ▶). Furthermore, when monitored by fluorescence, the folding of FGF-1 exhibits “roll over” in the low denaturant concentration region (i.e., <0.6 M), indicative of non-two-state folding behavior (Kim et al. 2003). To address the issue of the short folding arm, we sampled a finer increment of denaturant concentration in comparison to the unfolding arm. However, given the larger magnitude of the kinetic mf value in comparison with mu, Δlnkobs for each denaturant concentration sampled is essentially equivalent for both arms of the chevron plot (Fig. 2 ▶). Protein samples were dialyzed against 20 mM ADA, 100 mM NaCl, 2 mM DTT, and 2.5 M GuHCl (pH 6.60) overnight at 298K prior to data collection. WT and mutant proteins were essentially completely denatured at this GuHCl concentration (Blaber et al. 1999). Folding was initiated by a 1 : 10 dilution of this protein solution into 20 mM ADA, 100 mM NaCl, and 2m M DTT (pH 6.60) containing 0.05M increments of GuHCl near to the midpoint of denaturation as determined by isothermal equilibrium denaturation experiments. All data were collected using a KinTek SF-2000 stopped-flow system (KinTek) and monitoring the quenching of the Trp 107 fluorescence on folding. Data collection times for each protein were designed so as to quantitate the fluorescence signal over five half-lives, or >96% of the total expected amplitude.
Kinetic analysis
Both folding and unfolding kinetic data were collected in triplicate at each GuHCl buffer condition. In all cases, data from at least three separate experiments were averaged. The kinetic rates and amplitudes versus denaturant concentration were calculated from the time-dependent change in tryptophan fluorescence using a single exponential model:
 |
(3) |
where I(t) is the intensity of fluorescent signal at time t, A is the corresponding amplitude, k is the observed rate constant for the reaction and C is a constant that is the asymptote of the fluorescence signal.
Folding and unfolding rate constant data were fit to a global function describing the contribution of both rate constants to the observed kinetics as a function of denaturant (chevron plot), as described by Fersht (1999a):
 |
(4) |
where kf0 and ku0 are the folding and unfolding rate constants, respectively, extrapolated to 0 M denaturant; mf and mu are the slopes of the linear functions relating ln(kf) and ln(ku), respectively, to denaturant concentration; and D is denaturant concentration. Changes in activation barriers on mutation were calculated using a modified version of transition state theory (Fersht 1999b):
 |
(5) |
 |
(6) |
where kf0Mut, ku0Mut, kf0WT, and ku0WT are the folding and unfolding rates for mutant and WT, respectively, in water. ΔΔG≠-D and ΔΔG≠-N are the changes in the activation barrier for folding and unfolding, respectively, between mutation and WT. The βT value, that describes the solvent exposure of the rate-limiting TSE in relationship to the native state, was calculated from the kinetic m values according to:
 |
(7) |
Acknowledgments
We thank Drs. Ewa Bienkiewicz of the Physical Biochemistry Facility and T. Somasundaram of the X-ray Facility, Kasha Laboratory, Institute of Molecular Biophysics, for helpful comments. We also thank Mr. Matthew Bernett, Mr. Gurunathan Laxmikanthan, Ms. Ginger Spielmann, and Ms. Joanna Balsamo for technical assistance. This work was supported by grants GM54429–01 from the U.S. Public Health Service/NIH and MCB 0314740 from the NSF.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
FGF-1, human acidic fibroblast growth factor
GuHCl, guanidinium hydrochloride
ADA, N-(2-acetamido)iminodiacetic acid
DTT, dithiothreitol
ANS, 1-anilinonapthalene-8-sulfonic acid
TIM, triose phosphate isomerase
DSC, differential scanning calorimetry
DTNB, dithionitrobenzoate
TSE, transition state ensemble
WT, wild type
r.m.s., root mean square
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03374903.
References
- Bergdoll, M., Eltis, L.D., Cameron, A.D., Dumas, P., and Bolin, J.T. 1998. All in the family: Structural and evolutionary relationships among three modular proteins with diverse functions and variable assembly. Protein Sci. 7 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz, S.F. and DeGrado, W.F. 1996. Controlling topology and native-like behavior of de novo-designed peptides: Design and characterization of antiparallel four-stranded coiled coils. Biochemistry 35 6955–6962. [DOI] [PubMed] [Google Scholar]
- Betz, S.F., Liebman, P.A., and DeGrado, W.F. 1997. De novo design of native proteins: Characterization of proteins intended to fold into antiparallel, rop-like, four-helix bundles. Biochemistry 36 2450–2458. [DOI] [PubMed] [Google Scholar]
- Blaber, M., DiSalvo, J., and Thomas, K.A. 1996. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry 35 2086–2094. [DOI] [PubMed] [Google Scholar]
- Blaber, S.I., Culajay, J.F., Khurana, A., and Blaber, M. 1999. Reversible thermal denaturation of human FGF-1 induced by low concentrations of guanidine hydrochloride. Biophys. J. 77 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brych, S.R., Blaber, S.I., Logan, T.M., and Blaber, M. 2001. Structure and stability effects of mutations designed to increase the primary sequence symmetry within the core region of a b-trefoil. Protein Sci. 10 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson, J.W., Betz, S.F., Lu, H.S., Suich, D.J., Zhou, H.X., O’Neil, K.T., and DeGrado, W.F. 1995. Protein design: A hierarchic approach. Science 270 935–941. [DOI] [PubMed] [Google Scholar]
- Buckle, A.M., Henrick, K., and Fersht, A.R. 1993. Crystal structural analysis of mutations in the hydrophobic cores of barnase. J. Mol. Biol. 234 847–860. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Ivaninskii, S., and Lustig, A. 2002. Improving coiled-coil stability by optimizing ionic interactions. J. Mol. Biol. 318 901–910. [DOI] [PubMed] [Google Scholar]
- Chen, J. and Stites, W.E. 2001. Packing is a key selection factor in the evolution of protein hydrophobic cores. Biochemistry 40 15280–15289. [DOI] [PubMed] [Google Scholar]
- Chi, Y., Kumar, T.K., Wang, H.M., Ho, M.C., Chiu, I.M., and Yu, C. 2001. Thermodynamic characterization of the human acidic fibroblast growth factor: Evidence for cold denaturation. Biochemistry 40 7746–7753. [DOI] [PubMed] [Google Scholar]
- Copeland, R.A., Ji, H., Halfpenny, A.J., Williams, R.W., Thompson, K.C., Herber, W.K., Thomas, K.A., Bruner, M.W., Ryan, J.A., Marquis-Omer, D., et al. 1991. The structure of human acidic fibroblast growth factor and its interaction with heparin. Arch. Biochem. Biophys. 289 53–61. [DOI] [PubMed] [Google Scholar]
- Culajay, J.F., Blaber, S.I., Khurana, A., and Blaber, M. 2000. Thermodynamic characterization of mutants of human fibroblast growth factor 1 with an increased physiological half-life. Biochemistry 39 7153–7158. [DOI] [PubMed] [Google Scholar]
- De Vos, S., Backmann, J., Prevost, M., Steyaert, J., and Loris, R. 2001. Hydrophobic core manipulations in ribonuclease T1. Biochemistry 40 10140–10149. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1994. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 66 482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley, P., Fotinou, C., Black, I., Fairweather, N.F., Charles, I.G., Watts, C., Hewitt, E., and Isaacs, N.W. 2000. The structures of the H(C) fragment of tetanus toxin with carbohydrate subunit complexes provide insight into ganglioside binding. J. Biol. Chem. 275 8889–8894. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., and Matthews, B.W. 1993. Similar hydrophobic replacements of Leu99 and Phe153 within the core of T4 lysozyme have different structural and thermodynamic consequences. J. Mol. Biol. 229 747–769. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. 1999a. Kinetics of protein folding. W.H. Freeman and Co., New York.
- ———. 1999b. Structure and mechanism in protein science. W.H. Freeman and Co., New York.
- Friel, C.T., Capaldi, A.P., and Radford, S.E. 2003. Structural analysis of the rate-limiting transition states in the folding of Im7 and Im9: Similarities and differences in the folding of homologous proteins. J. Mol. Biol. 326 293–305. [DOI] [PubMed] [Google Scholar]
- Gimenez-Gallego, G., Conn, G., Hatcher, V.B., and Thomas, K.A. 1986. The complete amino acid sequence of human brain-derived acidic fibroblast growth factor. Biochem. Biophys. Res. Commun. 128 611–617. [DOI] [PubMed] [Google Scholar]
- Grek, S.B., Davis, J.K., and Blaber, M. 2001. An efficient, flexible-model program for the analysis of differential scanning calorimetry protein denaturation data. Protein Pept. Lett. 8 429–436. [Google Scholar]
- Habazettl, J., Gondol, D., Wiltscheck, R., Otlewski, J., Schleicher, M., and Holak, T.A. 1992. Structure of hisactophilin is similar to interleukin-1b and fibroblast growth factor. Nature 359 855–858. [DOI] [PubMed] [Google Scholar]
- Hecht, M.H., Richardson, J.S., Richardson, D.C., and Ogden, R.C. 1990. De novo design, expression, and characterization of Felix: A four-helix bundle protein of native-like sequence. Science 249 884–891. [DOI] [PubMed] [Google Scholar]
- Holder, J.B., Bennett, A.F., Chen, J., Spencer, D.S., Byrne, M.P., and Stites, W.E. 2001. Energetics of side chain packing in staphylococcal nuclease assessed by exchange of valines, isoleucines, and leucines. Biochemistry 40 13998–14003. [DOI] [PubMed] [Google Scholar]
- Holm, I., Ollo, R., Panthier, J.J., and Rougeon, F. 1984. Evolution of aspartyl proteases by gene duplication: The mouse renin gene is organized in two homologous clusters of four exons. EMBO J. 3 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley, J.H., Baase, W.A., and Matthews, B.W. 1992. Design and structural analysis of alternative hydrophobic core packing arrangements in bacteriophage T4 lysozyme. J. Mol. Biol. 224 1143–1159. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K., Nakamura, H., Morikawa, K., and Kanaya, S. 1993. Stabilization of Escherichia coli ribonuclease HI by cavity-filling mutations within a hydrophobic core. Biochemistry 32 6171–6178. [PubMed] [Google Scholar]
- Itzhaki, L.S., Otzen, D.E., and Fersht, A.R. 1995. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: Evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 254 260–288. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kaneko, S., Kuno, A., Fujimoto, Z., Shimizu, D., Machida, S., Sato, Y., Yura, K., Go, M., Mizuno, H., Taira, K., et al. 1999. An investigation of the nature and function of module 10 in a family F/10 xylanase FXYN of Streptomyces olivaceoviridis E-86 by module shuffling with the Cex of Cellulomonas fimi and by site-directed mutagenesis. FEBS Lett. 460 61–66. [DOI] [PubMed] [Google Scholar]
- Karpusas, M., Baase, W.A., Matsumura, M., and Matthews, B.W. 1989. Hydrophobic packing in T4 lysozyme probed by cavity-filling mutants. Proc. Natl. Acad. Sci. 86 8237–8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., Blaber, S.I., and Blaber, M. 2002. Alternative type I and I′ turn conformations in the β8/β9 β-hairpin of human acidic fibroblast growth factor. Protein Sci. 11 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., Brych, S.R., Lee, J., Logan, T.M., and Blaber, M. 2003. Identification of a key structural element for protein folding within β-hairpin turns. J. Mol. Biol. 328 951–961. [DOI] [PubMed] [Google Scholar]
- Lacy, D.B., Tepp, W., Cohen, A.C., DasGupta, B.R., and Stevens, R.C. 1998. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 5 898–902. [DOI] [PubMed] [Google Scholar]
- Lang, D., Thoma, R., Henn-Sax, M., Sterner, R., and Wilmanns, M. 2000. Structural evidence for evolution of the β/α barrel scaffold by gene duplication and fusion. Science 289 1546–1550. [DOI] [PubMed] [Google Scholar]
- Linemeyer, D.L., Kelly, L.J., Menke, J.G., Gimenez-Gallego, G., DiSalvo, J., and Thomas, K.A. 1987. Expression in Escherichia coli of a chemically synthesized gene for biologically active bovine acidic fibroblast growth factor. Biotechnology 5 960–965. [Google Scholar]
- Linemeyer, D.L., Menke, J.G., Kelly, L.J., Disalvo, J., Soderman, D., Schaeffer, M.-T., Ortega, S., Gimenez-Gallego, G., and Thomas, K.A. 1990. Disulfide bonds are neither required, present, nor compatible with full activity of human recombinant acidic fibroblast growth factor. Growth Factors 3 287–298. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Chirino, A.J., Misulovin, Z., Leteux, C., Feizi, T., Nussenzweig, M.C., and Bjorkman, P.J. 2000. Crystal structure of the cysteine-rich domain of mannose receptor complexed with a sulfated carbohydrate ligand. J. Exp. Med. 191 1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu, M.G. and Fersht, A.R. 1999. Mutually compensatory mutations during evolution of the tetramerization domain of tumor suppressor p53 lead to impaired hetero-oligomerization. Proc. Natl. Acad. Sci. 96 3595–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan, A.D. 1979. Three-fold structural pattern in the soybean trypsin inhibitor (Kunitz). J. Mol. Biol. 133 557–563. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, D. 2000. The molecular evolutionary history of a winged bean α-chymotrypsin inhibitor and modeling of its mutations through structural analysis. J. Mol. Evol. 50 214–223. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Lesk, A.M., and Chothia, C. 1992. β-Trefoil fold. Patterns of structure and sequence in the kunitz inhibitors interleukins-1b and 1a and fibroblast growth factors. J. Mol. Biol. 223 531–543. [DOI] [PubMed] [Google Scholar]
- Offredi, F., Dubail, F., Kischel, P., Sarinski, K., Stern, A.S., Van de Weerdt, C., Hoch, J.C., Prosperi, C., Francois, J.M., Mayo, S.L., et al. 2003. De novo backbone and sequence design of an idealized α/β-barrel protein: Evidence of stable tertiary structure. J. Mol. Biol. 325 163–174. [DOI] [PubMed] [Google Scholar]
- Ohmura, T., Ueda, T., Ootsuka, K., Saito, M., and Imoto, T. 2001. Stabilization of hen egg white lysozyme by a cavity-filling mutation. Protein Sci. 10 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, C.A., Jones, D.T., and Thornton, J.M. 1994. Protein superfamilies and domain superfolds. Nature 372 631–634. [DOI] [PubMed] [Google Scholar]
- Ortega, S., Schaeffer, M.-T., Soderman, D., DiSalvo, J., Linemeyer, D.L., Gimenez-Gallego, G., and Thomas, K.A. 1991. Conversion of cysteine to serine residues alters the activity, stability, and heparin dependence of acidic fibroblast growth factor. J. Biol. Chem. 266 5842–5846. [PubMed] [Google Scholar]
- Otwinowski, Z. 1993. Oscillation data reduction program. In Proceedings of the CCP4 Study Weekend: Data collection and processing (eds. L. Sawyer et al.), pp. 56–62. SERC Daresbury Laboratory, Daresbury, England.
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure: A practical approach. (ed. T.E. Creighton), pp. 299–321. Oxford University Press, Oxford.
- Pellegrini, L., Burke, D.F., von Delft, F., Mulloy, B., and Blundell, T.L. 2000. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 407 1029–1034. [DOI] [PubMed] [Google Scholar]
- Ponting, C.P. and Russell, R.B. 2000. Identification of distant homologues of fibroblast growth factors suggests a common ancestor for all β-trefoil proteins. J. Mol. Biol. 302 1041–1047. [DOI] [PubMed] [Google Scholar]
- Priestle, J.P., Schar, H.-P., and Grutter, M.G. 1989. Crystallographic refinement of interleukin 1b at 2.0 Å resolution. Proc. Natl. Acad. Sci. 86 9667–9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn, T.P., Tweedy, N.B., Williams, R.W., Richardson, J.S., and Richardson, D.C. 1994. Betadoublet: De novo design, synthesis, and characterization of a β-sandwich protein. Proc. Natl. Acad. Sci. 91 8747–8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan, L. and DeGrado, W.F. 1988. Characterization of a helical protein designed from first principles. Science 241 976–978. [DOI] [PubMed] [Google Scholar]
- Richardson, J. and Richardson, D.C. 1989. The de novo design of protein structures. Trends Biochem. Sci. 14 304–309. [DOI] [PubMed] [Google Scholar]
- Rutenber, E., Katzin, B.J., Ernst, S., Collins, E.J., Mlsna, D., Ready, M.P., and Robertus, J.D. 1991. Crystallographic refinement of ricin to 2.5Å. Proteins 10 240–250. [DOI] [PubMed] [Google Scholar]
- Samuel, D., Kumar, T.K.S., Balamurugan, K., Lin, W.-Y., Chin, D.-H., and Yu, C. 2001. Structural events during the refolding of an all β-sheet protein. J. Biol. Chem. 276 4134–4141. [DOI] [PubMed] [Google Scholar]
- Seddon, A.P., Aviezer, D., Li, L.-Y., Bohlen, P., and Yayon, A. 1995. Engineering of fibroblast growth factor: Alteration of receptor binding specificity. Biochemistry 34 731–736. [DOI] [PubMed] [Google Scholar]
- Springer, B.A., Pantoliano, M.W., Barbera, F.A., Gunyuzlu, P.L., Thompson, L.D., Herblin, W.F., Rosenfeld, S.A., and Book, G.W. 1994. Identification and concerted function of two receptor binding surfaces on basic fibroblast growth factor required for mitogenesis. J. Biol. Chem. 269 26879–26884. [PubMed] [Google Scholar]
- Sweet, R.M., Wright, H.T., Janin, J., Chothia, C.H., and Blow, D.M. 1974. Crystal structure of the complex of porcine trypsin with soybean trypsin inhibitor (Kunitz) at 2.6 Å resolution. Biochemistry 13 4212–4228. [DOI] [PubMed] [Google Scholar]
- Tahirov, T.H., Lu, T.H., Liaw, Y.C., Chen, Y.L., and Lin, J.Y. 1995. Crystal structure of abrin-a at 2.14 Å. J. Mol. Biol. 250 354–367. [DOI] [PubMed] [Google Scholar]
- Tang, J., James, M.N., Hsu, I.N., Jenkins, J.A., and Blundell, T.L. 1978. Structural evidence for gene duplication in the evolution of the acid proteases. Nature 271 618–621. [DOI] [PubMed] [Google Scholar]
- Thornton, J.M., Orengo, C.A., Todd, A.E., and Pearl, F.M. 1999. Protein folds, functions and evolution. J. Mol. Biol. 293 333–342. [DOI] [PubMed] [Google Scholar]
- Tronrud, D.E. 1992. Conjugate-direction minimization: An improved method for the refinement of macromolecules. Acta Crystallogr. A 48 912–916. [DOI] [PubMed] [Google Scholar]
- ———. 1996. Knowledge-based B-factor restraints for the refinement of proteins. Journal of Applied Crystallography 29 100–104. [Google Scholar]
- Tronrud, D.E, Ten Eyck, L.F, and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr. A 43 489–501. [Google Scholar]
- Vallee, F., Kadziola, A., Bourne, Y., Juy, M., Rodenburg, K.W., Svensson, B., and Haser, R. 1998. Barley α-amylase bound to its endogenous protein inhibitor BASI: Crystal structure of the complex at 1.9 Å resolution. Structure 6 649–659. [DOI] [PubMed] [Google Scholar]
- Ventura, S., Vega, M.C., Lacroix, E., Angrand, I., Spagnolo, L., and Serrano, L. 2002. Conformational strain in the hydrophobic core and its implications for protein folding and design. Nat. Struct. Biol. 9 485–493. [DOI] [PubMed] [Google Scholar]
- Volbeda, A. and Hol, W.G. 1989. Pseudo 2-fold symmetry in the copper-binding domain of arthropodan haemocyanins. Possible implications for the evolution of oxygen transport proteins. J. Mol. Biol. 206 531–546. [DOI] [PubMed] [Google Scholar]
- Walsh, S.T., Sukharev, V.I., Betz, S.F., Vekshin, N.L., and DeGrado, W.F. 2001. Hydrophobic core malleability of a de novo designed three-helix bundle protein. J. Mol. Biol. 305 361–373. [DOI] [PubMed] [Google Scholar]
- Wei, Y., Liu, T., Sazinsky, S.L., Moffet, D.A., Pelczer, I., and Hecht, M.H. 2003. Stably folded de novo proteins from a designed combinatorial library. Protein Sci. 12 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, M.W., Wang, W., Patterson, J., Mancias, J.D., Beasley, J.R., and Hecht, M.H. 1999. De novo amyloid proteins from designed combinatorial libraries. Proc. Natl. Acad. Sci. 96 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Y. and Erickson, B.W. 1994. Engineering of betabellin 14D: Disulfide-induced folding of a β-sheet protein. Protein Sci. 3 1069–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X., Komiya, H., Chirino, A., Faham, S., Fox, G.M., Arakawa, T., Hsu, B.T., and Rees, D.C. 1991. Three-dimensional structures of acidic and basic fibroblast growth factors. Science 251 90–93. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Hsu, B.T., and Rees, D.C. 1993. Structural studies of the binding of the anti-ulcer drug sucrose octasulfate to acidic fibroblast growth factor. Structure 1 27–34. [DOI] [PubMed] [Google Scholar]