

Abstract
The NS3 helicase of the hepatitis C virus (HCV) unwinds double-stranded (ds) nucleic acid (NA) in an NTP-dependent fashion. Mechanistic details of this process are, however, largely unknown for the HCV helicase. We have studied the binding of dsDNA to an engineered version of subdomain 2 of the HCV helicase (d2ΔNS3h) by NMR and circular dichroism. Binding of dsDNA to d2ΔNS3h induces a local unfolding of helix (α3), which includes residues of conserved helicase motif VI (Q460RxxRxxR467), and strands (β1 and β8) from the central β-sheet. This also occurs upon lowering the pH (4.4) and introducing an R461A point mutation, which disrupt salt bridges with Asp 412 and Asp 427 in the protein structure. NMR studies on d2ΔNS3h in the partially unfolded state at low pH map the dsDNA binding site to residues previously shown to be involved in single-stranded DNA binding. Sequence alignment and structural comparison suggest that these Arg–Asp interactions are highly conserved in SF2 DEx(D/H) proteins. Thus, modulation of these interactions by dsNA may allow SF2 helicases to switch between conformations required for helicase function.
Keywords: Nucleic acid-induced localized unfolding, HCV helicase, hepatitis C virus, structure–function relationships, DNA binding, helicase mechanism, NMR
The RNA virus hepatitis C (HCV) has chronically infected an estimated 170 million people worldwide, of whom 10%–20% will ultimately develop cirrhosis and 1%–5% will develop liver cancer (Lauer and Walker 2001). HCV contains a positive single-stranded (ss) RNA genome that encodes a polyprotein of about 3010 amino acids that is processed into structural (capsid, E1, and E2), p7, and nonstructural (NS2, NS3, NS4a, NS4b, NS5a, and NS5b) proteins by cellular and virus-encoded proteases (Bartenschlager 2002). The NS3 protein is a 631 amino acid residue bifunctional enzyme with a serine protease localized to the N-terminal 180 residues and an RNA helicase located in the C-terminal 451 residues (Reed and Rice 2000), and represents an attractive molecular target for drug development (Tan et al. 2002). Both protease and helicase domains retain their in vitro activity when expressed separately (Levin and Patel 2002). The helicase domain of NS3 (NS3h) is essential for viral RNA replication, which is carried out by a complex that includes the NS3 protease/helicase, the NS4a polypeptide, and the NS5b polymerase (Bartenschlager and Lohmann 2000).
Helicases are ubiquitous enzymes found in all organisms that catalyze the unwinding of double-stranded (ds) DNA and dsRNA in an NTP-dependent fashion during a wide variety of cellular processes such as replication, transcription, translation, splicing, recombination, and repair (von Hippel and Delagoutte 2001). At the sequence level, helicases have been divided into five main groups (Gorbalenya and Koonin 1993). Most of them are grouped into two major superfamilies (SF1 and SF2) based on the occurrence of seven so-called helicase motifs (I, Ia, II, III, IV, V, and VI; Hall and Matson 1999). In both superfamilies, motifs I and II are the highly conserved Walker A and B sequences characteristic of ATPases (Walker et al. 1982). The other sequence motifs are generally less highly conserved, and differ between the SF1 and SF2 proteins (Hall and Matson 1999), and their functional roles are less clear (Caruthers and McKay 2002). The HCV helicase is classified as a DExH protein of SF2 based on the consensus sequence of motif II. The HCV NS3h has unique properties because it is able to unwind dsDNA, dsRNA, or RNA/DNA heteroduplexes in a 3′ to 5′ direction using several NTPs and dNTPs as the energy source (Kwong et al. 2000; Pang et al. 2002). In in vitro experiments the HCV helicase requires a 3′ overhang region, which is proposed to provide an initiation for unwinding duplex NA (Tackett et al. 2001). It binds dsNA 300–1000-fold weaker than ssNA with equilibrium dissociation constants (Kd) in the μM range (Levin and Patel 2002). Several different mechanisms of unwinding have been proposed for helicases (Lohman and Bjornson 1996). Mechanistic details of how NTP binding and hydrolysis are coupled to the translocation of the enzyme along the NA and the unwinding of the dsNA are, however, largely unknown for the HCV helicase.
Crystal structures of the HCV helicase domain by itself (Yao et al. 1997; Cho et al. 1998), in complex with ssDNA (Kim et al. 1998), and in the bifunctional protease-helicase NS3 protein complexed with NS4a cofactor peptide (Yao et al. 1999) showed that it is composed of three nearly equal-sized subdomains. Subdomain 1 (residues 181–326 of NS3) and subdomain 2 (residues 327–481 of NS3) have little sequence identity, but share the same structure composed of a large central β-sheet flanked by α-helices, and are homologous in structure to the central region of the RecA protein (Korolev et al. 1998). Subdomain 3 (residues 482–631 of NS3) is mostly α-helical, and contains part of the ssNA binding site (Kim et al. 1998). All conserved motifs of the HCV helicase are located within subdomains 1 and 2. Motifs I, Ia, II, and III, as well as an additional motif, TxGx (Pause and Sonenberg 1992), reside within subdomain 1; motifs IV, V, and VI are located within subdomain 2. The crystal structure of the complex between NS3h and ssDNA revealed that ssNA binds to the groove between subdomain 1, 2, and 3, mainly through nonhydrophobic interactions involving the phosphates of the nucleic acid backbone and interacts with some residues within motifs TxGx, Ia, IV, and V (Kim et al. 1998). In contrast, little is known about the dsNA binding site of the HCV helicase.
To gain further insights into the structure–function relationships of the HCV helicase, we have designed a novel HCV NS3h construct of subdomain 2 (d2ΔNS3h) which includes the helicase motifs IV, V, and VI (Gesell et al. 2001). NMR studies of d2ΔNS3h showed that the engineered construct retained its native structure in solution (Liu et al. 2001). NMR relaxation studies indicated that residues within motif V are highly dynamic in solution undergoing chemical exchange on the millisecond to microsecond timescale. In addition, hydrogen/deuterium (H/D) exchange NMR data revealed that residues with slowest exchange rates map to the well-packed hydrophobic core region of the protein, which is oriented away from the subdomain interfaces. In contrast, regions facing the subdomain interfaces, including residues in β-strands, β1 and β8, and within the α-helix, α3, showed fast H/D exchange rates suggesting that this part of the structure experiences a localized rapid unfolding and refolding process. In this study, we used NMR spectroscopy to map the dsDNA binding site of d2ΔNS3h and to detect a novel reversible local conformational change that involves the unfolding of two β-strands and an α-helix (motif VI) upon protein–dsDNA interaction. We show that this localized unfolding is caused by a disruption of salt bridges in the protein structure that are highly conserved in SF2 DEx(D/H) proteins. Based on an unwinding model (Kim et al. 1998; Lin and Kim 1999), residues in motif VI may be involved in coupling NTP hydrolysis to nucleic acid unwinding and translocation. We speculate that modulation of these interactions by dsNA may allow the HCV helicase to switch between conformations required for helicase function.
Results
Partial unfolding of d2ΔNS3h caused by interaction with dsDNA
We carried out NMR titration experiments using dsDNA (HSD2, GGCCTAAGCG-TAT-CGCTTAGGCC) and HCV helicase constructs containing either subdomain 1 (d1NS3h) or subdomain 2 (d2ΔNS3h ) to gain insight into the dsNA–protein interaction. Little spectral change was observed in the NMR spectra of d1NS3h during the titration (data not shown), indicating that little or no interaction occurs between subdomain 1 of the HCV helicase and HSD2. In contrast, dsDNA clearly interacts with subdomain 2 as evidenced by substantial spectral changes in the NMR spectra of d2ΔNS3h during the NMR titration experiments with HSD2 (Fig. 1A ▶). Different exchange behaviors were observed for different peaks during the titration. A few peaks gradually shifted their position with increasing HSD2 concentration (fast exchange on the NMR time scale), whereas many others became weaker in intensity and completely disappeared upon the addition of excess amounts of HSD2 (slow exchange). In addition, the disappearance of peaks in regions in the 2D 1H-15N heteronuclear single-quantum coherence (HSQC) NMR spectrum that are characteristic of “structured” protein and appearance of sharp peaks in spectral regions characteristic of unfolded protein strongly suggested a dsDNA-induced local unfolding of d2ΔNS3h. This was further supported by the observation that most of the well-dispersed arginine NɛH peaks were replaced with broad peaks corresponding to nonstructured arginine side chains.
Figure 1.

Interaction with dsDNA causes a reversible local unfolding of d2ΔNS3h. (A) Overlay of 2D 15N-HSQC NMR spectra of uniformly [15N]-labeled d2ΔNS3h acquired at different HSD2/[15N]d2ΔNS3h ratios: 0.0 (black), 0.4 (magenta), 0.62 (yellow), 1.32 (blue). (B) Overlay of 2D 15N-HSQC NMR spectra of purified [15N]d2ΔNS3h–HSD2 complex (blue) and after the dissociation of the complex in the presence of 0.3 M NaCl (red). Isolated peaks in the 15N-HSQC spectrum of purified [15N]d2ΔNS3h–HSD2 complex (blue) are labeled with the assigned one-letter amino acid code and residue number. Labels of additional peaks in crowded regions that have been assigned were omitted for clarity. Also indicated with an arrow and black labels are the chemical shift changes for three peaks, one within each of the three secondary structure elements (β1, α3, β8) that undergo transition from the “unfolded” state (blue: random coil chemical shift in the d2ΔNS3h–HSD2 complex) to the “folded” state (red: dissociated d2ΔNS3h–HSD2 complex upon addition of salt), to further illustrate the localized unfolding of these regions of d2ΔNS3h directly on the dsDNA complex (see text).
We were able to purify the d2ΔNS3h–HSD2 complex using size-exclusion chromatography (Fig. 1B ▶), confirming a strong interaction between the protein and dsDNA. Mass spectrometry data (data not shown) confirmed that d2ΔNS3h and HSD2 were both present in the same fraction. To test if this interaction is salt sensitive and the dsDNA-induced structural changes are reversible, aliquots of 5-M NaCl solution were added into the d2ΔNS3h–HSD2 NMR sample. The 15N-HSQC spectrum of the protein–dsDNA complex in the presence of 0.3 M NaCl became highly similar to that of the free protein (Fig. 1B ▶), suggesting that the structural changes induced by the protein–dsDNA interaction are indeed reversible, and this interaction is mainly of electrostatic nature. However, severe line broadening prevented the use of routine solution NMR techniques to determine the 3D structure of the d2ΔNS3h–HSD2 complex. Therefore, we used a combination of mutational data, CD, and NMR to reveal the nature and structural details of the local unfolding and approximate the dsDNA binding site of the protein by uncoupling dsDNA binding from protein local unfolding.
Partial unfolding of d2ΔNS3h at low pH
We noticed that the 15N-HSQC spectrum of the d2ΔNS3h–HSD2 complex resembled an earlier 15N-HSQC spectrum of free d2ΔNS3h at lower pH acquired during our optimization of NMR sample buffer conditions. To confirm this observation a pH titration experiment was carried out in which 15N-HSQC spectra of free d2ΔNS3h were acquired at pH 6.2, 5.8, 5.2, and 4.4 in a MES buffer system (Fig. 2A ▶). The NMR spectra did not change significantly until the pH was lowered to 4.4 and the original spectrum was obtained by increasing the pH back to 6.5. Interestingly, the overall patterns of these pH-dependent spectral changes were strikingly similar to those observed when forming the d2ΔNS3h–HSD2 complex at pH 6.5 (compare Fig. 1B ▶ with Fig. 2A ▶) with many of the dispersed peaks becoming clustered towards the “center” of the spectrum. The resulting NMR spectra were similar, suggesting that the locally unfolded states of d2ΔNS3h induced by either dsDNA binding or lowering the pH to 4.4 are related and reversible.
Figure 2.
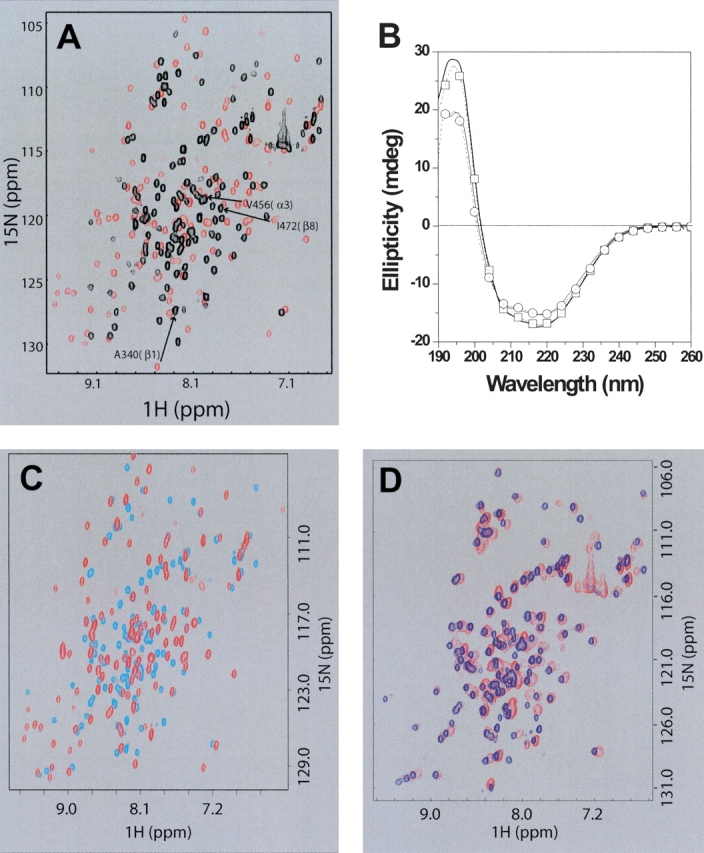
Similar local unfolding of d2ΔNS3h occurs at low pH and in the R461A mutant. (A) Overlay of 2D 15N-HSQC NMR spectra of [15N]d2ΔNS3h at different buffer pH: 6.2 (red) and 4.4 (black). Also indicated with an arrow and black labels are the chemical shift changes for the three peaks that are labeled in Figure 1B ▶, one within each of the three secondary structure elements (β1, α3, β8) that undergo transition from the “folded” state (red: d2ΔNS3h–HSD2 at pH 6.2) to the “unfolded” state (black: d2ΔNS3h–HSD2 at pH 4.4). (B) Far-UV CD spectra of wild-type d2ΔNS3h and mutants d2ΔNS3h–R461A and d2ΔNS3h–R462A. Wild-type d2ΔNS3h, pH 6.5(thick line); mutant d2ΔNS3h–R462A, pH 6.5 (open squares); wild-type d2ΔNS3h, pH 4.5 (thin line); mutant d2ΔNS3h–R461A, pH 6.5 (open circles). (C) Overlay of 2D 15N-HSQC NMR spectra of [15N]d2ΔNS3h–R461A mutant (cyan) and wild-type [15N]d2ΔNS3h (red) at pH 6.5. (D) Overlay of 2D 15N-HSQC NMR spectra of [15N]d2ΔNS3h–R461A mutant (blue) and purified [15N]d2ΔNS3h–HSD2 complex (red) at pH 6.5.
The partial unfolding of d2ΔNS3h at low pH was further confirmed from a pH titration study using CD. The far-UV CD spectrum of d2ΔNS3h at pH 6.5 (Fig. 2B ▶) has a minimum at 220 nm and a maximum at 195 nm, and is consistent with a protein containing a significant content of the α-helix secondary structure. The CD spectrum of d2ΔNS3h at pH 4.5 is also shown in Figure 2B ▶, and the decrease in ellipticity observed at both 190 and 220 nm suggest that the pH-dependent unfolding involves a partial loss of α-helical secondary structure. The observations that d2ΔNS3h undergoes a partial unfolding at pH 4.5 by CD and the similarity of NMR spectra between the d2ΔNS3h–HSD2 dsDNA complex and apo d2ΔNS3h at pH 4.4 indicated that additional studies could be used to further explore the structural reasons for the local unfolding and to map the dsDNA binding site of d2ΔNS3h.
Localization of regions becoming unfolded upon dsDNA binding and at low pH
Sequence-specific backbone resonance assignments of the d2ΔNS3h–HSD2 complex and d2ΔNS3h at pH 4.4 were obtained to localize those regions that become unstructured upon addition of dsDNA and at low pH. Complete assignments of the d2ΔNS3h–DNA complex were not feasible due to extensive line broadening caused by chemical exchange. However, sequence-specific HN/15N and 13Cα backbone resonance assignments have been obtained for 82 residues (64% of the 129 residues with nonlabile HN) in the d2ΔNS3h–HSD2 complex (Fig. 1B ▶). In contrast, nearly complete backbone resonance assignments were obtained for d2ΔNS3h at pH 4.4. In addition, some information derived from the low pH condition could be applied to the d2ΔNS3h–HSD2 complex, because the NMR spectra of d2Δ NS3h in the dsDNA complex and at pH 4.4 were similar. Limited chemical shift index (CSI; Wishart and Sykes 1994) data clearly suggested the unfolding of at least the β1 strand and the α3 helix of d2ΔNS3h in the dsDNA complex, whereas the core of d2ΔNS3h (β4–α2–β5–β6) remains structured upon binding of dsDNA (Fig. 3A ▶). This was confirmed by the higher quality NMR data available on d2ΔNS3h at pH 4.4. The regions with large chemical shift changes were easily identified by comparing CSI data of d2ΔNS3h at pH 6.5 and 4.4 (Fig. 3A ▶). These include residues 336–340 (β1), 472–475 (β8; from β-strand to random coil), and 454–462 (α3; from helical to random coil). The local unfolding of these regions is consistent with the backbone NOE information obtained from a 3D 15N-edited NOESY spectrum of d2ΔNS3h at pH 4.4 (data not shown). In addition, residues in these regions showed much lower {1H}-15N heteronuclear NOEs at pH 4.4 (data not shown) compared to pH 6.5, further confirming that they become unstructured and adopt a random coil conformation.
Figure 3.
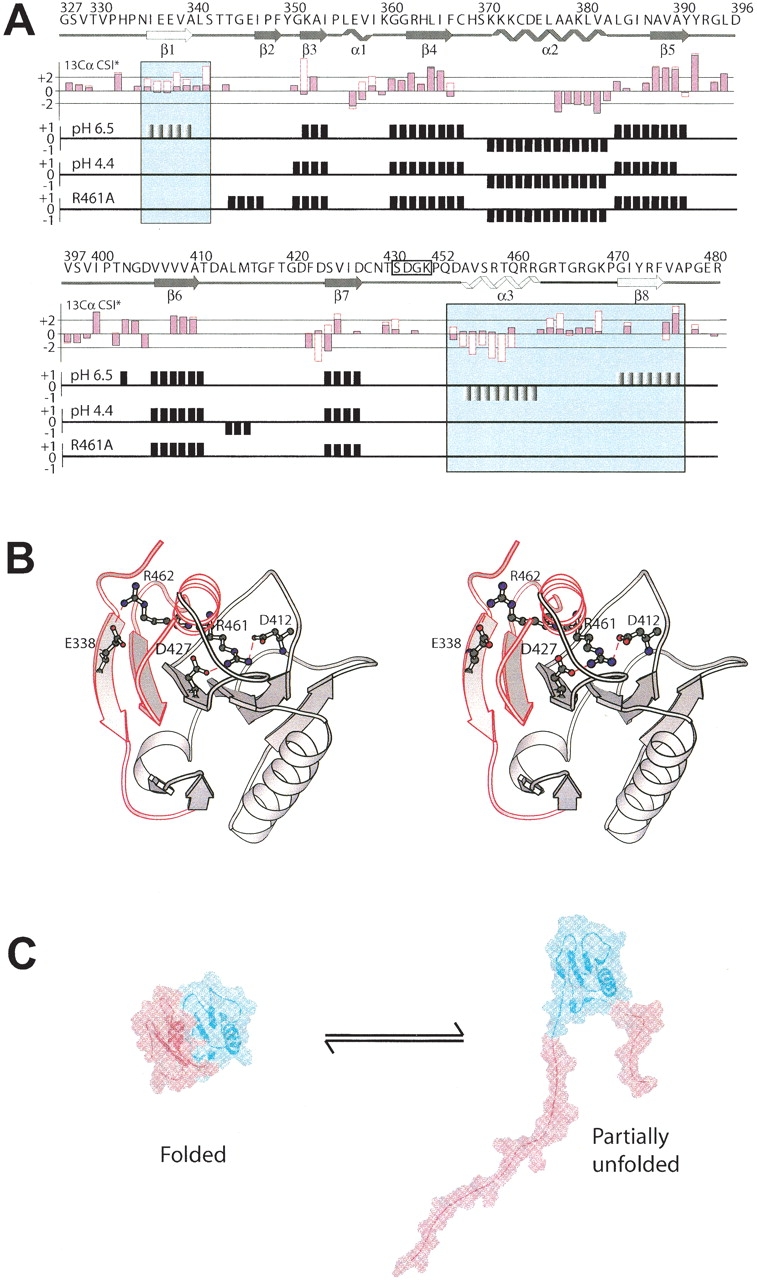
Interaction with dsDNA, lowering the pH to 4.4, or introducing an R461A point mutation all induce a local unfolding of the secondary structure elements β1, β8, and α3 (motif VI) in d2ΔNS3h. (A) Top panel: The primary sequence and the secondary structure of wild-type d2ΔNS3h, as observed in the solution structure at pH 6.5 (Liu et al. 2001) is indicated above the CSI data with the secondary structure elements β1, β8, and α3 highlighted in the panels. Second panel: Comparison of the secondary 13Cα CSI data between d2ΔNS3h–HSD2 complex (empty red boxes) and wild-type d2ΔNS3h (pink filled black boxes) at pH 6.5 for residues whose 13Cα could be assigned in the complex. CSI data of wild-type d2ΔNS3h at pH 6.5 (third panel) and pH 4.4 (fourth panel), and mutant d2ΔNS3h–R461A (fifth panel). (B) Ribbon structure of wild-type d2ΔNS3h at pH 6.5 (Liu et al. 2001) with the side chains of Glu 338, Asp 412, Asp 427, Arg 461, and Arg 462 indicated and labeled. The region which undergoes an order-to-disorder transition is highlighted in red and labeled (β1, β8, and α3). (C) Interaction with dsDNA, lowering the pH to 4.4, or introducing an R461A point mutation all cause the localized unfolding of the region highlighted in red.
Arg 461 to Ala mutation within conserved sequence motif VI induces unfolding of same regions
Next, we searched for possible reasons for the localized unfolding of the N- and C-terminal portions of d2ΔNS3h on a structural level. The fact that this localized unfolding occurs upon lowering the sample pH to 4.4 suggested the involvement of acidic amino acid side chains such as Glu and Asp residues, whose pKa values typically are in that range. Therefore, we speculated that the partial unfolding of d2ΔNS3h might be caused by the protonation of acidic amino acid side chains at pH 4.4, which would disrupt charge–charge interactions important for maintaining a stable fold of d2ΔNS3h. Inspection of the solution structure of d2ΔNS3h (Liu et al. 2001) and the crystal structure of the HCV helicase domain of NS3 (Yao et al. 1997) revealed two pairs of charge–charge interactions that may be important, Arg 461–Asp 412/Asp 427 and Arg 462–Glu 338 (Fig. 3B ▶). Two mutants, R461A and R462A, were constructed to test if disruptions of the Arg 461–Asp 412/Asp 427 and/or Arg 462–Glu 338 interactions were responsible for the observed structural changes.
Comparison of the 15N-HSQC spectra at pH 6.5 between the d2ΔNS3h–R461A mutant and wild-type d2ΔNS3h showed many spectral changes indicative of partial unfolding of the mutant protein (Fig. 2C ▶). In contrast, the d2ΔNS3h–R462A mutant did not have any significant spectral changes compared to wild-type d2ΔNS3h at pH 6.5 (data not shown), demonstrating that disruption of the Arg 462–Glu 338 interaction does not cause unfolding of d2ΔNS3h. A similar comparison was performed using circular dichroism to detect mutation-induced conformational changes. Figure 2B ▶ shows that the spectrum of wild-type d2ΔNS3h at pH 6.5 is nearly identical to the d2ΔNS3h–R462A mutant at pH 6.5. This suggests that the disruption of the Arg 462–Glu 338 interaction does not significantly perturb the secondary structure of this domain. In contrast, the far-UV CD spectrum of mutant d2ΔNS3h–R461A at pH 6.5 changed significantly throughout the far-UV CD region compared to the wild-type d2ΔNS3h pH 6.5 spectrum. The decrease in ellipticity at 195 and 220 nm suggests that disruption of the R461–Asp 412/Asp 427 interaction is critical, and causes a decrease in α-helical content. The pH 6.5 spectrum of d2ΔNS3h–R461A is very similar to the wild-type d2ΔNS3h spectrum at pH 4.5. It is likely that the conformations of both proteins are similar since each condition is expected to disrupt stabilizing charge–charge interactions such as the R461–Asp 412/Asp 427 interactions.
Interestingly, the 15N-HSQC spectrum of d2ΔNS3h–R461A is strikingly similar to those of d2ΔNS3h in the dsDNA complex at pH 6.5 (Fig. 2D ▶) and at pH 4.4 in the absence of dsDNA. In addition, nearly complete backbone resonance assignments of the d2ΔNS3h–R461A mutant were achieved. The CSI and NOE data of the mutant were consistent with the β1 and β8 strands and α3 helix unfolding (Fig. 3A ▶). This suggests that the Arg 461–Asp 412/Asp 427 interactions are critical for stabilizing the local conformation of d2ΔNS3h. The disruption of this secondary structure region upon dsDNA binding, at low pH, or by mutation induces a structural change in d2ΔNS3h that involves the unfolding of 20 and 30 N- and C-terminal residues, respectively (Fig. 3C ▶). These conformational changes involve nearly 30% of the residues located within regular secondary structural elements. The observed local unfolding of d2ΔNS3h at pH 4.4 by NMR and similar spectra in the presence of dsDNA at pH 6.5 may be structurally and functionally significant in the unwinding mechanism and will be discussed below.
Mapping the dsDNA binding site of d2ΔNS3h
The low quality NMR data attainable for the d2ΔNS3h–dsDNA complex prevented determination of its 3D structure. Moreover, due to the surprisingly large structural changes of d2ΔNS3h occurring upon addition of HSD2, it was not feasible to use NMR chemical shift perturbations (CSP) to map the dsDNA binding site of the protein. Because a stable d2ΔNS3h–dsDNA complex could be isolated using size-exclusion chromatography, it was reasonable to assume that the dsDNA binding site was located within the stable, structured regions of the protein. Therefore, we repeated the NMR titration of d2ΔNS3h with HSD2 at pH 4.4 where the protein is partially unfolded. Addition of dsDNA caused moderate CSP of backbone 1H-15N resonances of Ser 370, Lys 371, Lys 372, and Lys 373 in motif IV, and Thr 411 and Gly 417 in motif V of the HCV helicase (magenta in Fig. 4 ▶). In the crystal structure of a complex between the HCV helicase domain of NS3 and ssDNA (Kim et al. 1998), these residues are located within the ssDNA binding site at the 5′-terminal end near Val 432, which was found to disrupt nucleotide base stacking in the bound ssDNA. Notably, the location of these residues is consistent with an unwinding model (Kim et al. 1998; Lin and Kim 1999), which was proposed based on this crystal structure and site-directed mutagenesis data (Fig. 4 ▶).
Figure 4.
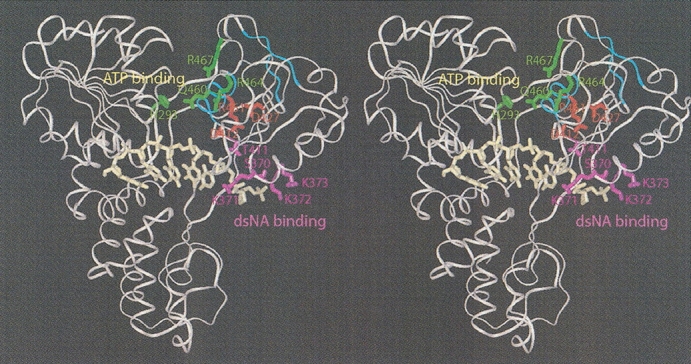
Mapping the dsDNA interaction site of d2ΔNS3h on the HCV NS3 helicase domain structure using chemical shift perturbation data. Stereoribbon representation of the crystal structure of the NS3h–ssDNA complex (PDB 1A1V; Kim et al. 1998) showing in magenta side chains of residues affected by dsDNA binding as determined in this study. Combined chemical shift changes ([(Δδ{1H})2 + (Δδ{15N}*0.2)2]1/2) of those residues were as follows: Ser 370 (0.019 ppm), Lys 371 (0.083 ppm), Lys 372 (0.034 ppm), Lys 373 (0.044 ppm), Thr 411 (0.034 ppm), Gly 417 (0.033 ppm). The location of these residues is consistent with a proposed unwinding model in which the NA duplex would interact with subdomain 2 and be unwound at the position of Val 432, which disrupts nucleotide base stacking of the bound ssNA at the 5′ end of the ssNA binding site (Kim et al. 1998; Lin and Kim 1999). Shown in green are side chains of residues which, as proposed in this model, would be important for the closure and opening of the cleft between subdomain 1 and 2 upon ATP binding and hydrolysis, respectively. The Arg–Asp interactions which are disrupted upon dsDNA binding in d2ΔNS3h are colored red, whereas the secondary structure elements β1, β8, and α3 (motif VI), which become unfolded, are highlighted in cyan. These interactions may provide a structural link between the residues involved in NTP binding and hydrolysis and those involved in NA binding.
Discussion
Collectively, our data suggest that dsDNA interacts with residues in motifs IV and V of the HCV helicase and destabilizes the Arg 461–Asp 412/Asp 427 interactions, which are critical for the packing of the α-helix (α3) against the central β-sheet leading to a reversible local unfolding of two β-strands (β1 and β8) and one α-helix (α3) in d2ΔNS3h. Apparently, the Arg 461–Asp 412/Asp 427 interactions are structurally important, and can be modulated by dsDNA in d2ΔNS3h. The regions that undergo this local unfolding were previously identified in NMR H/D exchange experiments as experiencing a localized rapid unfolding and refolding process (Liu et al. 2001). Therefore, these regions of the protein are in equilibrium between a well-folded state, as seen in both the NMR structure of d2ΔNS3h (Liu et al. 2001) and the crystal structure of the HCV helicase domain (Yao et al. 1997), and an unfolded state as observed in this study. This equilibrium is shifted toward the partially unfolded state upon dsDNA binding, at pH 4.4, and in the d2ΔNS3h–R461A mutant as a result of the destabilization of the Arg 461–Asp 412/Asp 427 interactions. These interactions may, therefore, serve as an internal switch for the protein to shift its conformational equilibrium in solution between a mainly well-folded state (interactions are intact) and a partially unfolded state (interactions are disrupted). It is conceivable that dsDNA binding can activate this internal switch through a transient interaction with Arg 461, thereby shifting the population of d2ΔNS3h from the mainly folded conformation to the predominantly locally unfolded conformation. In d2ΔNS3h this conformational switch may have a relatively low energy barrier and shift at low pH or upon introducing the R461A mutation, both of which would disrupt the Arg 461–Asp 412/Asp 427 interactions. Although the energy barrier may be different in the context of the intact HCV helicase domain or the full-length HCV NS3 bifunctional enzyme, this finding is likely to be an intrinsic property of the HCV helicase. In this regard, it has been argued that the function of a protein and its properties are not only decided by the static folded 3D structure, but also by the distribution of its conformational states, and in particular, by the redistributions of the populations under different environments (Kumar et al. 2000).
Arg 461 and Asp 412 are located within the conserved helicase motifs VI and V, respectively, whereas Asp 427 is part of the β7 strand in the central β-sheet (Liu et al. 2001). Sequence alignment of nineteen known viral SF2 DExH helicases and seven yeast DEAH-box proteins (http://www.expasy.ch/linder/helicases_list.html) reveals a high degree of conservation of these Arg–Asp interactions (Fig. 5A ▶): Arg 461 and Asp 427 of the HCV helicase are strictly conserved, and Asp 412 shows a conservative Asn substitution in four of the viral proteins and in all yeast proteins. Such a high degree of sequence conservation in related proteins implies potential structural and/or functional roles of the Arg 461–Asp 412/Asp 427 interactions. This hypothesis is further supported structurally by the recent crystal structures of two SF2 DEAD-box proteins, the yeast RNA helicase eIF4A (Arg 346–Asp 317/Asn 333) (PDB 1HV8; Story et al. 2001), and a putative RNA helicase, mj0669 (Arg 325–Asp 296/Asn 312) (PDB 1FUU; Caruthers et al. 2000), in which these interactions are also observed (Fig. 5B ▶).
Figure 5.
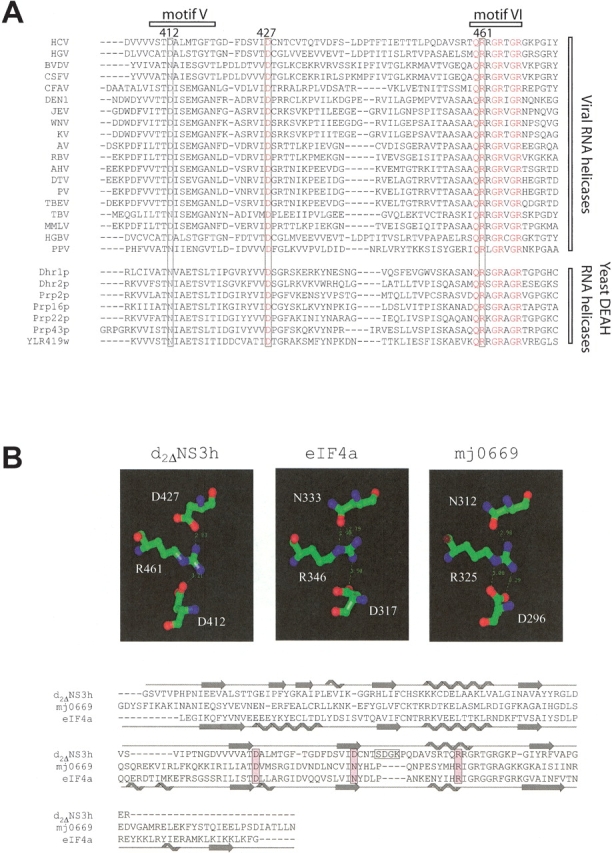
The Arg 461–Asp 412/Asp 427 interactions are conserved both on a sequence and structural level. (A) Sequence alignment of several viral SF2 helicases, which are homologous to the HCV NS3 helicase (HCV). These proteins include the homologous NS3 proteins of hepatitis G virus (HGV), two pestiviruses (bovine viral diarrhea virus [BVDV] and classical swine fever virus [CSFV]), and 14 flaviviruses (cell fusing agent virus [CFAV], dengue virus type 1 [DEN1], Japanese encephalitis virus [JEV], West Nile virus [WNV], kunjin virus [KV], apoi virus [AV], Rio Bravo virus [RBV], alkhurma virus [AHV], deer tick virus [DTV], powassan virus [PV], tick-borne encephalitis virus [TBEV], Tamana bat virus [TBV], Montana myotis leukoencephalitis virus [MMLV], and marmoset hepatitis GB virus [HGBV]). Also shown is the sequence of the CI helicase of a plant potyvirus, plum pox virus (PPV), and those of seven DEAH-box proteins from the yeast Saccharomyces cerevisiae, which are classified as RNA helicases (http://www.expasy.ch/linder/helicases_list.html). (B) Structural comparison of three SF2 helicases (d2ΔNS3h; mj0669, a DEAD-box putative RNA helicase from Methanococcus jannaschi; and eIF4a [eukaryotic translation initiation factor 4A] from yeast, a DEAD-box RNA helicase) illustrating the conservation of the Arg 461–Asp 412/Asp 427 interactions of the HCV helicase in eIF4a and mj0669. The secondary structure elements are indicated above and below the sequence alignment for d2ΔNS3h and eIF4a, respectively.
The local unfolding observed for d2ΔNS3h in this study may only represent one of several dynamic conformational changes required for the unwinding mechanism of the HCV helicase. The physical mechanism by which helicases move along an ss or ds stretch of NA has been extensively studied by both structural and biochemical methods (Caruthers and McKay 2002; Singleton and Wigley 2002). Large-scale relative rotations of the two RecA-like subdomains of the SF1 Rep helicase were found to be central for nucleic acid stimulated NTPase and translocase activities for this enzyme (Korolev et al. 1998). A rigid body movement involving the Arg-rich subdomain 2, which is flexibly linked to the remainder of the protein, is also observed for the HCV helicase when comparing different crystal forms of this domain (Yao et al. 1997; Cho et al. 1998; Kim et al. 1998). In addition to these large-scale relative rotations of subdomain 2, solution NMR studies of d2ΔNS3h revealed distinctive dynamic behavior of residues near the interface of subdomains 1 and 2 (Liu et al. 2001). For example, most residues within motif V exhibited slow conformational exchange on the millisecond-to-microsecond time scale, and many of these residues including those of motif VI displayed faster than average local folding and unfolding rates. The intrinsic conformational flexibility of these regions likely facilitates conformational changes required for helicase function. The localized unfolded state of the HCV helicase as a result of the disruption of the Arg 461–Asp 412/Asp 427 interactions upon dsDNA binding could reflect an intermediate state on the path of the catalyzed reaction in which hydrolysis of an NTP is coupled to the separation of an NA duplex. It is conceivable that Arg 461 in the HCV helicase plays a central structural role in allowing modulation of conformational changes of the Arg-rich motif VI through dsDNA binding. Consistent with this, R461A and R461Q mutants of HCV NS3 abolished helicase unwinding activity (Kwong et al. 2000; Tai et al. 2001). This may also be true for related helicases, because mutations of the corresponding residue in related RNA helicases of SF2, eIF4A-R346Q (Pause et al. 1993) and NPH-II-R492A (Gross and Shuman 1996), also abolished and significantly reduced, respectively, helicase activity. Interestingly, the R461A mutant of HCV NS3 increased basal ATPase activity (140% compared to weight), whereas its nucleic acid stimulated ATPase (33% compared to weight) and ssRNA binding (57% compared to weight) activities were impaired (Kwong et al. 2000), further suggesting that the Arg 461–Asp 412/Asp 427 interactions may provide a structural link between residues in motif VI, which are presumably involved in NTP binding and hydrolysis (Lin and Kim 1999; Caruthers and McKay 2002), with those in motifs IV and V, which are involved in NA binding (Kim et al. 1998; Fig. 4 ▶).
It is well documented that NA–protein recognition can induce large conformational changes in both protein and NA (for examples, see Chang and Varani 1997; Frankel and Smith 1998; Ciubotaru et al. 1999; Yin and Steitz 2002). Although more generally NA binding induces folding of the protein structure (for examples, see Spolar and Record Jr. 1994; Chang and Varani 1997; Frankel and Smith 1998), there are some known examples of DNA-induced partial unfolding of proteins (Newman et al. 1995; Petersen et al. 1995; Nandi et al. 2002). For example, in the transcription factor, Est-1, DNA binding induces unfolding of an α-helix in the N-terminal inhibitory region to relieve autoinhibition (Petersen et al. 1995). In one extreme case of remodeling, the BamHI endonuclease undergoes large-scale domain rearrangements upon DNA binding, folding of the disordered N-terminal part, restructuring of other segments, and even an unfolding of C-terminal α-helices to form partially disordered arms that bind the minor groove (Newman et al. 1995). In the prion protein, on the other hand, DNA-induced partial unfolding leads to its polymerization to amyloid (Nandi et al. 2002). The same physical phenomenon is likely used in the HCV helicase to more easily switch between intermediate states during the catalytic events. It will be interesting to further pursue this hypothesis in the context of the full-length HCV NS3 protein.
Materials and methods
Expression, purification, and mutant construction of d2ΔNS3h
Design, plasmid construction, and protein production of engineered HCV helicase subdomain 2 (d2ΔNS3h) have been described in detail (Gesell et al. 2001). The d2ΔNS3h–R461A and d2ΔNS3h–R462A mutants were constructed by Quickchange PCR using pNS3d2Δ as the template (Stratagene). Mutants were confirmed by DNA sequence. Expression and purification of isotopically labeled proteins for NMR studies were carried out as described (Liu et al. 2001).
NMR spectroscopy
NMR spectra were acquired at 600 MHz at 25°C and processed with FELIX 2000 (Accelrys). Typically, the NMR samples contained 0.55 mL of 0.2~0.5 mM proteins. Three NMR buffer systems were used for the current study: (1) 75 mM KiPO4, pH 6.5; (2) 10 mM KiPO4, pH 6.5; and (3) 50 mM MES, 50 mM KCl at various pH values during the pH titration with 1 M HCl; 5 mM DTT, 0.015% (w/v) sodium azide and 5%–10% (v/v) D2O were included in all the buffers. The dsDNA (HSD2, GGCCTAAGCG-TAT-CGCTTAGGCC) was used for the binding studies. The DNA sequence was designed to have a 10 base pair stem, which represents part of the HCV NS3 helicase substrate, and a three base loop. We expect this DNA hairpin structure to be primarily formed under the low salt solution conditions of the NMR study. The following 3D experiments were acquired to achieve backbone resonance assignments: HNCO, HNCACB, CBCACONH, 15N-NOESY-HSQC, 15N-TOCSY-HSQC (Liu et al. 2001). The heteronuclear {1H}-15N NOE values for d2ΔNS3h–R461A and d2ΔNS3h at pH 4.4 were obtained as previously described (Liu et al. 2001).
Circular dichroism studies
Far-UV CD spectra were recorded using a Jasco 810 spectropolarimeter with protein concentrations of 0.16 mg/mL. Samples of wild-type and mutant d2ΔNS3h were prepared in either 37 mM potassium phosphate (pH 6.5) or 37 mM sodium acetate (pH 4.5) buffer containing 1.0 mM DTT. The spectra were recorded using a 1-m path length cell at a scan rate of 50 nm/min, time constant of 1 sec, bandwidth of 1 nm. Each spectrum was averaged from eight scans, recorded at 20°C and corrected by subtracting a buffer blank.
Acknowledgments
We thank Prasanti Repaka for her work on protein purification and Dr. Yu-Sen Wang for his assistance in NMR data collection.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CSI, chemical shift index
CSP, chemical shift perturbations
ds, double-stranded
d2ΔNS3h, engineered version of subdomain 2 of the HCV NS3 helicase
HCV, hepatitis C virus
HSD2, dsDNA: GGCCT AAGCG-TAT-CGCTTAGGCC
HSQC, heteronuclear single quantum coherence
NA, nucleic acid
NS3h, NS3 helicase
ss, single-stranded
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03280803.
References
- Bartenschlager, R. 2002. Hepatitis C virus replicons: Potential role for drug development. Nat. Rev. Drug Discov. 1 911–916. [DOI] [PubMed] [Google Scholar]
- Bartenschlager, R. and Lohmann, V. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81 1631–1648. [DOI] [PubMed] [Google Scholar]
- Caruthers, J.M. and McKay, D.B. 2002. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 12 123–133. [DOI] [PubMed] [Google Scholar]
- Caruthers, J.M., Johnson, E.R., and McKay, D.B. 2000. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl. Acad. Sci. 97 13080–13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, K.Y. and Varani, G. 1997. Nucleic acids structure and recognition. Nat. Struct. Biol. Suppl: 854–858. [PubMed]
- Cho, H.S., Ha, N.C., Kang, L.W., Chung, K.M., Back, S.H., Jang, S.K., and Oh, B.H. 1998. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. A feasible mechanism of unwinding duplex RNA. J. Biol. Chem. 273 15045–15052. [DOI] [PubMed] [Google Scholar]
- Ciubotaru, M., Bright, F.V., Ingersoll, C.M., and Koudelka, G.B. 1999. DNA-induced conformational changes in bacteriophage 434 repressor. J. Mol. Biol. 294 859–873. [DOI] [PubMed] [Google Scholar]
- Frankel, A.D. and Smith, C.A. 1998. Induced folding in RNA–protein recognition: More than a simple molecular handshake. Cell 92 149–151. [DOI] [PubMed] [Google Scholar]
- Gesell, J.J., Liu, D., Madison, V.S., Hesson, T., Wang, Y.-S., Weber, P.C., and Wyss, D.F. 2001. Design, high-level expression, purification and characterization of soluble fragments of the hepatitis C virus NS3 RNA helicase suitable for NMR-based drug discovery methods and mechanistic studies. Protein Eng. 14 573–582. [DOI] [PubMed] [Google Scholar]
- Gorbalenya, A.E. and Koonin, E.V. 1993. Helicases: Amino acid sequence comparisons and structure–function relationships. Curr. Opin. Struct. Biol. 3 419–429. [Google Scholar]
- Gross, C.H. and Shuman, S. 1996. The QRxGRxGRxxxG motif of the vaccinia virus DExH box RNA helicase NPH- II is required for ATP hydrolysis and RNA unwinding but not for RNA binding. J. Virol. 70 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, M.C. and Matson, S.W. 1999. Helicase motifs: The engine that powers DNA unwinding. Mol. Microbiol. 34 867–877. [DOI] [PubMed] [Google Scholar]
- Kim, J.L., Morgenstern, K.A., Griffith, J.P., Dwyer, M.D., Thomson, J.A., Murcko, M.A., Lin, C., and Caron, P.R. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: The crystal structure provides insights into the mode of unwinding. Structure 6 89–100. [DOI] [PubMed] [Google Scholar]
- Korolev, S., Yao, N., Lohman, T.M., Weber, P.C., and Waksman, G. 1998. Comparisons between the structures of HCV and Rep helicases reveal structural similarities between SF1 and SF2 super-families of helicases. Protein Sci. 7 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., Ma, B., Tsai, C.J., Sinha, N., and Nussinov, R. 2000. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 9 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong, A.D., Kim, J.L., and Lin, C. 2000. Structure and function of hepatitis C virus NS3 helicase. Curr. Top. Microbiol. Immunol. 242 171–196. [DOI] [PubMed] [Google Scholar]
- Lauer, G.M. and Walker, B.D. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345 41–52. [DOI] [PubMed] [Google Scholar]
- Levin, M.K. and Patel, S.S. 2002. Helicase from hepatitis C virus, energetics of DNA binding. J. Biol. Chem. 277 29377–29385. [DOI] [PubMed] [Google Scholar]
- Lin, C. and Kim, J.L. 1999. Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 73 8798–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D., Wang, Y.-S., Gesell, J.J., and Wyss, D.F. 2001. Solution structure and backbone dynamics of an engineered arginine-rich subdomain 2 of the hepatitis C virus NS3 RNA helicase. J. Mol. Biol. 314 543–561. [DOI] [PubMed] [Google Scholar]
- Lohman, T.M. and Bjornson, K.P. 1996. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 65 169–214. [DOI] [PubMed] [Google Scholar]
- Nandi, P.K., Leclerc, E., Nicole, J.-C., and Takahashi, M. 2002. DNA-induced partial unfolding of prion protein leads to its polymerisation to amyloid. J. Mol. Biol. 322 153–161. [DOI] [PubMed] [Google Scholar]
- Newman, M., Strzelecka, T., Dorner, L.F., Schildkraut, I., and Aggarwal, A.K. 1995. Structure of Bam HI endonuclease bound to DNA: Partial folding and unfolding on DNA binding. Science 269 656–663. [DOI] [PubMed] [Google Scholar]
- Pang, P.S., Jankowsky, E., Planet, P.J., and Pyle, A.M. 2002. The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J. 21 1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause, A. and Sonenberg, N. 1992. Mutational analysis of a DEAD box RNA helicase: The mammalian translation initiation factor eIF-4A. EMBO J. 11 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause, A., Methot, N., and Sonenberg, N. 1993. The HRIGRXXR region of the DEAD box RNA helicase eukaryotic translation initiation factor 4A is required for RNA binding and ATP hydrolysis. Mol. Cell. Biol. 13 6789–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, J.M., Skalicky, J.J., Donaldson, L.W., McIntosh, L.P., Alber, T., and Graves, B.J. 1995. Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an α helix. Science 269 1866–1869. [DOI] [PubMed] [Google Scholar]
- Reed, K.E. and Rice, C.M. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242 55–84. [DOI] [PubMed] [Google Scholar]
- Singleton, M.R. and Wigley, D.B. 2002. Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol. 184 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolar, R.S. and Record Jr., M.T. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science 263 777–784. [DOI] [PubMed] [Google Scholar]
- Story, R.M., Li, H., and Abelson, J.N. 2001. Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii. Proc. Natl. Acad. Sci. 98 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackett, A.J., Wei, L., Cameron, C.E., and Raney, K.D. 2001. Unwinding of nucleic acids by HCV NS3 helicase is sensitive to the structure of the duplex. Nucleic Acids Res. 29 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, C.L., Pan, W.C., Liaw, S.H., Yang, U.C., Hwang, L.H., and Chen, D.S. 2001. Structure-based mutational analysis of the hepatitis C virus NS3 helicase. J. Virol. 75 8289–8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, S.L., Pause, A., Shi, Y., and Sonenberg, N. 2002. Hepatitis C therapeutics: Current status and emerging strategies. Nat. Rev. Drug Discov. 1 867–881. [DOI] [PubMed] [Google Scholar]
- von Hippel, P.H. and Delagoutte, E. 2001. A general model for nucleic acid helicases and their “coupling” within macromolecular machines. Cell 104 177–190. [DOI] [PubMed] [Google Scholar]
- Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994. Chemical shifts as a tool for structure determination. Methods Enzymol. 239 363–392. [DOI] [PubMed] [Google Scholar]
- Yao, N., Hesson, T., Cable, M., Hong, Z., Kwong, A.D., Le, H.V., and Weber, P.C. 1997. Structure of the hepatitis C virus RNA helicase domain. Nat. Struct. Biol. 4 463–467. [DOI] [PubMed] [Google Scholar]
- Yao, N., Reichert, P., Taremi, S.S., Prosise, W.W., and Weber, P.C. 1999. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 7 1353–1363. [DOI] [PubMed] [Google Scholar]
- Yin, Y.W. and Steitz, T.A. 2002. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science 298 1387–1395. [DOI] [PubMed] [Google Scholar]