

Abstract
As part of a program to discover improved glycoside hydrolase family 12 (GH 12) endoglucanases, we have extended our previous work on the structural and biochemical diversity of GH 12 homologs to include the most stable fungal GH 12 found, Humicola grisea Cel12A. The H. grisea enzyme was much more stable to irreversible thermal denaturation than the Trichoderma reesei enzyme. It had an apparent denaturation midpoint (Tm) of 68.7°C, 14.3°C higher than the T. reesei enzyme. There are an additional three cysteines found in the H. grisea Cel12A enzyme. To determine their importance for thermal stability, we constructed three H. grisea Cel12A single mutants in which these cysteines were exchanged with the corresponding residues in the T. reesei enzyme. We also introduced these cysteine residues into the T. reesei enzyme. The thermal stability of these variants was determined. Substitutions at any of the three positions affected stability, with the largest effect seen in H. grisea C206P, which has a Tm 9.1°C lower than that of the wild type. The T. reesei cysteine variant that gave the largest increase in stability, with a Tm 3.9°C higher than wild type, was the P201C mutation, the converse of the destabilizing C206P mutation in H. grisea. To help rationalize the results, we have determined the crystal structure of the H. grisea enzyme and of the most stable T. reesei cysteine variant, P201C. The three cysteines in H. grisea Cel12A play an important role in the thermal stability of this protein, although they are not involved in a disulfide bond.
Keywords: Thermal stability, cellulase, cellulose, endoglucanase, homolog, protein crystal structure
Bacterial and fungal cellulases are widely used in the detergent, textile, and food industries, and there is continuing interest in their potential use in the conversion of cellulosic biomass to fermentable sugars. Cellulases are glycoside hydrolases found in at least 12 families of this very large group of enzymes (Henrissat and Davies 2000), and consist of cellobiohydrolases (or exoglucanases, EC 3.2.1.91) and endoglucanases (EC 3.2.1.4). Glycoside hydrolase family 12 (GH 12) members hydrolyse the β-1,4-glycosidic bond in cellulose via a double displacement reaction and a glycosyl-enzyme intermediate that results in retention of the anomeric configuration in the product (Schulein 1997; Birsan et al. 1998). Structures of GH 12 endoglucanases from both bacterial (Sulzenbacher et al. 1997; Crennell et al. 2002) and fungal (Sandgren et al. 2001, 2003; Khademi et al. 2002) sources have now been determined, and these provide the structural framework for the whole family. As part of a program to discover cellulases with improved properties, many novel GH 12 endoglucanases have been cloned and expressed (van Solingen et al. 2001; Goedegebuur et al. 2002). Previously, we studied the biochemical and structural diversity of several of these to better understand the sequence–structure–function relationships within the GH 12 family. Residue 35 (Trichoderma reesei Cel12A numbering) was identified as important in stability, and the introduction of a valine at that position led to an increase in the midpoint of thermal denaturation (Tm) of 7.7°C (Sandgren et al. 2003). The present work extends these studies to include the most stable GH 12 found in this program, Humicola grisea Cel12A (GenBank AF435071).
H. grisea is a thermophilic fungus with a growth temperature maximum above 50°C, now thought to be a member of the species Scytalidium thermophilum. This species produces a number of thermostable enzymes including hemicellulases and cellulases (Maheshwari et al. 2000). The secreted product of the H. grisea Cel12A gene is identical to that from the GH 12 gene reported for Humicola insolens (GenBank A22907; Dalboge and Heldt-Hansen 1994). Some enzymatic properties of the Cel12 from H. insolens have been reported (Schulein 1997).
In this work, we have found the H. grisea enzyme to be much more thermally stable than the T. reesei enzyme; it has a Tm that is 14.3°C higher than that of the T. reesei enzyme (Table 1). The H. grisea Cel12A shares 44% sequence identity with the T. reesei enzyme (93 residues out of 218). This low homology makes it difficult to identify residues that contribute to the stability difference between these homologs. We have focused on one marked difference, the three cysteines in the H. grisea Cel12A that are not found in the T. reesei protein. To determine if they are important for stability, we have introduced these cysteines into the corresponding positions in the T. reesei Cel12A enzyme as single, double, and triple mutations. We have also constructed six H. grisea Cel12A single mutants in which we have exchanged these cysteines with the corresponding residues in the T. reesei enzyme and with serine. The thermal stability of these variants has been determined. The crystal structures of the H. grisea enzyme and of the most stable T. reesei Cel12A cysteine variant, P201C, were determined and provide a structural basis for the observed effects of the mutations on stability.
Table 1.
Thermal denaturation data and relative specific enzyme activity
| GH 12 homolog | Variant | ΔTma | Tm (°C)b | Fit errorc | Activityd |
| T. reesei Ce112A | WT | 0.0 | 54.4e | 0.2 | 1.0 |
| H. grisea Ce112A | WT | 14.3 | 68.7e | 0.3 | 0.2 |
| T. reesei Ce112A | G170C | 2.1 | 56.5e | 0.2 | 1.2 |
| P201C | 3.9 | 58.3e | 0.2 | 0.8 | |
| V210C | 0.1 | 54.5e | 0.1 | 1.8 | |
| G170C/P201C | 0.7 | 55.1e | 0.1 | 0.4 | |
| P201C/V210C | 0.7 | 55.1 | 0.1 | 0.5 | |
| G170C/V210C | ND | ND | ND | 1.4 | |
| G170C/P201C/V210C | 0.0 | 54.5 | 0.3 | 0.1 | |
| H. grisea Ce112A | C175G | 1.3 | 69.9 | 0.2 | 0.7 |
| C175S | 0.2 | 68.9 | 0.1 | 0.7 | |
| C206P | −9.1 | 59.5 | 0.2 | 0.9 | |
| C206S | −5.4 | 63.3 | 0.1 | 1.5 | |
| C216V | 0.8 | 69.5 | 0.2 | 0.7 | |
| C216S | −5.5 | 63.1 | 0.2 | 0.7 |
a ΔTm values are relative to T. reesei Ce112A WT, H. grisea Ce112A WT.
b The thermal denaturation experiments were performed at 217 nm, in 0.05 M Bis-Tris propane, 0.05 M ammonium acetate (pH 8.0), and by increasing the temperature from 30°C to 90°C with data collected every 2°. The midpoint of the transition (Tm) is an apparent value because the thermal denaturation is not reversible.
cTm fit error corresponds to one standard deviation.
d The specific enzyme activities was measured after 10 min incubation with oNPC, at 40°C, pH 5.5. The specific activities given are relative to T. reesei Ce112A WT, H. grisea Ce112A WT.
e Two to five replicates were run with errors corresponding to one standard deviation between 0.15° and 0.42°.
ND, not determined.
Although none of the three cysteines were observed to be in a disulfide bond, the residues at these positions were found to be important for the stability of the protein. This is especially true for the residue at position 201 in T. reesei Cel12A (position 206 in H. grisea Cel12A).
Results
Stability determination
The circular dichroism (CD) signal at 217 nm as a function of temperature was collected for the two homologs and the mutants and fitted to two-state models (Fig. 1A ▶). The parameters from those fits were used to calculate a curve representing the apparent fraction of unfolded protein, Fapp, and the fitted curve for each sample (Fig. 1B ▶). All stability determinations reported for T. reesei Cel12A enzymes were on proteins deglycosylated with endoglucanase H. For the wild-type T. reesei Cel12A, this results in a form with the same Tm as the native protein expressed in T. reesei (data not shown). This is consistent with the direct observation of an identical single NAG residue on Asn 164 in the native T. reesei Cel12A (Sandgren et al. 2001) and in the deglycosylated recombinant variants. The H. grisea Cel12A proteins did not contain any N-linked glycosylation sites.
Figure 1.


The raw thermal denaturation data (A) are shown for a subset of proteins examined. These data and similar traces for all of the proteins listed in Table 1 were fitted to two-state models and used to produce a fraction apparent unfolded (Fapp) plot (B). By this convention, 0 represents native and 1 represents unfolded states for the protein. The protein concentration for all experiments was between 10 and 20 μM.
The pH of maximum thermal stability for T. reesei Cel12A is pH 5 (Fig. 2 ▶) as seen for the urea denaturation (Arunachalam and Kellis Jr. 1996). The pH of maximum thermal stability for H. grisea Cel12A is pH 7 (Fig. 2 ▶). None of the mutations studied shifts the pH of maximum stability compared to the wild types (data not shown).
Figure 2.

The thermal denaturation as a function of pH. Apparent midpoints (Tm) of the thermal transitions of H. grisea Cel12A (filled squares) and T. reesei Cel12A (filled diamonds).
The apparent Tm for each protein at pH 8.0 is listed in Table 1. The ΔTm values given in Table 1 have a standard deviation of ~0.2°C, and differences of less than 0.5°C are not considered significant. The H. grisea Cel12A enzyme has a Tm (68.7°C) that is 14.3°C higher than the T. reesei enzyme. The T. reesei Cel12A cysteine variants recruited from H. grisea Cel12A have Tm changes ranging from zero to an increase of 3.9°C for the most stable variant, P201C. The H. grisea Cel12A variants have Tm changes ranging from a decrease of 9.1°C for the least stable variant, C206P, to an increase of 1.3°C for the most stable variant, C175G.
Relative enzyme activity
The specific activity of wild-type and mutant enzymes was determined with oNPC as substrate. Their relative activities are presented in Table 1. The H. grisea enzyme has only about 20% of the activity of the T. reesei Cel12A at 40°C on oNPC (Table 1) and on hydroxyethyl-cellulose (data not shown). The identical Cel12 enzyme from H. insolens is reported to have very low activity on similar substrates (Schulein 1997). None of the single mutations in H. grisea Cel12A or in the T. reesei enzyme have large effects on oNPC activity. Decreased activity is seen in two of the double mutations and, especially, the triple mutation (G170C/P201C/V210C) in T. reesei Cel12A.
The oNPC activity of the H. grisea Cel12A as a function of temperature (Fig. 3 ▶) was determined, and corresponds to an apparent activation energy of 7.1 kcal/mole, slightly less than the 10 kcal/mole reported for T. reesei Cel12A (Sandgren et al. 2003). The H. grisea enzyme shows a continual increase in activity over the full temperature range from 25°C to 60°C.
Figure 3.

The temperature dependence of enzyme activity was assayed by hydrolysis of the chromogenic substrate oNPC (ortho-nitrophenyl cellobioside) in 0.05 M Bis-Tris propane, 0.05 M ammonium acetate (pH 5.5) over 10 min at 25°C, 40°C, 50°C, and 60 °C. Data are shown for H. grisea Cel12A (filled squares) and T. reesei Cel12A (filled diamonds; Sandgren et al. 2003). It is expressed as a molar specific activity relative to T. reesei Cel12A at 40°C.
DTNB assays
Both wild-type proteins and the single mutations in T. reesei Cel12A were assayed for accessible thiols. The thiol contents measured for the denatured proteins (in 8 M urea) correspond to the expected free thiols. T. reesei Cel12A wild type has no detectable cysteines, all single mutations (G170C, P201C, V210C) show one cysteine per mole protein, and the H. grisea Cel12A has three cysteines. In the native state, only 0.3 thiols per mole are titratable in H. grisea Cel12A. In the native T. reesei Cel12A proteins P201C and V210C, the introduced thiols are partially available, at 0.5 titratable thiols per mole. In T. reesei Cel12A G170C, the introduced cysteine is fully available even in the native state.
Three-dimensional structures
To shed some light on the structural basis for the thermal stability differences between the H. grisea and T. reesei Cel12A enzymes, we have solved the structures of the H. grisea WT and the T. reesei cysteine variant (P201C) with the largest increase in Tm (3.9°C). Statistics on data collection, refinement, and the final models are given in Tables 2a and 2b. Both structures have the expected fold of a GH family 12 enzyme. They consist mainly of 15 β-strands building up two β-sheets stacked on top of one another to form a β-sandwich (Fig. 4 ▶). The concave surface of the larger β-sheet B produces the 35-Å-long substrate-binding cleft that runs across one face of the enzyme (Sulzenbacher et al. 1997; Sandgren et al. 2001). The catalytic nucleophile and the Brønsted acid/base are the two glutamyl residues 120 and 205 in H. grisea Cel12A and 116 and 200 in T. reesei Cel12A.
Table 2a.
X-ray data collection and processing statistics
| GH 12 homolog | H. grisea WT | T. reesei P201C |
| Collected | Max-Laba | ESRFb |
| Beamline | I711 | ID14:EH1 |
| Detector | MAR IP | MAR CCD 165 |
| Wavelength (Å) | 1.09 | 0.93 |
| Oscillation range | 0.5° | 0.5° |
| Space group | P43212 | P31 |
| Cell parameters (Å) | 49.2, 49.2, 165.5 | 70.6, 70.6, 69.1 |
| Resolution range (Å) | 30–1.22 | 20–1.70 |
| Resolution range outer shell | 1.24–1.22 | 1.73–1.70 |
| No. of observed reflections | 321,815 | 184,371 |
| No. of unique reflections | 58,847 | 42,832 |
| Average multiplicity | 5.5 | 4.3 |
| Completeness (%)c | 94.6 (84.4) | 99.8 (99.6) |
| Rmerge (%)d | 7.6 (35.2) | 7.4 (39.0) |
| I/σ (I) | 24.1 (4.0) | 18.3 (2.8) |
a Swedish National Electron Accelerator Laboratory for Nuclear Physics and Synchrotron Radiation Research (Max-Lab), Lund, Sweden.
b European Synchrotron Radiation Facility (ESRF), Grenoble, France.
c Numbers in parentheses are for the highest resolution bins.
dRmerge = ΣhklΣi|I − 〈I〉|/ΣhklΣi|I|.
Table 2b.
Structure refinement and final model statistics
| GH 12 homolog | H. grisea WT | T. reesei P201C |
| PDB access codes | 1olr | 1olq |
| Resolution used in refinement (Å) | 30–1.22 | 20–1.70 |
| Reflections in | ||
| Working set | 56,977 | 40,981 |
| Test set | 1793 | 1331 |
| R & Rfree factor (%) | 13.5; 15.1 | 20.9; 25.1 |
| Protein molecules in AU | 1 | 2 |
| Residues in protein | 224 | 218 |
| Protein atoms | 1832 | 3320 |
| Waters | 300 | 286 |
| Residues with double conformations | 17 | — |
| N-glycosylation (NAG) residues | — | 2 |
| 〈B〉 (Å2) | 13.1 | 15.4 |
| Protein 〈B〉 (Å2) | 11.4 | 14.8 |
| Water 〈B〉 (Å2) | 23.3 | 23.0 |
| RMSD bond lengths (Å)a | 0.013 | 0.015 |
| RMSD bond angles (°)a | 1.6 | 1.7 |
| RMSD ΔB on bonded atoms (Å2) | 1.5 | 1.3 |
| Average RMSD NCS Cα (Å) | — | 0.5 |
| Average RMSD NCS all atoms (Å) | — | 0.6 |
| Stringent Ramachandran outliersb (%) | 1.0 | 0.7 |
Values were calculated with O (Jones et al. 1991; Jones and Kjeldgaard 1997), CNS (Brünger et al. 1998), MOLEMAN (Kleywegt and Jones 1996b), LSQMAN (Kleywegt and Jones 1997), and Refmac 5.0 (Murshudov et al. 1997).
a From Engh and Huber 1991.
b According to the stringent boundary definition (Kleywegt and Jones 1996a).
Figure 4.
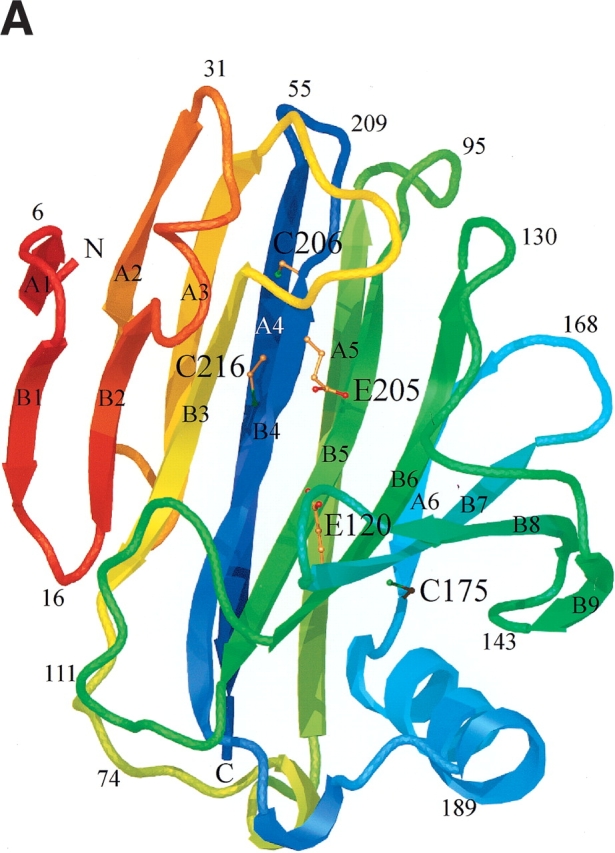

Schematic ribbon diagram showing top (A) and side views (B) of the H. grisea Cel12A crystal structure, color-ramped according to residue number, starting with red at the N terminus and ending with blue at the C terminus of the structure. The structure has the expected fold of a GH family 12 enzyme. It consists mainly of 15 β-strands building up two β-sheets, A and B, A consisting of six β-strands and B of nine β-strands, that stack on top of one another revealing a β-sandwich. The individual β-strands are labeled (A1–A6 and B1–B9) according to their positions in the two β-sheets. The structure has side chains drawn for the three cysteine residues (C175, C206, and C216), and the two catalytic residues (E120 and E205). Figures 4 ▶, 6 ▶, and 7 ▶ were prepared using O (Jones et al. 1991), and rendered with Molray (Harris and Jones 2001).
H. grisea Cel12A WT structure
The final model of the H. grisea enzyme contains the complete sequence of 224 amino acids. The H. grisea Cel12A shares 44% sequence identity (93 residues out of 218) with the T. reesei Cel12A enzyme (Fig. 5 ▶). Equivalent Cα atoms from the two structures can be superimposed with pairwise root-mean-square deviations (RMSDs) in the range of 0.3–0.5 Å. There are no large insertions or deletions in the H. grisea structure, compared with the T. reesei structure. The extra six residues in the H. grisea structure are distributed over the molecule as single amino acid insertions. Two of the extra residues are located at the N terminus. Some of the biggest main-chain differences between the two structures can be found in the four loops that each contain an extra residue in the H. grisea structure. These loops correspond to residues 29–32, 40–47, 92–96, and 165–169, and connect β-strands B2 to A2, A2 to A3, A5 to B5, and A6 to B7, respectively.
Figure 5.

Structure-based sequence alignment of five glycosyl hydrolase family 12 amino acid sequences with known protein structure. The secondary structure elements of the proteins, color-ramped from red at the N terminus to blue at the C terminus, are drawn at the top of the alignment. The position of the nucleophile and the acid–base in the sequences are indicated with filled and open arrows, respectively. The aligned protein sequences, with their GenBank or PDB access codes indicated in parentheses, are: Streptomyces lividans CelB2 (U04629, 2NLR); Streptomyces sp. 11AG8 Cel12A (AF233376, 1OA4); Humicola grisea Cel12A (AF435071, 1OLR); Trichoderma reesei Cel12A (AB003694, 1H8V); Hypocrea schweinitzii Cel12A (AF435068, 1OA3).
The three cysteine residues, C175, C206, and C216, are located on β-strands A6, B4, and A4, respectively (Fig. 4 ▶). Their side chains point into the core between the two β-sheets, where they form extensive interactions with surrounding residues (Fig. 4A ▶).
Cysteine residue 175 is on β-strand A6, on the smaller β-sheet, A, that creates the convex outer surface of the enzyme, close to the only α helix in the structure (Fig. 4 ▶). The side chain points into the core between the two β-sheets, where it has a set of van der Waals interactions with the side chains of six residues; T85, I123, A144, F173, I177, and F180 (interatomic separations in the range of 3.5–4.1 Å; Fig. 6A ▶). The cysteine at residue 206 is next to the catalytic acid/base (Glu 205) on β-strand B4 (Fig. 4 ▶). The side chain points into the β-sheet core, where it has an extensive network of interactions with the side chains of eight residues; Q34, W52, W54, S63, P65, Y91, A98, and T204, with interatomic separations in the range of 3.3–4.9 Å (Fig. 6B ▶). Many of the interactions with Sγ are polar in nature. In particular, the contact with the indole Nɛ1 of W52 is 3.3 Å, indicative of a short S-HN hydrogen bond. The third cysteine is residue 216 on β-strand A4 in β-sheet A, the sheet that creates the outer convex surface of the enzyme. This cysteine residue has its side chain pointing into the core region below the active site, where it interacts with the side chains of five residues; W48, V87, W89, F214, and F219, with interatomic separations in the range of 3.3–4.8 Å (Fig. 6C ▶).
Figure 6.
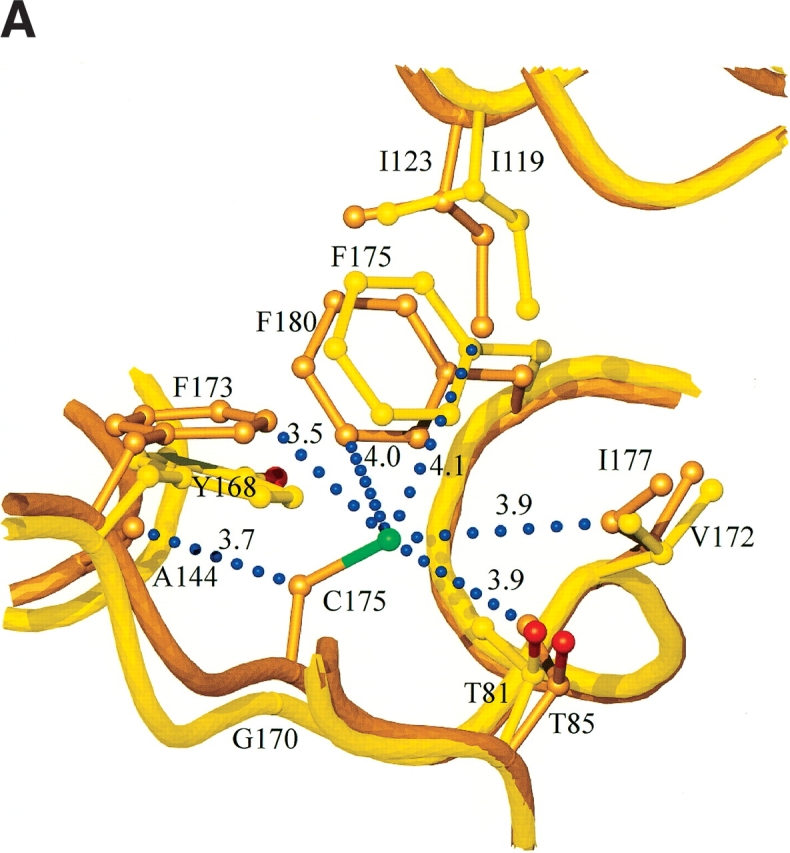
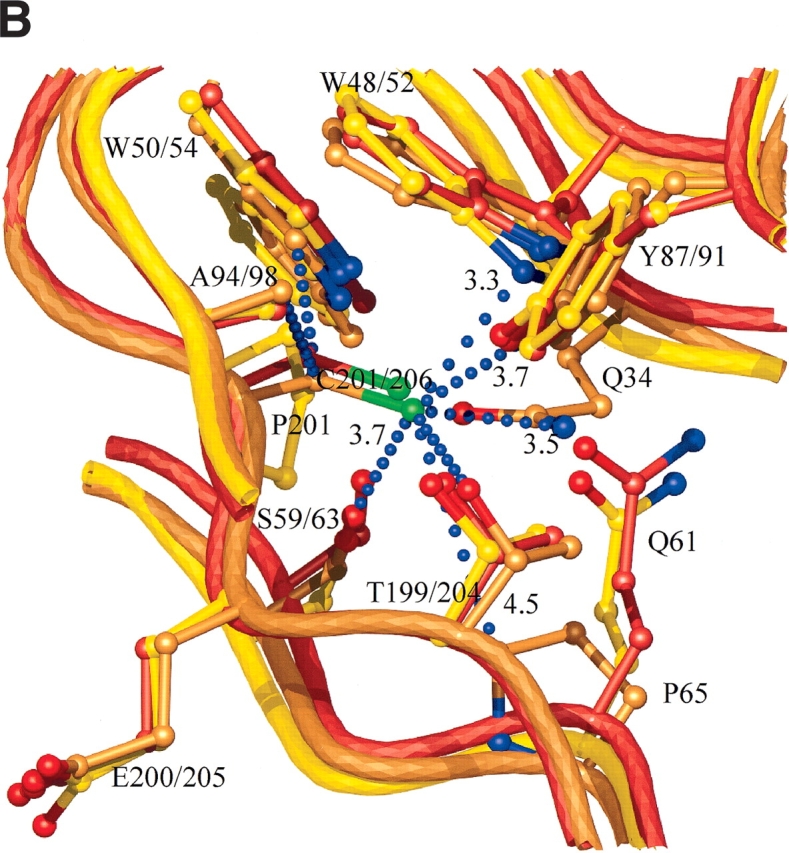
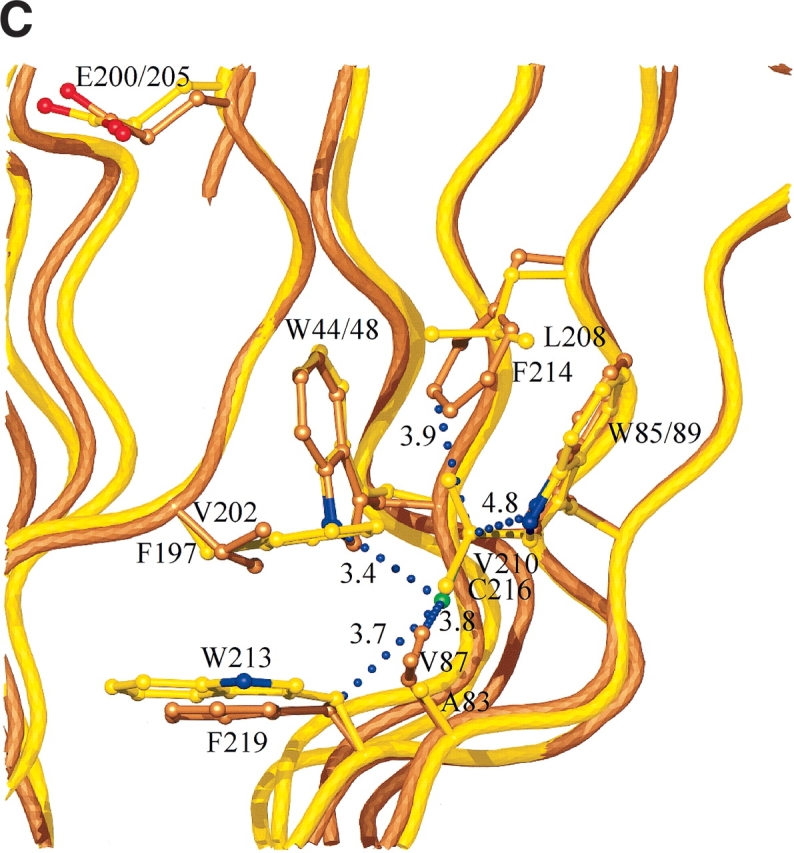
(A) Interactions and conformational changes close to residues 175 of the H. grisea Cel12A and residue 170 the T. reesei WT Cel12A structures. (B) Interactions and conformational changes close to residue 206 of the H. grisea Cel12A structure, and residue 201 of the T. reesei wild-type and P201C Cel12A structures. (C) Interactions and conformational changes close to residue 216 of the H. grisea Cel12A and 210 of the T. reesei wild-type Cel12A structures. The H. grisea Cel12A structures have carbon atoms colored goldenrod, and the T. reesei wild-type and P201C Cel12A structures have carbon atoms colored yellow and orange, respectively. The blue bubbles in A, B, and C indicate contacts to the free cysteine residues in H. grisea Cel12A.
T. reesei Cel12A P201C structure
The T. reesei Cel12A P201C variant crystallizes in the space group P31, with two noncrystallographic symmetry (NCS)-related protein molecules in the asymmetric unit, forming a structural dimer. The two NCS molecules pack to block their substrate binding clefts (Fig. 7 ▶), as in previous T. reesei Cel12A structures (Sandgren et al. 2001, 2003). They can be superimposed with an RMSD of 0.5 Å on Cα atoms. The areas in the structure that are most affected by crystal contacts are mostly loops, in particular those in the region of residues 26, 37, 111, and 153. Pairwise comparisons of the two NCS molecules with the six NCS molecules in the wild-type T. reesei Cel12A structure have RMSDs in the range of 0.3–0.6 Å. Residue 201 in T. reesei Cel12A is located on β-strand B4, a strand on the bigger β-sheet B, one residue from the catalytic acid/base (E200). Figure 6B ▶ shows the environment around residue 201 in the wild-type and mutant T. reesei Cel12A structures. The introduction of a cysteine results in only small local changes and does not cause any major conformational change in the variant compared to the wild-type structure. The effect of introducing the P201C mutation on the solvent accessibility of the interacting residues is very small.
Figure 7.

The Cα drawing of two crystallographic interacting molecules from the H. grisea Cel12A structure (A), colored blue, and two NCS interacting molecules from the T. reesei P201C mutant structure (B), colored red, showing that the packing of the two NCS molecules in the T. reesei crystals prevents any potential ligand binding in the catalytic cleft, but that the packing of the molecules in the H. grisea crystal potentially allows binding of a ligand.
Discussion
The Cel12A from the thermophilic fungus H. grisea has a Tm that is 14.3°C higher than that of Cel12A from the mesophilic fungus T. reesei (Table 1). However, it has low activity compared to the mesophilic enzyme (Table 1; Schulein 1997). This is true even at temperatures the thermophilic fungus prefers, from 40°C to 50°C. The H. grisea enzyme shows a continual increase in activity over the temperature range from 25°C to 60°C (Fig. 3 ▶), whereas T. reesei Cel12A showed a decrease in activity at the highest temperature (Sandgren et al. 2003), presumably due to thermal inactivation. Despite this, the T. reesei enzyme always has the higher activity. We would like to understand how to improve the stability of the T. reesei Cel12A.
The H. grisea Cel12A is more stable than any GH family 12 member in our previous study (Sandgren et al. 2003), thus extending the observed wide variation in temperature stability to give a range in the Tm, from the least to the most stable Cel12 of 23°C. Cel12 enzymes of even greater stability have been identified in a hyperthermophilic bacterium (Bok et al. 1998) and archaeon (Bauer et al. 1999) and in a thermophilic eubacterium (Wicher at al. 2001). There has long been an interest in identifying the sources of such stability differences between homologous proteins (Jaenicke 2000). However, no generally reliable rules have emerged to predict stabilizing changes from comparisons of sequences or, even, modeled structures. Although we find a high level of structural homology between the T. reesei Cel12A and the more stable H. grisea enzyme, the low sequence identity of 44% makes it difficult to judge which changes are needed to stabilize the T. reesei protein. An examination of the GH family 12 sequence alignment (Fig. 5 ▶) shows one marked difference to be the presence of three additional Cys residues near the C terminus of the H. grisea enzyme, at positions 175, 206, and 216. Gliocladium roseum Cel12C (GeneBank AF435065; Goedegebuur et al. 2002) has one additional cysteine at the position equivalent to the last of these. We focused on these three sites to see what role they may play in enzyme stability. A possible simple explanation for the increased stability of the H. grisea enzyme would be that two of these Cys residues are involved in an additional disulfide bond. Such bonds are well known to be important for protein stability (Thornton 1981; Betz 1993). Specifically, in GH 12, the totally conserved disulfide bond between Cys residues 4 and 32 (T. reesei Cel12A numbering), is very important for stability (Arunachalam and Kellis Jr. 1996), and a T. reesei Cel12A C32A mutation greatly destabilizes the enzyme (data not shown). However, DTNB analysis showed three free thiols in H. grisea Cel12A, and solution of the crystal structure showed no disulfide other than the expected bond between Cys6 and Cys35. It is unusual for a secreted protein to include so many free cysteines (Thornton 1981), unless they have a catalytic function, and they can lead to chemical lability (Tomazic and Klibanov 1988), or to misformation of essential disulfides (e.g., Cys6–35; Volkin and Klibanov 1987). The three cysteine residues in H. grisea Cel12A, C175, C206 and C216, are located on β-strands A6, B4, and A4, respectively (Fig. 4 ▶). Their side chains point into the core between the two β-sheets, where they form extensive interactions with surrounding residues (Fig. 6A ▶). We explored the role of these residues by mutating the three cysteines in the H. grisea Cel12A enzyme, one by one, to a serine (a conservative substitution) or to the equivalent amino acid at these positions in the T. reesei Cel12A enzyme. We also introduced these cysteines into T. reesei Cel12A as single, double, and triple mutations. Most of these mutations did, indeed, have significant effects on stability (Table 1).
The Cel12A residue at position 201 in T. reesei, 206 in H. grisea, has the largest influence on stability. In H. grisea, C206 is next to the catalytic acid/base (Glu 205), but the effects of mutations at this position in H. grisea and in T. reesei (Table 1) show that C206 alone is not responsible for the low activity of the H. grisea enzyme. The side chain of C206 points into the β-sheet core to make extensive interactions with the side chains of eight residues (Fig. 6B ▶). Many of the interactions with Sγ are polar in nature, including an apparent S-HN hydrogen bond with the indole Nɛ1 of W52. Given this complex network of interactions, it is easy to understand how changing residue 206 leads to large decreases in stability. Mutating C206 in H. grisea Cel12A to a proline (the residue at the equivalent position in the T. reesei enzyme) or to a serine causes a reduction in the Tm of 9.1°C and 5.4°C, respectively, compared to the parent. The P201C mutation at the equivalent position in T. reesei increases Tm by 3.9°C compared to the wild type. One reason for the increased Tm of the P201C variant is the filling of a small cavity by the introduced cysteine side chain, resulting in a new set of van der Waals interactions. However, not all of the interactions present in the H. grisea enzyme (Fig. 6B ▶) are made in the T. reesei P201C enzyme. In particular, the equivalent of the Q34–C206 interaction is missing. This is consistent with the stabilizing effect of the P201C mutation in T. reesei Cel12A being smaller than the destabilizing effect of the C206P mutation in the H. grisea enzyme.
The Cel12A residue at position 210 in T. reesei, 216 in H. grisea, is also important for stability. The side chain of C216 in H. grisea points into the core region below the active site, interacting with the side chains of five hydrophobic residues (Fig. 6C ▶). These are all van der Waals contacts, so mutating this residue to a valine in H. grisea C216V or from a valine to cysteine in T. reesei V210C causes little change in stability. In contrast, in H. grisea C216S, the introduction of the polar serine residue into a region with no possibility for hydrogen bonding has a large destabilizing effect (Table 1).
The residue at position 170/175 has least, but still significant, impact on stability. In H. grisea, the C175 side chain points into the core between the two β-sheets, where it makes a set of van der Waals interactions with the side chains of six residues (T85, I123, A144, F173, I177, and F180; Fig. 6A ▶). Surprisingly, mutation of this residue to serine has no effect on stability, and mutation to glycine is slightly stabilizing. The converse G170C mutation in the T. reesei Cel12A enzyme is surprisingly stabilizing (Table 1). The effects of mutations at the other two sites appear relatively simple to understand, at least qualitatively. The more puzzling effects at this position (170/175) are a reminder that the environment of the same amino acid at equivalent positions in two sequence-diverse proteins may well be different. For instance, by DTNB analysis, all three cysteine thiols are almost completely inaccessible in native H. grisea, whereas in the T. reesei context, cysteines 201 and 210 are about 50% accessible and that at 170 is completely accessible.
It is often possible to combine mutations to give additive effects on stability, for example, in the case of T4 lysozyme (Zhang et al. 1995). No additivity is observed here, however. The two double mutants analyzed, G170C/P201C and P201C/V210C, are only moderately more stable than wild type, and the triple mutation G170C/P201C/V210C was no more stable than wild type (Table 1). It is probable that mutations at these three sites do not act as independent and additive because they are clustered in the same region of the protein (Fig. 4 ▶). There may also be some complexity due to the fact that stability is being measured as Tms for irreversible denaturation rather than as the preferred thermodynamic free energy values: Despite extensive efforts, we could not find conditions under which we could compare the reversible denaturation of T. reesei Cel12A wild type with Cel12A proteins of different stability. Nonadditivity is also seen with respect to activity. None of the single mutations in the T. reesei enzyme has large effects on oNPC activity, but decreased activity is seen in two of the double mutations and, especially, the triple mutation (G170C/P201C/V210C), which has activity as low as that of H. grisea Cel12A (Table 1).
The effects of the mutations studied here do not explain the total difference in thermal stability between the Cel12A homologs from H. grisea and T. reesei. Some mutations studied previously in T. reesei Cel12A (Sandgren et al. 2003) represent substitutions to the residue found in H. grisea homolog (W7Y, S39N, S63V, N91D, S143T, T163S) and had relatively little effect on stability. In a series of recent mutations (Q162P, Y168F, N174D, and V192L), only N174D had any effect, raising Tm by 1.2°C (data not shown). Obviously these mutations represent a sparse sampling of the differences between the two wild-type enzymes, and there are potentially many contributors to the stability of the H. grisea protein (Jaenicke 2000). Increased hydrophobic packing (Golovanov et al. 2000) plays a large part in the specific stabilizing effects of all three cysteines, but there are no significant global differences in packing or compactness seen between H. grisea and T. reesei Cel12A structures. Reduced length of surface loops is thought to be stabilizing (Crennell et al. 2002), but it is the H. grisea protein that has four such loops with an additional residue compared to T. reesei Cel12A. There is also no significant difference in the number of surface ion pairs (Kumar et al. 2000) between the two structures that can explain the stability difference.
We find that the three additional cysteines found in H. grisea Cel12A play an important role in the thermal stability of this protein, although they are not involved in a disulfide bond. The residue at position 201 in T.reesei Cel12A (position 206 in H. grisea Cel12A) is particularly important. We note that the C175G and C216V mutations in H. grisea Cel12A are more stable than the wild type, showing that cysteines are not necessarily the most stabilizing amino acid to have in all these positions. Also, the observation of more open access to the active-site cleft in the H. grisea structure has led us to attempt to obtain cocrystals of this enzyme with substrates and substrate analogs, the topic of ongoing work.
Materials and methods
Protein expression and purification
The DNA encoding H. grisea Cel12A (Goedegebuur et al. 2002) was amplified from genomic DNA using PCR primers that introduced a BglII restriction endonuclease site at the 5′ end of the Cel12A gene (immediately upstream of the first ATG codon) and an XbaI site at the 3′ end (immediately downstream from the stop codon). The amplified fragment was then subcloned as a BglII-XbaI fragment into pLITMUS 28 vector. The DNA encoding T. reesei Cel12A was amplified from a genomic DNA clone (Ward et al. 1993) and subcloned in an identical manner but for the use of an AgeI site at the 3′ end.
All H. grisea and T. reesei Cel12A point mutants were made in the resultant vector using QuikChange mutagenesis methods (Stratagene, La Jolla, CA). The wild-type or variant gene was then subcloned as a BglII-XbaI fragment into the Aspergillus expression vector pGPT-pyrG, and the sequence-verified vector was transformed into Aspergillus niger (Berka and Barnett 1989). The resultant strain was grown in either shake-flasks or a fermentor. The culture supernatants containing T. reesei Cel12A proteins were treated overnight with 0.18 mg/mL of endoglucanase H at 37°C. Ammonium sulfate was added to the supernatants to a final concentration of 0.5 M and they were centrifuged. The Cel12A proteins were purified from these supernatants by column chromatography. Butyl Sepharose 4 Fast Flow resin (Amersham Biosciences, Piscataway, NJ) was loaded and equilibrated in a disposable drip column with 0.05 M Bis Tris Propane, 0.05 M ammonium acetate (pH 8) containing 0.5 M ammonium sulfate. The supernatants were loaded at about 10 mg of Cel12A protein per milliliter of resin and the column washed with three volumes of equilibration buffer before elution with three volumes of the same buffer without ammonium sulfate. Each column volume was collected as a separate fraction with the pure Cel12A proteins appearing in the second elution fraction. The identity of the purified proteins was confirmed by chymotryptic mapping using HPLC mass spectrometry.
Chymotryptic digestion
Fifty micrograms of Cel12A enzyme in 300 to 500 μL water were chilled on ice. One molar HCl was added to a final nominal concentration of 0.1 M. After 10 to 15 min, 50% trichloroacetic acid (w/v) was added to a final concentration of 20%. The sample was centrifuged at 13,000 rpm for two min and the precipitate washed with 90% (v/v) acetone (−20°C), to remove acid and soluble contaminants. The sample was centrifuged again, supernatant removed, and the pellet air dried. The pellet was resuspended in 50 μL of 8 M urea, 0.5 M NH4HCO3, then 4 μL of 0.2 M dithiothreitol was added and the sample was incubated for 15 min at 50°C. Next, 4 μL of 0.44 M iodoacetamide was added to the sample, which was then kept in the dark for 15 min. The sample was diluted slowly fivefold with water containing 0.1% (w/v) n-octyl-β-D-glucopyranoside. Four microliters of α-chymotrypsin (TLCK-treated; 10 mg per milliliter in 1 mM HCl) were then added. Following digestion at 37°C for 30–60 min, the reaction was stopped with 10% (v/v) of trifluoracetic acid (TFA), final concentration 1%. A 20-μL aliquot was then loaded onto an HPLC column.
Peptide separation and identification
Peptides were separated on a reversed-phase C18 column (2.1 × 150 mm, 300 Å pore size; Vydac) at 50°C, using a model 1090 HPLC (Hewlett Packard). Solvent system A was 0.1% TFA and solvent B was 0.08% TFA in acetonitrile. A complex step gradient increased the concentration of solvent B to 37% in 50 min, which was followed by a jump to 80% of B in 10 min. Total ion current chromatograms and UV spectra were recorded by a single-quadrupole mass spectrometer (model 5989B, Hewlett Packard) equipped with an API electrospray source (model 59987A, Hewlett-Packard) using software provided by the manufacturer.
Column fractions were dried in vacuo and resuspended in 0.1% glacial acetic acid, 50% methanol, 50% acetonitrile. The sequence of peptides was then confirmed by MS-MS analysis using an iontrap mass spectrometer (model LCQ, Finnigan) and matching data to a customized database using the SEQUEST search engine (Yates 3rd 1998).
These methods gave complete sequence coverage of the Cel12A proteins. Once the peptide map was established and every relevant peak assigned to the given sequence, variant proteins were screened by mass analysis of only those peptides that did not match the pattern of the parent molecule.
Thermal denaturation experiments
Circular dichroism experiments were performed on an Aviv 62ADS spectrophotometer (Protein Solutions, Lakewood, NJ), equipped with a five-position thermoelectric cell holder supplied by Aviv. Buffer conditions were 0.05 M Bis-Tris propane, 0.05 M ammonium acetate, adjusted to pH 8.0, unless noted otherwise, with acetic acid. The final protein concentration for each experiment was in the range of 10 to 20 μM. All T. reesei Cel12A proteins were deglycosylated with endoglucanase H prior to stability determinations. Data were collected in a 0.1-cm path length cell. The thermal denaturation experiments were performed at 217 nm, the wavelength in the far-UV spectra with maximum signal difference, as expected for a predominantly β-sheet protein. The temperature was increased from 30° to 90°C at 1°C min−1, with data collected every 2°. The equilibration time at each temperature was 0.1 min and data were collected for 4 sec per sample. Thus the total time for a denaturation experiment was 90 min. The thermal denaturation data were fitted to a two-state transition (Chen et al. 1992) using Savuka software provided by Dr. Osman Bilsel (University of Massachusetts Medical School). The midpoint of the transition (Tm) is an apparent value because the thermal denaturation of all the Cel12A proteins studied was not reversible. Because of this and a slow kinetic component due to aggregation, sufficiently fast scan rates were chosen and experiments were carefully controlled for time spent in the thermal denaturation transition, so that small temporal variations did not lead to apparent Tm differences.
The pH of maximum thermal stability for the Cel12A proteins studied was below pH 8 (Fig. 2 ▶). A suboptimal condition of pH 8.0 was used to facilitate detection of thermal stability differences.
Specific enzyme activity
To evaluate specific enzyme activity, and to monitor expression and purification of all the Cel12 proteins, an o-nitrophenyl β-D-cellobioside (oNPC, Sigma N 4764) hydrolysis assay was used. In a microtiter plate, 100 μL 0.05 M sodium acetate (pH 5.5) and 20 μL 25 mg/mL oNPC in assay buffer was added. Once equilibrated, 10-μL cellulase was added and the plate incubated at 40°C for 10 min, unless otherwise specified. To stop the reaction, 70 μL of 0.2 M glycine (pH 10.0) was added. The plate was then read in a microtiter plate reader at 410 nm. As a reference, 10 μL of a 0.1 mg/mL solution of T. reesei Cel12A enzyme provided an O.D. of around 0.3. The concentration of the Cel12A enzymes was determined by the absorbance at 280 nm, using an extinction coefficient for T. reesei Cel12A of 78,711 M−1 cm−1 (3.352 g/L−1), determined experimentally by the method of Edelhoch as described in Pace et al. (1995). Extinction coefficients for the H. grisea Cel12A proteins were calculated on the basis of their amino acid compositions (Pace et al. 1995).
DTNB assays
Thiol content of Cel12A proteins was measured by standard methods, using Ellman’s reagent 5,5′-dithiobis-2-nitrobenzoic acid (DTNB; Ellman 1958). Two to 6 μM protein was reacted with 400 μM DTNB in 0.05 M Tris, 0.01 M EDTA (pH 8.0). For determinations of accessible thiols in denatured proteins, Cel12A enzymes were incubated in 8 M urea, 0.05 M Tris, 0.01 M EDTA (pH 8.0) overnight at room temperature before the addition of DTNB.
Protein crystallization
The supernatant from a fermentation of an A. niger strain expressing H. grisea Cel12A was concentrated approximately sevenfold by ultrafiltration. Ammonium sulfate was added to the concentrate to a final concentration of 1 M. The suspension was stirred at 4°C for 60 min, then centrifuged. The pellet was redissolved in the original volume of buffer (0.05 M Bis Tris Propane and 0.05 M ammonium acetate at pH 8). Thirty milliliters of this sample was purified on a 10-mL Butyl Sepharose column, as described above, and eluted as an approximately 1 mg/mL solution in buffer. The long rod-shaped (0.05 × 0.1 × 1.0 mm) H. grisea Cel12A crystals were found to grow in the protein stock upon standing undisturbed at 4°C for 1–3 d. The crystals belong to space group P41212 or P43212 with cell dimensions: a = 49.2 Å, b = 49.2 Å, c = 165.5 Å, and have a calculated Vm of 2.0 (Matthews 1968) with one molecule in the asymmetric unit.
The T. reesei Cel12A P201C variant was crystallized under conditions similar to the wild-type enzyme (Sandgren et al. 2001), using 0.02 M cacodylate buffer (pH 6.0), 0.02 M calcium acetate, and 10%–30% (w/w) mono-methyl-ether (mme) polyethylene glycol (PEG) 2000, at 20–24°C using hanging and sitting drops (McPherson 1982). Crystallization drops were prepared by mixing equal amounts of protein solution (20 mg/mL) and crystallization agent. Large single, wedge-shaped crystals grow to a maximum size of 0.5 mm in all directions within 1–2 d. The crystals belong to the space group P31 or P32 with cell dimensions a = 70.6 Å, b = 70.6 Å, c = 69.1 Å, and have a calculated Vm of 1.9 (Matthews 1968) with two molecules in the asymmetric unit.
X-ray data collection
The crystals were equilibrated in 25%–40% mme PEG 2000, 0.02 M sodium cacodylate (pH 5.0), mounted in a cryo-loop, and plunge frozen in liquid nitrogen prior to transportation to the synchrotron. All X-ray data sets were collected from single crystals at 100 K. Data collection and processing statistics for the structures are given in Table 2a. The data sets were processed and scaled with DENZO and SCALEPACK (Otwinowski and Minor 1997). All subsequent data processing, after image integration, was performed using the CCP4 package (Collaborative Computational Project Number 4 1994), unless otherwise stated. A set of 3% of the reflections from each data set was used for monitoring the R-free (Brünger 1992).
Structure solution and refinement
Both structures were solved by molecular replacement with Amore (Navaza 1994), using the T. reesei Cel12A structure as the search model. The space groups were determined to be P43212 and P31 for the H. grisea Cel12A and the T. reesei Cel12A P201C structure, respectively.
The structures were refined with alternating cycles of model building using O (Jones et al. 1991) and maximum likelihood refinement using CNS version 1.0 (Brünger et al. 1998). Final rounds of refinement used Refmac 5.0 (Murshudov et al. 1997), and included anisotropic temperature factors for the H. grisea Cel12A structure.
Most water molecules in the structure models were located automatically by using the water-picking protocols in the refinement programs, and then manually selected or discarded by inspection. A summary of refinement statistics is given in Table 2b.
All structural comparisons were made with O (Jones et al. 1991), and figures were prepared with O and rendered with MolRay (Harris and Jones 2001). Coordinates and structure-factor amplitudes have been deposited with the RCSB Protein Data Bank (Bernstein et al. 1977), and have access codes 1olr and 1olq for the H. grisea Cel12A and T. reesei P210C Cel12A enzymes, respectively. The final electron density maps are available for viewing as part of the electron density server (EDS) service at http://ashtray.bmc.uu.se/eds.
Acknowledgments
We thank Steve Kim for help in protein expression, Dr. Osman Bilsel for providing Savuka, and Dr. Dave Lambright for his contributions to an early version of that software. We also thank Dr. Roopa Ghirnikar for critical reading of the manuscript. This work was supported in part by a subcontract from The Office of Biomass Program, within the U.S. DOE Office of Energy Efficiency and Renewable Energy.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
DTNB, 5,5′-dithiobis-2-nitrobenzoic acid
GH, glycoside hydrolase
HPLC, high-pressure liquid chromatography
mme, mono-methyl-ether
NCS, noncrystallographic symmetry
PEG, polyethylene glycol
RMSD, root-mean-square deviation
TFA, trifluoracetic acid
Tm, the midpoint of thermal denaturation
WT, wild type
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03220403.
References
- Arunachalam, U. and Kellis Jr., J.T. 1996. Folding and stability of endoglucanase III, a single-domain cellulase from Trichoderma reesei. Biochemistry 35 11379–11385. [DOI] [PubMed] [Google Scholar]
- Bauer, M.W., Driskill, L.E., Callen, W., Snead, M.A., Mathur, E.J., and Kelly, R.M. 1999. An endoglucanase, EglA, from the hyperthermophilic archaeon Pyrococcus furiosus hydrolyzes β-1,4 bonds in mixed-linkage (1–3),(1–4)-β-D-glucans and cellulose. J. Bacteriol. 181 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berka, R.M. and Barnett, C.C. 1989. The development of gene expression systems for filamentous fungi. Biotechnol. Adv. 7 127–154. [DOI] [PubMed] [Google Scholar]
- Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer Jr., E.T., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Betz, S.F. 1993. Disulfide bonds and the stability of globular proteins. Protein Sci. 2 1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsan, C., Johnson, P., Joshi, M., MacLeod, A., McIntosh, L., Monem, V., Nitz, M., Rose, D.R., Tull, D., Wakarchuck, W.W., et al. 1998. Mechanisms of cellulases and xylanases. Biochem. Soc. Trans. 26 156–160. [DOI] [PubMed] [Google Scholar]
- Bok, J.-D., Yernool, D.A., and Eveleigh, D.E. 1998. Purification, characterization, and molecular analysis of thermostable cellulases CelA and CelB from Thermotoga neapolitana. Appl. Environ. Microbiol. 64 4774–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. Free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system (CNS): A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Chen, B.L., Baase, W.A., Nicholson, H., and Schellman, J.A. 1992. Folding kinetics of T4 lysozyme and nine mutants at 12 degrees C. Biochemistry 31 1464–1476. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Crennell, S.J., Hreggvidsson, G.O., and Nordberg Karlsson, E. 2002. The structure of Rhodothermus marinus Cel12A, a highly thermostable family 12 endoglucanase, at 1.8 Å resolution. J. Mol. Biol. 320 883–897. [DOI] [PubMed] [Google Scholar]
- Dalboge, H. and Heldt-Hansen, H.P. 1994. A novel method for efficient expression cloning of fungal enzyme genes. Mol. Gen. Genet. 243 253–260. [DOI] [PubMed] [Google Scholar]
- Ellman, G.L. 1958. A colorimetric method for determining low concentrations of mercaptans. Arch. Biochem. Biophys. 74 443–450. [DOI] [PubMed] [Google Scholar]
- Engh, R.A. and Huber, R. 1991. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A 47 392–400. [Google Scholar]
- Goedegebuur, F., Fowler, T., Phillips, J., Van Der Kley, P., Van Solingen, P., Dankmeyer, L., and Power, S.D. 2002. Cloning and relational analysis of 15 novel fungal endoglucanases from family 12 glycosyl hydrolase. Curr. Genet. 41 89–98. [DOI] [PubMed] [Google Scholar]
- Golovanov, A.P., Vergoten, G., and Arseniev, A.S. 2000. Stabilization of proteins by enhancement of inter-residue hydrophobic contacts: Lessons of T4 lysozyme and barnase. J. Biomol. Struct. Dyn. 18 477–491. [DOI] [PubMed] [Google Scholar]
- Harris, M. and Jones, T.A. 2001. Molray—A web interface between O and the POV-Ray ray tracer. Acta Crystallogr. D 57 1201–1203. [DOI] [PubMed] [Google Scholar]
- Henrissat, B. and Davies, G.J. 2000. Glycoside hydrolases and glycosyltransferases. Families, modules, and implications for genomics. Plant Physiol. 124 1515–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke, R. 2000. Stability and stabilization of globular proteins in solution. J. Biotechnol. 79 193–203. [DOI] [PubMed] [Google Scholar]
- Jones, T.A. and Kjeldgaard, M.O. 1997. Electron-density map interpretation. Methods Enzymol. 277 173–208. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Khademi, S., Zhang, D., Swanson, S.M., Wartenberg, A., Witte, K., and Meyer, E.F. 2002. Determination of the structure of an endoglucanase from Aspergillus niger and its mode of inhibition by palladium chloride. Acta Crystallogr. D 58 660–667. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. and Jones, T.A. 1996a. Phi/Psi-chology: Ramachandran revisited. Structure 4 1395–1400. [DOI] [PubMed] [Google Scholar]
- ———. 1996b. xdlMAPMAN and xdlDATAMAN—Programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr. 52 826–828. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Detecting folding motifs and similarities in protein structures. Methods Enzymol. 277 525–545. [DOI] [PubMed] [Google Scholar]
- Kumar, S., Tsai, C.J., and Nussinov, R. 2000. Factors enhancing protein thermostability. Protein Eng. 13 179–191. [DOI] [PubMed] [Google Scholar]
- Maheshwari, R., Bharadwaj, G., and Bhat, M.K. 2000. Thermophilic fungi: Their physiology and enzymes. Microbiol. Mol. Biol. Rev. 64 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- McPherson, A.J. 1982. Preparation and analysis of protein crystals. John Wiley and Sons, New York.
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren, M., Shaw, A., Ropp, T.H., Wu, S., Bott, R., Cameron, A.D., Ståhlberg, J., Mitchinson, C., and Jones, T.A. 2001. The X-ray crystal structure of the Trichoderma reesei family 12 endoglucanase 3, Cel12A, at 1.9 Å resolution. J. Mol. Biol. 308 295–310. [DOI] [PubMed] [Google Scholar]
- Sandgren, M., Gualfetti, P.J., Shaw, A., Gross, L.S., Saldajeno, M., Day, A.G., Jones, T.A., and Mitchinson, C. 2003. Comparison of family 12 glycoside hydrolases and recruited substitutions important for thermal stability. Protein Sci. 12 848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulein, M. 1997. Enzymatic properties of cellulases from Humicola insolens. J. Biotechnol. 57 71–81. [DOI] [PubMed] [Google Scholar]
- Sulzenbacher, G., Shareck, F., Morosoli, R., Dupont, C., and Davies, G.J. 1997. The Streptomyces lividans family 12 endoglucanase: Construction of the catalytic core, expression, and X-ray structure at 1.75 Å resolution. Biochemistry 36 16032–16039. [DOI] [PubMed] [Google Scholar]
- Thornton, J.M. 1981. Disulphide bridges in globular proteins. J. Mol. Biol. 151 261–287. [DOI] [PubMed] [Google Scholar]
- Tomazic, S.J. and Klibanov, A.M. 1988. Mechanisms of irreversible thermal inactivation of Bacillus α-amylases. J. Biol. Chem. 263 3086–3091. [PubMed] [Google Scholar]
- van Solingen, P., Meijer, D., van der Kleij, W.A., Barnett, C., Bolle, R., Power, S.D., and Jones, B.E. 2001. Cloning and expression of an endocellulase gene from a novel streptomycete isolated from an East African soda lake. Extremophiles 5 333–341. [DOI] [PubMed] [Google Scholar]
- Volkin, D.B. and Klibanov, A.M. 1987. Thermal destruction processes in proteins involving cystine residues. J. Biol. Chem. 262 2945–2950. [PubMed] [Google Scholar]
- Ward, M., Wu, S., Dauberman, J., Weiss, G., Larenas, E., Bower, B., Rey, M., Clarkson, K., and Bott, R. 1993. Cloning, sequence and preliminary structural analysis of a small, high pI endoglucanase (EGIII) from Trichoderma reesei. In The Tricell 93 symposium (eds. P. Suominen and T. Reinikainen), pp. 153–158. Foundation for Biotechnical and Industrial Fermentation Research, Espoo, Finland.
- Wicher, K.B., Abou-Hachem, M., Halldorsdottir, S., Thorbjarnadottir, S.H., Eggertsson, G., Hreggvidsson, G.O., Karlsson, E.N., and Holst, O. 2001. Deletion of a cytotoxic, N-terminal putative signal peptide results in a siginificant increase in production yields in Escherichia coli and improved specific activity of Cel12A from Rhodothermus marinus. Appl. Microbiol. Biotechnol. 55 578–584. [DOI] [PubMed] [Google Scholar]
- Yates 3rd, J.R. 1998. Database searching using mass spectrometry data. Electrophoresis 19 893–900. [DOI] [PubMed] [Google Scholar]
- Zhang, X.J., Baase, W.A., Shoichet, B.K., Wilson, K.P., and Matthews, B.W. 1995. Enhancement of protein stability by the combination of point mutations in T4 lysozyme is additive. Protein Eng. 8 1017–1022. [DOI] [PubMed] [Google Scholar]