

Abstract
The Sac10b family consists of a group of highly conserved DNA binding proteins from both the euryarchaeotal and the crenarchaeotal branches of Archaea. The proteins have been suggested to play an architectural role in the chromosomal organization in these organisms. Previous studies have mainly focused on the Sac10b proteins from the crenarchaeota. Here, we report the 2.0 Å resolution crystal structure of Mja10b from the euryarchaeon Methanococcus jannaschii. The model of Mja10b has been refined to an R-factor of 20.9%. The crystal structure of an Mja10b monomer reveals an α/β structure of four β-strands and two α-helices, and Mja10b assembles into a dimer via an extensive hydrophobic interface. Mja10b has a similar topology to that of its crenarchaeota counterpart Sso10b (also known as Alba). Structural comparison between the two proteins suggests that structural features such as hydrophobic inner core, acetylation sites, dimer interface, and DNA binding surface are conserved among Sac10b proteins. Structural differences between the two proteins were found in the loops. To understand the structural basis for the thermostability of Mja10b, the Mja10b structure was compared to other proteins with similar topology. Our data suggest that extensive ion-pair networks, optimized accessible surface area and the dimerization via hydrophobic interactions may contribute to the enhanced thermostability of Mja10b.
Keywords: DNA binding protein, thermostability, archaea, chromosomal organization, crystal structure
All living organisms can be divided into three primary domains: eukarya, bacteria, and archaea (Woese et al. 1990). Archaea are often found in various extreme environments, and can be classified into two kingdoms: the euryarchaeota, and the crenarchaeota. Archaea are thought to include the most ancient organisms on earth, and to have some biological features of both bacteria and eukarya (Eeling and Doolittle 1995). Homologs of eukaryotic histones have been found in the euryarchaeota (Sandman et al. 1990; Grayling et al. 1996), and these archaeal histones are capable of organizing DNA into nucleosomes both in vivo and in vitro (Pereira et al. 1997; Balley et al. 1999). Structural studies have shown that archaeal histones share a similar fold with their eukaryotic counterparts (Tarich et al. 1996; Luger et al. 1997; Decanniere et al. 2000). No histone has so far been identified in the crenarchaeota, but small abundant DNA binding proteins (classified into 7-kD, 8-kD, and 10-kD molecular mass classes) have been found in the genus Sulfolobus (Dick and Reinhardt 1986; Choli et al. 1988; McAfee et al. 1995; Edmonson and Shriver 2001). Among these proteins, the 7-kD proteins have been most extensively studied. Based on the crystal structure of the Sac7d/DNA complex, a model for the chromosomal organization in Sulfolobus has been proposed (Robinson et al. 1998).
In 1998, Forterre et al. showed that Sac10b, a 10-kD protein from Sulfolobus acidocaldarius, has at least one homolog in each of the hyperthermophilic archaea whose genomes have been sequenced (Foterre et al. 1999). The Sac10b family includes members from both euryarchaeota and the crenarchaeota. Given their ubiquity, these proteins may play an important physiological role in these organisms. Biochemical studies have been performed on three crenarchaeotal members of the Sac10b family, that is, Sac10b from S. acidocaldarius, Ssh10b from S. shibatae and Sso10b from S. solfataricus. Sac10b exists as a dimer in solution and binds cooperatively to DNA. Although this protein does not compact DNA, it is capable of enveloping two strands of duplex DNA into a helix protein structure (Lurz et al. 1986). Ssh10b possesses the ability to constrain negative DNA supercoils in a temperature-dependent fashion, suggesting that the Sac10b proteins are involved in chromosomal organization in archaea (Xue et al. 2000). Recently, Sso10b (or Alba) was found to form a stable complex with the silencing protein Sir2, which has histone deacetylase activity. Acetylation of Sso10b at Lys16 strongly reduces the affinity of the protein for DNA. Deacetylation of Sso10b by Sir2 results in transcriptional repression in a reconstituted in vitro transcription system (Bell et al. 2002). Despite these in vitro findings, the function of the Sac10b proteins remains to be determined.
To understand how Sac10b proteins interact with DNA, considerable efforts have been made to determine their crystal structure. Recently, the crystal structure of Sso10b was resolved at 2.6 Å (Wardleworth et al. 2002). A model for the interaction of this crenarchaeotal protein with DNA was also proposed. In this article, we present the 2.0 Å resolution crystal structure of Mja10b, the smallest known member of the Sac10b family, from the euryarchaeon Methanococcus jannaschii (Jones et al. 1983). By comparing the crystal structure of Mja10b with that of Sso10b, we show the evolutionarily conserved structural features as well as structural variations of the Sac10b family. We also describe the structural features of the two Sac10b proteins that may contribute to their thermostability.
Results and Discussion
Structure of Mja10b
The crystal structure of the Mja10b monomer consists of four β-strands (β1, β2, β3, and β4) and two α-helices (α1 and α2; Fig. 1 ▶). This structural model includes 80 (of the 87 total) amino acid residues and 24 ordered water molecules. All seven residues that were not visible in the electron density map (Val68 to Arg74) belong to the disordered loop that connects strands β3 and β4. Superimposition of the structure of Mja10b on that of Sso10b shows that the two proteins share a very similar overall fold with a root-mean-square deviation of 0.7 Å for 80 equivalent residues. Compared with Sso10b, Mja10b lacks the first seven residues at the N terminus, and the seven residues are not included in the model of Sso10b deposited in the PDB. The main differences between the two structures occur in loop regions, where the sequences are not conserved. Mja10b shares 56% identity at the amino acid sequence level with Sso10b and the conservation occurs throughout the primary sequence of the proteins. Comparison of the structures of the two proteins would permit the identification of the common structural features that are of importance for the function of the Sac10b family.
Figure 1.
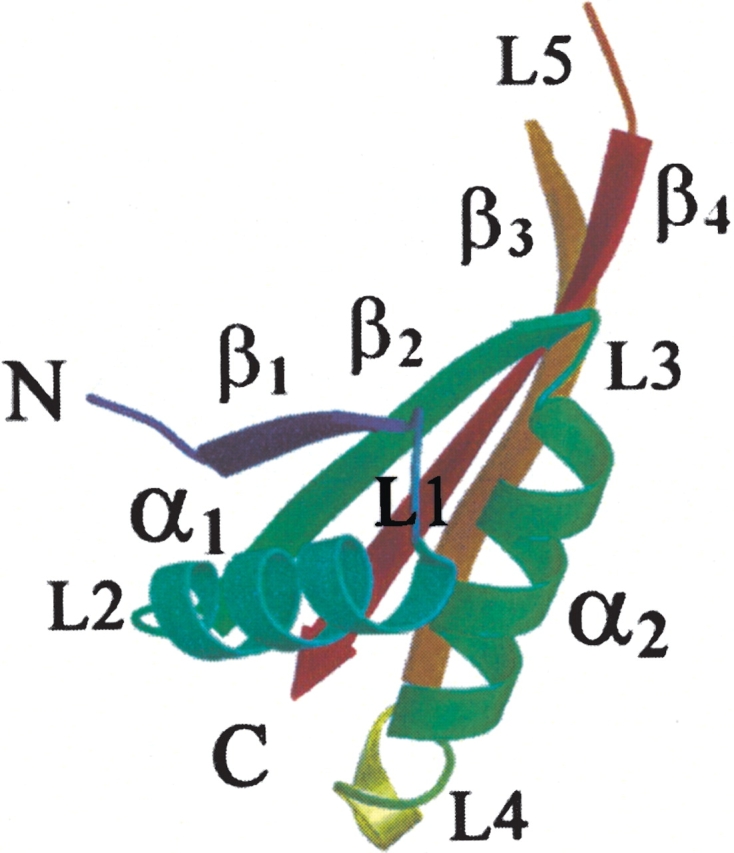
Cartoon representation of the Mja10b monomer. The two α-helices are labeled α1 and α2, the four β-strands are labeled β1–β4, and the five loops are labeled L1–L5.
Conserved motifs
The Mja10b sequence includes a conserved motif spanning residues 8–16 (Fig. 2 ▶). In this motif, Lys9 is equivalent to Lys16 of Sso10b and is the potential acetylation site (Wardleworth et al. 2002). Both Lys9 and Lys10 lie on the top of loop L1, which is stabilized by flanking residues with the following hydrogen bonds: Gly8 N—Tyr15 OH, Gly8 O—Lys40 NZ, Lys10 N—Try15 OH, Lys10 NZ—Asn14 ND2, and Lys10 O—Lys40 NZ. These interactions are also observed in the Sso10b structure. The residue of Pro11, strictly conserved in the Sac10b family, provides extra stability to loop L1 (Watanabe et al. 1991). In addition, a total of 12 hydrophobic residues are involved in the formation of a hydrophobic core in the structure of Mja10b, and nine of these residues are strictly conserved in all members of the Sac10b family. These hydrophobic interactions, along with those from other residues, appear to be the main force for the stabilization of the Mja10b structure. Furthermore, seven hydrophobic residues (Ile38, Val42, Ile58, Ile61, Ile63, Ile81, and Ile83) form a hydrophobic interface for dimer formation. All seven residues are conserved in the Sac10b family, though complementary substitutions are observed in some proteins.
Figure 2.

Sequence alignment of the Sac10b family. Mja10b, Methanococcus jannaschii (Q57665); Ssh10b, Sulfolobu shibatae; Sso10b, S. solfataricus (P74761); Pho10b, Pyrococcus horikoshi (O74101); Pab10b, P. abyssi (Q9VIN3); Afu10b1, Afu10b2, Archaeoglobus fulgidus (O28323,O29195); Tac10b, Thermoplasma acidophilum (Q9HJQ5); Tvo10b, T. Volcanium (Q97955); Mth10b, Methanobacterium thermoautotrophicum (O27527); Ape10b1, Ape10b2, Aeropyrum pernix (Q9YAX2, Q9YAW1). The secondary structure of Mja10b derived from the crystal structure is shown above the alignment. The residues in the conserved motif are marked with an “*.” Residues involved in hydrophobic interactions between the two monomers at the dimer interface are marked with a “+.”
Dimer structure
Proteins of the Sac10b family have been shown to exist as dimers or higher oligomers in solution in vitro (Lurz et al. 1986; Xue et al. 2000). The crystal structure of Sso10b reveals a dimer in the asymmetric unit (Wardleworth et al. 2002). The present study shows that Mja10b exists as a monomer in the asymmetric unit. Two monomers in adjacent asymmetric units interact, forming a dimer related by twofold symmetry. Helix α2 and strands β3 and β4 in each monomer are involved in dimer formation (Fig. 3A ▶). Twelve hydrogen bonds are formed at the interface between the two interacting monomers (3.5 Å cutoff). Additional contacts include a number of tightly hydrophobic interactions. The residues Ile38, Val42, Ile58, Ile61, Ile63, Ile81, and Ile83 in each monomer form a hydrophobic surface, which is conserved among the Sac10b family. These residues are presumably essential for the maintenance of the dimer structure. A total solvent-exposed surface area of 1454 Å2 (or 727 Å2 per monomer) would be buried by the formation of a dimer. In a systematic study of noncovalent protein–protein interactions, Janin showed that the buried surface area per monomer as a result of oligomerization is typically larger than 700 Å2 (Janin 1997). It appears, therefore, that the total buried surface area of 1454 Å2 reflects a true dimer interface in Mja10b.
Figure 3.
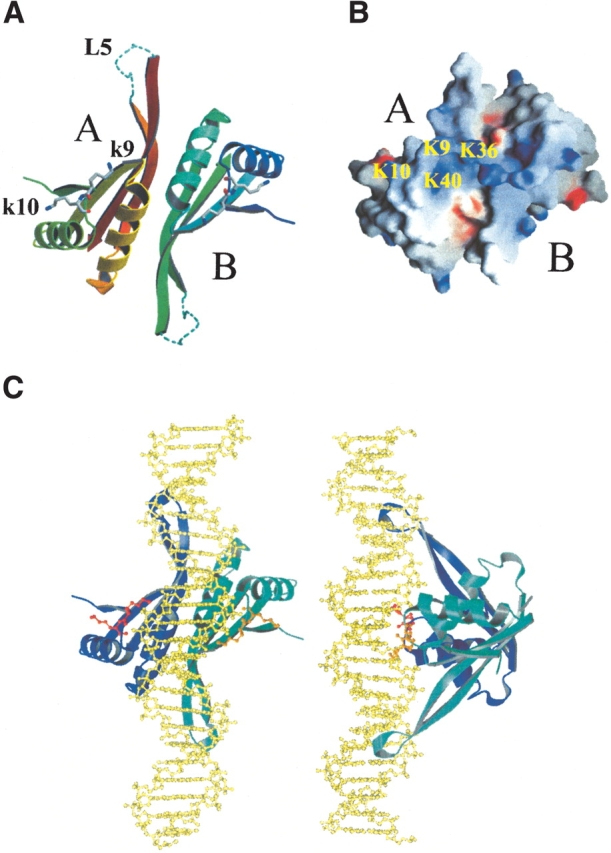
(A) The DNA binding surface of the Mja10b dimer. Loop L5 was modeled using the Sso10b structure and is shown as a dashed line. The residues of Lys9 and Lys10 are shown as ball and stick. (B) The electrostatic potential surface of the DNA binding surface of an Mja10b dimer. Positively and negatively charged residues are indicated in blue and red, respectively. Four lysines (Lys9, Lys10, Lys36, and Lys40) are labeled in molecule A. (C) A DNA binding model for the Mja10b dimer. B-form DNA is colored gold and is shown as ball and stick. The Mja10b dimer is shown in blue/cyan. Residues K9 and K10 of each subunit are shown as ball and stick, in red and orange, respectively.
The structure of Sso10b was observed to have two different types of monomer association (Wardleworth et al. 2002). The first type results in a compact dimer in which 690 Å2 surface area is buried per monomer, while the second type yields a less compact dimer with only 460 Å2 buried. The Mja10b dimer shown in Figure 3A ▶ is similar to the more compact form of the Sso10b dimer (r.m.s.d. 0.81 Å), albeit with a larger buried surface area. The second type of monomer association, involving helices α1 and α2 of each molecule, is also observed in the Mja10b crystal structure. However, this second type of dimer is considerably less compact that the equivalent Sso10b dimer and buries only 290 Å2 surface area per monomer.
DNA binding surface
Proteins of the Sac10b family bind to double-stranded DNA with little sequence specificity in vitro (Lurz et al. 1986; Foterre et al. 1999; Xue et al. 2000). However, it remains unclear how the proteins interact with DNA. Structural analysis of Sso10b suggests a model for the interaction of a protein dimer with B-form DNA (Wardleworth et al. 2002). As shown for Sso10b, a putative DNA binding surface of Mja10b is formed by loop L1, helix α2, loop L3, strands β3 and β4 and loop L5 in each monomer. It is proposed that, in a dimer, loops L5 interact with the minor groove of the DNA (Bell et al. 2002) so that the beginning portions of helices α2 would be placed to interact with the major groove and loops L1 would grasp the sides of the DNA double helix (Fig. 3A ▶). Figure 3B ▶ shows the electrostatic potential map of the putative DNA binding surface. Positively charged residues are mainly located in the middle area of the binding surface. There are four lysine residues (Lys9, Lys10, Lys36, and Lys40) from each monomer, which are strictly conserved in the Sac10b family. Residues Lys9 and Lys10 at the top of loop L1 are equivalent to Lys16 and Lys17 of Sso10b, which are known from site-directed mutagenesis studies to be important for DNA binding (Bell et al. 2002).
Gel retardation assays have shown that Ssh10b has two distinctively different modes of DNA binding, depending on the binding density (Xue et al. 2000). In the low-binding density mode, Ssh10b exhibits a binding size of 10–12 bp of DNA, whereas, in the high-binding density mode, the protein appears to bind DNA with smaller binding sizes (R. Guo, H. Xue, and L. Huang, unpubl.). In agreement with the binding size of Ssh10b in the low-binding density mode, the protein was found to bind with similar affinity to DNA duplexes of 10 bp or larger, but only weakly to an 8-bp duplex (R. Guo, H. Xue, and L. Huang, unpubl.). To account for the two DNA binding modes, we propose two corresponding models for the interaction of Sac10b proteins with DNA on the basis of the structural data for Mja10b and Sso10b. In the low-binding density mode, dimers of the protein bind B-form DNA in a head-to-tail fashion along the axis of the double DNA helix (a head-to-tail model, Fig. 3C ▶). Each dimer covers an area slightly larger than a complete turn of the DNA. Only minor distortion in DNA may occur to accommodate steric clashes between the DNA and the protein in this model. In the high-binding density mode, the protein binds to the DNA in a side-by-side fashion and forms a right-handed spiral (a side-by-side model). In this model, three consecutive dimers can complete a turn around the DNA helix, as suggested previously (Wardleworth et al. 2002). A binding stoichiometry of ~5 bp may be achieved in this model. Significant conformational changes in the protein-bound DNA appear to be required to avoid steric clashes in the side-by-side model. This is consistent with the finding that Ssh10b is capable of significantly constraining negative DNA supercoils at elevated temperatures only when a critical protein/DNA mass ratio is reached (Xue et al. 2000). Sac7b forms a 1:1 complex with DNA and causes a sharp kink in the bound DNA (Robinson et al. 1998). In contrast, Sac10b proteins appear to form a dimer for DNA binding. Furthermore, from the models discussed here, it seems unlikely that the DNA would be kinked on binding to Sac10b proteins. Clearly, the issue of the interaction of the Sac10b proteins with DNA needs to be addressed through a structural analysis of a protein–DNA cocrystal.
Loop comparison
Despite the overall structural similarity between Mja10b and Sso10b, the two proteins show variations in loops (Fig. 4 ▶). There are five loops in Mja10b. Among them, loops L1 and L3, both of which are involved in DNA binding, are very similar to the corresponding loops in Sso10b. However, Mja10b differs from Sso10b in other loops, especially loop L2. Loop L2 in Sso10b consists almost entirely of polar residues (Asn, Gln, Gly, Ser), and is tilted away from the N terminus. By comparison, the corresponding loop in Mja10b is shorter by a few residues. The Methanococcus protein does not have a residue equivalent to Val34 in Sso10b, and residue Ser35 in Sso10b is replaced with an Asp27 in Mja10b. Residues Asp31, Gln32, and Gly33 in Sso10b are also substituted in Mja10b, and form part of the α1 helix. Loop L4 is of similar length in the two proteins, but residue Lys64 in Sso10b has no equivalent in Mja10b and Pro63 in Sso10b is replaced with Lys54 in Mja10b. Loop L5 is suggested to interact with target DNA. By structural comparison and primary sequence alignment, we observe that residues Ser79 and Gln80 in this region of Sso10b are replaced by Asn70 and Pro71, respectively, in Mja10b.
Figure 4.

Superposition of Mja10b and Sso10b. Cα traces of Mja10b (red) and Sso10b (blue) are shown. The five loops are labeled as L1–L5.
It is worth noting that loop L5 is absent in the Mja10b structure owing to its poor electron density, which suggests that the loop may be highly flexible compared to the rest of the structure. A similar observation was made on one monomer (molecule B) of the hexagonal crystal form of an Sso10b dimer, in which the average B-factor of the loop was two times higher than the average B-factor of the entire monomer (Wardleworth et al. 2002). However, the loop of monomer A of Sso10b is ordered, with lower than average B-factors, presumably as a result of stabilization by crystal packing because the loop has extensive contacts with symmetry-related monomers. Structural comparison shows that loop L5 apparently has different conformation in the two molecules of Sso10b. Several DNA binding proteins have been shown to employ flexible loops or arms in their interaction with double-stranded DNA. Escherichia coli histone-like protein HU possesses two flexible arms that embrace one helical turn of B-DNA over the minor groove (Pinson et al. 1999). Therefore, it appears that the flexibility of loop L5 may be required for a Sac10b protein to dock onto its target DNA.
In general, loops L1, L3, and L5 play an important role in interaction with target DNA; therefore, residues in loops L1 and L3 are strictly conserved. In loop L5, there are residue substitutions but not deletion. However, both residue substitution and deletion were observed in the loops L2 and L4. It seemed that the functional portions of Sac10b proteins are structurally conserved, and other loops are more variable in the evolution.
Thermostability
Several structural features have been suggested to contribute to the stability of thermostable proteins at high temperature. These features include the increase in the number of hydrogen bonds and ion pairs (Vogt et al. 1997), a decrease of accessible surface area (Chan et al. 1995), and oligomerization (Walden et al. 2001). To learn more about the structural basis of the thermal stability of Mja10b, we compared the structure of the protein to those of other proteins with similar topology using the program DALI (Holm and Sander 1993). Targets were chosen on the basis of Z-score, with Z < 2.0 being considered structurally dissimilar. In addition to Sso10b (PDB ID: 1H0X; Wardleworth et al. 2002), the C-terminal domain of the translation initiation factor IF3 from Bacillus stearothermophilus (PDB ID: 1TIG; Biou et al. 1995) and the N-terminal domain of DNase I (PDB ID: 2DNJ; Suck et al. 1988) were employed in our comparison. IF3 and DNase I had respective DALI Z-scores of 7.9 and 5.5. All of these proteins are thermostable, with the exception of DNase I-N (Moore 1981).
Ion pairs
Recent studies on the high-resolution crystal structures of thermostable proteins have provided a considerable insight into the role of ionic interactions in protein stabilization. Thermostable proteins appear to have more ionic interactions and more extensive ion-pair networks. We determined the number of ion pairs in Mja10b, Sso10b, IF3-C, and DNase I-N using a cutoff distance of 4.0 Å between oppositely charged residues (Barlow and Thornton 1983). Although the four proteins are similar in size, they vary in amino acid composition. The three thermostable proteins contain more charged residues than DNase I-N. The numbers of ion pairs per residue are 0.063, 0.079, 0.057, and 0.012 for Mja10b, Sso10b, IF3-C, and DNase I-N, respectively. The numbers for the three thermostable proteins are significantly higher than the average value (~0.04) for proteins (Barlow and Thornton 1983). An increase in the number of ion pairs has been observed in aldehyde ferredoxin oxidoreductase from Pyrococcus furiosus (Chan et al. 1995) and xylose isomerase from Thermus caldophilus (Chang et al. 1999). Taken together, these data suggest that ionic interactions may play a key role in the stabilization of a protein at elevated temperatures.
It has been reported that the helical conformation is stabilized by (i to i + 4) or (i to i + 3) glutamate–lysine intrahelix ion pairs in a short model peptide system (Marquee and Baldwin 1987). In Mja10b, an intrahelix ion pair (Glu46–Arg49) exists in the helix α2, which will contribute to reinforce the stability of helix α2 (Fig. 5 ▶). Interestingly, both Glu46 and Arg49 are strictly conserved among members of the Sac10b family. Lys60 and Glu62 form an intrastrand ion pair in strand β3. These two amino acid residues are conserved in all Sac10b proteins except for Mth10b and Ape10b2. Remarkably, the covariance is observed in other Sac10b proteins that Lys60 and Glu62 are replaced by the glutamic acid (or aspartic acid) and the arginine (or lysine), respectively. This ion pair serves to stabilize strand β3. The presence of a selective pressure to maintain this ion pair suggests its importance to the structural and functional stability of Mja10b.
Figure 5.

A stereo figure showing ion-pair interactions in Mja10b. An intrahelix ion-pair is located on helix α2 between E46–R49. The ion-pairs of K59–E28 and R34–D66 connect strands β2 and β3. The ion-pair network of E62–K60–E82 connects strands β3 and β4.
There are three other ion pairs connecting secondary structures in Mja10b. Glu28–Lys59 and Arg34–Asp66 are interstrand ion pairs between the β2 and β3 strands (Fig. 5 ▶). The ion pair of Glu28–Lys59 is conserved in most Sac10b proteins, and the covariance is observed in Tac10b and Tvo10b. The Arg34–Asp66 ion pair does not exist in Ssh10b, Sso10b, and Ape10b1, which is formed in other members of the Sac10b family with Asp66 replaced by the glutamic acid. The ion pair between Lys60–Glu82 is an ion pair between the β3 and β4 strands, and is unique to Mja10b. In fact, an ion-pair network is formed among residues of Glu82, Lys60, and Glu62.
Accessible surface area (ASA)
In general, a decrease in the accessible surface area (ASA) of a protein is favorable to the stability of the protein (Chan et al. 1995). The ASA of Mja10b was compared with those of the other three proteins (Table 1). These proteins vary in the size of accessible surface area, and hydrophobic, charged and polar ASAs of the four proteins were calculated and compared. In the four proteins analyzed in this study, charged and polar residues account for 66%–78% of the accessible surface. It is noteworthy that, compared to DNase I-N, the proteins from the thermophililes have fewer polar residues and more charged residues that are exposed to solvent. Therefore, the solvation effect on the surface will be enhanced in the thermostable proteins. Otherwise, a relative increase in charged surface area is consistent with the finding that thermostable proteins possess more surface ion pairs than the thermolabile protein. Our data suggest that not only the size of accessible surface area but also the property of ASA are close related to the stability of Mja10b.
Table 1.
Comparison of Mja10b with three proteins with similar topology
| Mja10b | Sso10b | IF3-C | DNase I-N | |
| Total no. of residues | 80 | 89 | 87 | 86 |
| Amino acid composition | ||||
| % of charged residues | 30.0 | 26.9 | 36.8 | 24.4 |
| % of hydrophobic residues | 47.5 | 44.9 | 41.4 | 43.0 |
| % of polar residues | 22.5 | 28.2 | 21.8 | 32.6 |
| Accessible surface area (Å2)a | 50/70 | 5880 | 5400 | 5520 |
| Surface area of charged residues | 2390 (47.1%) | 2630 (44.6%) | 3230 (59.7%) | 2000 (36.1%) |
| Surface area or hydrophobic residues | 1680 (33.2%) | 1450 (24.6%) | 1190 (21.9%) | 1500 (27.1%) |
| Surface area of polar residues | 1000 (19.3%) | 1820 (30.8%) | 1000 (18.4%) | 2030 (36.8%) |
| No. of ion pairs | 5 | 7 | 5 | 1 |
| No. of ion pairs/residue | 0.063 | 0.079 | 0.057 | 0.012 |
| No. of hydrogen bonds (3.5 Å cutoff) | ||||
| Total | 129 | 141 | 149 | 157 |
| Main chain to main chain | 100 | 119 | 120 | 110 |
| Main chain to side chain | 26 | 18 | 22 | 37 |
| Side chain to side chain | 3 | 4 | 7 | 10 |
a Numbers in parentheses correspond to the percentage of total surface area.
Dimerization
Strong hydrophobic interactions at the dimer interface have been shown by site-directed mutagenesis to be important determinants of thermostability of dimeric 3-isopropylmalate dehydrogenases from thermus thermophilus (Kirino et al. 1994). In addition, the tetramerization of triosephosphate isomerase from Pyrococcus woesei was found to bury a significant portion of hydrophobic surface area (Walden et al. 2001). It appears that dimer association in Mja10b via strong hydrophobic interactions may contribute to the thermostability of the protein. First, the buried area in each monomer is equal to one-third of the hydrophobic ASA, which will be favorable to the stability of Mja10b. Second, formation of an Mja10b dimer with an interaction surface area of 1454 Å2 minimizes the solvent-accessible surface area to volume ratio by 16% (0.271 Å−1 versus 0.317 Å−1 for the monomeric Mja10b), a feature that has been related to thermostability in the dimeric aldehyde ferredoxin reductase from Pyrococcus furiosus (Chan et al. 1995).
Hydrogen bonds
An increase in the number of hydrogen bonds has been reported in various thermophilic proteins (Vogt et al. 1997). As shown in Table 1, the numbers of hydrogen bonds in Mja10b, Sso10b, IF3-C, and DNase I-N are 129, 141, 149, and 157, respectively. Among these hydrogen bonds, those formed between main-chain atoms are 100, 119, 120, and 110, those between main-chain and side-chain atoms are 26, 18, 22, and 37, and those between side-chain atoms are 3, 4, 7, and 10 for Mja10b, Sso10b, IF3-C, and DNase I-N, respectively. Therefore, the present study suggests that hydrogen bonds are unlikely to be the major factor contributing to the enhanced thermostability of the three thermophilic proteins.
Materials and methods
Crystallization and X-ray data collection
The purification and preliminary X-ray crystallography analysis of Mja10b has been described elsewhere (Wang et al. 2002). A set of data was collected on beamline 19-ID of the Advanced Photon Source (APS), Argonne, to 2.0 Å resolution. All data were integrated using DENZO/HKL and scaled and merged with the SCALEPACK package (Otwinowski and Minor 1997). Crystal parameters and data-collection statistics are summarized in Table 2.
Table 2.
Data collection, refinement and model statistics
| Data Collection | |
| Space group | P6522 |
| Unit cell parameters (Å, °) | a=b=51.32, c=125.02, γ=120 |
| Solvent content (%) | 49.1 |
| Resolution (Å) | 50–2.0 (2.06–2.0) |
| Total observations | 91002.1 |
| Unique reflections | 6946.1 |
| Redundancy | 13.1 |
| Average l/σ(I) | 27.0 (6.2) |
| Rmergea (%) | 4.9 (31.1) |
| Completeness (%) | 96.6 (99.1) |
| Refinement | |
| Reflections | 5539 (work set) |
| 678 (test set) | |
| Resolution range (Å) | 20.0–2.0 |
| Number of atoms (Average B-values [A2]) | |
| Protein | 619 (37.7) |
| Water | 24 (38) |
| R-factor (%) work | 20.9 |
| R-factor (%) freeb | 26.4 |
| RMSD bonds (Å) | 0.009 |
| RMSD angles (°) | 1.405 |
| Ramachandran plot (%) | |
| Most favored regions | 91.7% |
| Additional allowed regions | 8.3% |
aRmerge = Σ | I − <I> |/ΣI; Numbers in parentheses correspond to the outer shell.
bRfree = Σ | | Fo | − | Fc | |/Σ | Fo |, where Fo and Fc are the observed and calculated structure factor amplitudes, and Rfree was calculated with 10% of the reflections not used during refinement.
Structure determination and refinement
The structure of Mja10b was determined by molecular replacement method using the Sso10b structure as a search model (PDB number 1h0x). Cross-rotation and translation searches were performed using CNS (Brünger et al. 1998), and the model was built into the electron density map using the program O (Jones et al. 1991). The Mja10b structure was initially subjected to simulated-annealing refinement using a starting temperature of 5000 K. A total of 10% of the reflections were randomly selected prior to the refinement to calculate Rfree values for cross-validation. The model was then subjected to alternate cycles of manual rebuilding in O and the minimization using CNS. After extensive rebuilding with O, solvent molecules were added in the final rounds of refinement and individual temperature factors were refined. The final refinement statistics are summarized in Table 2. The final model shows good stereochemistry as defined by the Ramachandran plot calculated in PROCHECK (Laskowski et al. 1993), with 91.7% of the residues in the most favored regions and none in the disallowed regions.
Structural analysis
The exposed surface areas were calculated using the program GRASP (Nichol et al. 1991) with a probe radius of 1.4 Å. Molecular volumes were also calculated using GRASP. Hydrogen bonds and ion pairs between protein atoms were calculated using the CCP4 package (Collaborative Computational Project 4 1994) with the default parameters for distances and angles. Ion pairs were assigned when atoms of opposite charge were found within 4 Å of each other. Figures 1 ▶, 3A ▶, 3C ▶, 4 ▶, and 5 ▶ were created using BOBSCRIPT (Esnouf 1997). Figure 3B ▶ was produced with GRASP.
Acknowledgments
We are grateful to Dr. David Boone (Portland State University) for the generous gift of the genomic DNA from Methanococcus jannaschii. We are also grateful to Rongguang Zhang (APS) for help with data collection. This work was supported by the following grants: National Natural Science Foundation of China (NSFC) Nos. 39970155, 30170197; Project “863” No. 2001AA233011; Project “973” No. G1999075602.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03325103.
References
- Balley, K.A., Chow, C.S., and Reeve, J.N. 1999. Histone stoichiometry and DNA circularization in archaeal nucleosomes. Nucleic Acids Res. 27 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, D.J. and Thornton, J.M. 1983. Ion pairs in proteins. J. Mol. Biol. 168 867–885. [DOI] [PubMed] [Google Scholar]
- Bell, S.D., Botting, C.H., Wardleworth, B.N., Jackson, S.P., and White, M.F. 2002. The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science 296 148–151. [DOI] [PubMed] [Google Scholar]
- Biou, V., Shu, F., and Ramakrishnan, V. 1995. X-ray crystallography shows that translational initiation factor IF3 consists of two compact α/β domains linked by an α-helix. EMBO J. 14 4056–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., GrosseKunstleve, R., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S. et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Chan, M.K., Mukund, S., Kletzin, A., Adams, M.W.W., and Rees, D.C. 1995. Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science 267 1463–1469. [DOI] [PubMed] [Google Scholar]
- Chang, C.S., Park, B.C., Lee, D.S., and Suh, S.W. 1999. Crystal structure of thermostable xylose isomerases from Thermus caldophilus and Thermus thermophilus: Possible structural determinants of thermostability. J. Mol. Biol. 288 623–634. [DOI] [PubMed] [Google Scholar]
- Choli, T., Liebold, B.W., and Reihardt, R. 1988. Microsequence analysis of DNA-binding protein 7a, 7b, and 7e from the Archaebacterium Sulfolobus acidocaldrius. J. Biol. Chem. 263 7087–7093. [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Decanniere, K., Babu, A.M., Sandman, K., Reeve, J.N., and Heinemann, U. 2000. Crystal structures of recombinant histones HMfA and HMfB from the hyperthermophilic archaeon Methanothermus fervidus. J. Mol. Biol. 303 35–47. [DOI] [PubMed] [Google Scholar]
- Dick, J., and Reinhardt, R. 1986. The structure of DNA-binding proteins from eu- and archaebacteria. In Bacterial chromatin (eds. C.O. Gualerz and C.L. Pon), pp. 185–218. Springer-Verlag, New York.
- Edmondson, S.P. and Shriver, J.W. 2001. DNA binding proteins Sac7d and Sso7d from Sulfolobus. Methods Enzymol. 334 129–145. [DOI] [PubMed] [Google Scholar]
- Eeling, P.J. and Doolittle, W.F. 1995. Archaea: Narrowing the gap between prokaryotes and eukaryotes. Proc. Natl. Acad. Sci. 92 5761–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. BOBSCRIPT: An extensively modified version of MOLSCRIPT that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15 132–143. [DOI] [PubMed] [Google Scholar]
- Foterre, P, Confalonier, F., and Knapp, S. 1999. Identification of the gene encoding archeal-specific DNA-binding protein of the Sac10b family. Mol. Microbiol. 32 32669–32670. [DOI] [PubMed] [Google Scholar]
- Grayling, R.A., Sandman, K., and Reeve, J.N. 1996. Histone and chromatin structure in hyperthermophilic archaea. FEMS Microbiol. Rev. 18 203–213. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Janin, J. 1997. Specific versus non-specific contacts in protein crystals. Nat. Struct. Biol. 4 973–974. [DOI] [PubMed] [Google Scholar]
- Jones, W.J., Leugh, J.A., Mayer, F., Woese, C.R., and Wolfe, R.S. 1983. Methanococcus jannaschii sp. nov., an extremely thermarine hydrothermal vent. Arch. Microbiol. 136 254–261. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kirino, H., Aoki, M., Aoshima, M., Hayashi, Y., Ohba, M., Yamagishi, A., Wakagi, T., and Oshima, T. 1994. Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur. J. Biochem. 220 275–281. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Luger, K., Maeder, A.W., Richmond, R.K., Sargent, D.F., and Richmond, T.J. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389 251–260. [DOI] [PubMed] [Google Scholar]
- Lurz, R., Grote, M., Dijk, J., and Dobrinski, B. 1986. Electron microscopic study of DNA complexes with proteins from the archaebacterium Sulfolobus acidocaldarius. EMBO J. 5 3715–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqusee, S. and Baldwin, R.L. 1987. Helix stabilization by Glu−•••Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. 83 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee, J.G., Edmondson, S.R., Datta, P.K., Shriver, J.W., and Gupta, R. 1995. Gene cloning, expression, and characterization of the Sac7 protein from the hyperthermophile Sulfolobus acidocaladarius. Biochemistry 34 10063–10077. [DOI] [PubMed] [Google Scholar]
- Moore, S. 1981. Pancreatic Dnase. In The enzymes (ed. P.D Boyer), 3rd ed., pp. 281–296, Vol. 14. Academic Press, New York.
- Nichol, A., Sharp, K.A., and Honig B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pereira, S.L., Grayling, R.A., Lurz, R., and Reeve, J.N. 1997. Archaeal nucleosomes. Proc. Natl. Acad. Sci. 94 12633–12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinson, V., Takahashi, M., and Rouviere-Yaniv, J. 1999. Differential binding of the Escherichia coli HU, homodimeric forms and heterodimeric form to linear, gapped and cruciform DNA. J. Mol. Biol. 287 485–497. [DOI] [PubMed] [Google Scholar]
- Robinson, H., Gao, Y.G., McCrary, B.S., Edmondson, S.P., Shriver, J.W., and Wang, A.H.J. 1998. The hyperthermophile chromosomal protein Sac7d sharply kinks DNA. Nature 392 202–205. [DOI] [PubMed] [Google Scholar]
- Sandman, K., Krzycki, J.A., Dobrinski, B., Lurz, R., and Reeve, J.N. 1990. HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc. Natl. Acad. Sci. 87 5788–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suck, D., Lahm, A., and Oefner, C. 1988. Structure refined to 2 Å of a nicked DNA octanucleotide complex with DNase I. Nature 332 464–468. [DOI] [PubMed] [Google Scholar]
- Tarich, M.R., Sandman, K., Reeve, J.N., and Summers, M.F. 1996. NMR structure of HMfB from the hyperthermophile, Methanothermus fervidus, confirms that this Archaeal protein is a histone. J. Mol. Biol. 255 187–203. [DOI] [PubMed] [Google Scholar]
- Vogt, G., Woell, S., and Argos, P. 1997. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 269 631–643. [DOI] [PubMed] [Google Scholar]
- Walden, H., Bell, G.S., Russell, R.J.M., Siebers, B., Hensel, R., and Taylor, G.L. 2001. Tiny TIM: A small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol. 306 745–757. [DOI] [PubMed] [Google Scholar]
- Wang, G.G., Guo, R., Bartlam, M., Xue, H., Yang, H.T., Liu, Y.W., Huang, L., and Rao, Z.H. 2002. Expression, purification, crystallization and preliminary X-ray analysis of a DNA binding protein from Methanococcus jannaschii. Acta Crystallogr. D 58 1240–1242. [DOI] [PubMed] [Google Scholar]
- Wardleworth, B.N., Russell, R.J.M., Russell, S.D., Raylor, G.L., and White, M.F. 2002. Structure of Alba: An archaeal chromatin protein modulated by acetylation. EMBO J. 21 4654–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, K., Chishiro, K., Kitamura, K., and Suzuki, Y. 1991. Proline residues responsible for thermostability occur with high frequency in the loop regions of an extremely thermostable oligo-1, 6-glucosidase from Bacillus thermoglucosidasius KP1006. J. Biol. Chem. 266 24287–24294. [PubMed] [Google Scholar]
- Woese, C.R., Kandler, O., and Wheelis, M.L. 1990. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, Eucarya. Proc. Natl. Acad. Sci. 87 4576–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, H., Guo, R., Wen, Y.F., Liu, D.X., and Huang, L. 2000. An abundant DNA binding protein from the hyperthermophilic archaeon Sulfolobus shibatae affects DNA supercoiling in a temperature-dependent fashion. J. Bacteriol. 182 3929–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]