

Abstract
The ribosomal protein S28E from the archaeon Methanobacterium thermoautotrophicum is a component of the 30S ribosomal subunit. Sequence homologs of S28E are found only in archaea and eukaryotes. Here we report the three-dimensional solution structure of S28E by NMR spectroscopy. S28E contains a globular region and a long C-terminal tail protruding from the core. The globular region consists of four antiparallel β-strands that are arranged in a Greek-key topology. Unique features of S28E include an extended loop L2–3 that folds back onto the protein and a 12-residue charged C-terminal tail with no regular secondary structure and greater flexibility relative to the rest of the protein. The structural and surface resemblance to OB-fold family of proteins and the presence of highly conserved basic residues suggest that S28E may bind to RNA. A broad positively charged surface extending over one side of the β-barrel and into the flexible C terminus may present a putative binding site for RNA.
Keywords: Heteronuclear NMR, Methanobacterium thermoautotrophicum, ribosomal protein S28E, Northeast Structural Genomics Consortium
The open reading frame mth256 from Methanobacterium thermoautotrophicum encodes a 7.7 kD protein, S28E, a component of the 30S ribosomal subunit. S28E exhibits high sequence conservation among archaea, and homologs have been identified in yeast, fungi, plants, and mammals (Fig. 1 ▶). The sequence homolog of S28E proteins across species in archaea and eukaryotes implies a key role of S28E in protein synthesis throughout evolution. However, the function of S28E in the ribosome is unknown and genetic and biochemical data are scarce. A study in Prunus persica suggests that plant S28E might control expression of the mature mRNAs at the level of splicing and turnover of the precursor RNA (Giannino et al. 2000).
Figure 1.

Sequence alignment of S28E from various species. Archaea: Methanothermobacter thermautotrophicum (O26356), Pyrococcus horikoshii (O74014), Thermoplasma volcanium (Q97BK7), Aeropyrum pernix (Q9Y9A6), Archaeoglobus fulgidus (O29493), Methanocaldococcus jannaschii (P54065). Eukaryote: Kluyveromyces marxianus (P33286), Schizosaccharomyces pombe (Q10421), Saccharomyces cerevisiae (P02380), Zea mays (P46302), Homo sapiens (P25112), Ictalurus punctatus (Q90YP3). Identical and similar residues are highlighted in black and gray, respectively. The NMR-derived secondary structural elements of S28E are illustrated above the alignment.
Ribosomes are the essential machinery for protein synthesis in all organisms (Ramakrishnam 2002). A typical archaeal ribosome contains 68 different proteins (28 in the 30S subunit and 40 in the 50S subunit), as compared to 57 proteins in the bacterial ribosome and 78 proteins in a eukaryotic ribosome (Lecompte et al. 2002). Though the crystal structure of the 50S subunit from Haloarcula marismortui has been reported (Ban et al. 2000), a limited amount of structural information is available for the 30S archaeal subunit. Archaeal and eukaryotic S28E proteins range between 46 and 91 amino acids in length, have no sequence homologs with a known 3D structure and have no counterparts in bacteria, making them ideal and important targets for structural genomics. S28E from both M. thermoautotrophicum and Pyrococcus horikoshii were selected by the Northeast Structural Genomics Consortium (www.nesg.org) as targets for structure determination. The two proteins share 64% sequence identity. In this paper, we report the solution structure of S28E from M. thermoautotrophicum. Comparison of the structures from both species (Aramini et al. 2003) provides clues to its putative function characterization.
Results and Discussion
Assignment and structure determination
Sequence specific resonance assignments were obtained for all residues that appeared in the 15N-HSQC spectrum. Fifty-six out of 64 expected backbone peaks were observed in the 15N-HSQC recorded at 25°C and pH 6.5. The eight missing amide resonances were Asp 2, Gly 17, Met 18, Thr 19, Gly 20, Glu 21, Arg 33 and Leu 56. Ninety-six percent of the C and H resonances for all side chains have been assigned.
The three-dimensional structure of S28E was generated and refined using automated CANDID/DYANA iterative calculations followed by restrained molecular dynamic simulations in explicit water with the program CNS. Ninety-two percent of the manually picked NOE cross-peaks were assigned and a total of 1146 NOE distance restraints were used in DYANA refinement calculations. The value of the DYANA target function was typically between 0.47 Å2 and 0.56 Å2. The water refinement protocol improves the structure quality by removing interatomic steric and electrostatic clashes. The 20 best water-refined structures were selected to represent the S28E structure in solution, and the structural parameters are summarized in Table 1. The average root-mean-square deviation (r.m.s.d.) to the mean structure for the backbone of the ordered residues was 0.49 Å. None of these structures had NOE violations >0.5 Å or dihedral angle violations >5.0°. The Ramachandran plot of the φ and ϕ angles for the 20 structures shows 94.8% of the φ and ϕ angles to be in the most favored regions, 3.4% in the additional allowed regions, and 1.7% in the generously allowed regions.
Table 1.
Structural statistics for the ensemble calculated for S28E
| Distance restraints | |
| All | 1146 |
| Intraresidue | 266 |
| Sequential (|i − j| = 1) | 275 |
| Medium range (2 ≤ |i − j| ≤ 4) | 103 |
| Long range (|i − j| > 4) | 502 |
| Hydrogen bonds | 16 × 2 |
| Dihedral angle restraints | |
| All | 98 |
| φ | 49 |
| ϕ | 49 |
| DYANA target function, (Å2) | 0.53 ± 0.027 |
| Number of violations | |
| Distance restraints (>0.5 Å) | 0 |
| Dihedral angle restraints (>5°) | 0 |
| R.m.s.d. from experimental restraints | |
| Distance (Å) | 0.011 ± 0.0012 |
| Dihedral angle (°) | 0.54 ± 0.12 |
| R.m.s.d. from idealized covalent geometry | |
| Bond (Å) | 0.0037 ± 0.0001 |
| Bond angles (°) | 0.48 ± 0.016 |
| CNS energy (kcal/mole) | |
| Total | −2197 ± 78 |
| van der Waals | −234 ± 14 |
| Electrostatic | −2440 ± 84 |
| Pairwise r.m.s.d. | |
| All residuesa | |
| Backbone atoms | 0.95 ± 0.15 |
| All heavy atoms | 1.48 ± 0.13 |
| Ordered regionsb | |
| Backbone atoms | 0.49 ± 0.13 |
| All heavy atoms | 0.99 ± 0.11 |
| Ramachandran plot (%)c | |
| Residues in most favored regions | 94.8 |
| Residues in additional allowed regions | 3.40 |
| Residues in generously allowed regions | 1.70 |
| Residues in disallowed regions | 0.00 |
S28E consists of an ensemble of the 20 lowest energy structures out of 200 calculated.
a r.m.s.d. values for residues 4–56.
b Only residues in β-strands are included (residues 4–13, 22–29, 37–44, and 51–56).
c Dihedral angle characteristics from PROCHECK-NMR.
Overall fold
The solution structure of S28E is represented in Figure 2 ▶. The protein is composed of four β-strands connected by loops and a long disordered C-terminal tail. The strands are arranged with a Greek-key topology in the order 3–2–1–4 and constitute the central core of the protein. β-Strand 1 contains a β-bulge at residue Ile 10 that is protected from hydrogen exchange and is probably involved in hydrogen bond interaction. Protruding from the core are three loops of 8, 6, and 6 residues, respectively. Loop L1–2 is composed of residues Lys 14–Glu 21. Residues Gly 17–Glu 21 in loop L1–2 display no observable peaks in the 15N-HSQC spectrum, and assignments of Gly 17–Gly 20 are missing, likely due to line broadening arising from intermediate time scale conformational exchange. Thus, the atomic coordinates of the L1–2 loop are poorly defined (Fig. 2 ▶). Loop L2–3 consists of residues Asp 31–Gly 36 and curves back onto the β-barrel core. Its conformation is defined by distinct NOE contacts between Asp 34 and Arg 37. The loop is flanked by glycine residues (Gly 32 and Gly 36) that appear to act as hinges. Loop L3–4 consists of residue Pro 46–Gly 50 and packs onto the top of the barrel. The 12 residues in the C terminus (Asp 57–Pro 68) display chemical shifts close to random coil values, intraresidue NOEs only, exchange cross-peaks with water resonance, and no diagonal peaks in a 15N-edited NOESY spectrum. Consequently, this region appears to be disordered, and probably exhibits a greater level of internal motion than the folded core of the protein.
Figure 2.

Solution structure of S28E from Methanobacterium thermoautotrophicum. (A) Ribbon representation of the lowest-energy structure of S28E. (B) Stereoview of an ensemble of 20 refined structures represented in an orientation similar to A. The backbone atoms are colored blue for β-strands and gray for loops and terminus.
Although the extended C-terminal tail projects away from the core of the structure, the central domain is compact, with many hydrogen bonds. The well-defined hydrophobic core of S28E contains side chains mainly from β-strand: Thr 5, Ala 7, Val 9, and Val 12 of β-strand 1; Met 23, Val 25, and Ile 29 of β-strand 2; Val 43 of β-strand 3; and Leu 53 and Leu 55 of β-strand 4. The side chain of Ile 47 in loop L3–4 makes hydrophobic interactions with that of Val 9, Val 12, Val 25, Val 43, and Leu 53.
The Protein Data Bank (PDB) was searched to identify structures similar to S28E using the DALI server (Holm and Sander 1993). The fold of S28E is reminiscent of the well-known OB-fold involved in oligosaccharide and single-stranded nucleic acid binding. Figure 3 ▶ shows the similarity in topology among S28E, maltose transport protein malk (Diederichs et al. 2000), cytoplasmic molybdate-binding protein ModG (Delarbe et al. 2001), and molybdate/tungstate-binding protein Mop (Wagner et al. 2000). The best match for the globular core of S28E was obtained for maltose transport protein malk (PDB ID code 1G29, Z score 5.4), where the OB-fold motif could be superimposed with an r.m.s.d. for Cα atoms equal to 1.8 Å over 49 equivalent residues. These residues have 20% sequence identity. Mop has a Z score of 4.9 and an r.m.s.d. of 2.2 Å over 48 equivalent residues with 19% sequence identity. The OB-fold defines a five-stranded Greek-key β-barrel capped by an α-helix located between the third and the fourth strands (Murzin 1993). However, the topology of S28E differs significantly from that of the classical OB-fold family. The fold of S28E lacks both β-strand 5 and the α-helix connecting β-strand 3 and 4. The length and conformation of the three loops in S28E are not the same as the member of OB-fold family, and the L2–3 loop folds back onto the backbone, a feature that classical OB-fold does not share.
Figure 3.

Ribbon diagram depicting (A) S28E, (B) maltose transport protein malk (PDB accession number 1G29), (C) cytoplasmic molybdate-binding protein ModG (PDB accession number 1H9J), (D) molybdate/tungestate binding protein mop (PDB accession number 1FR3). The common OB-fold motif and α-helix are shown in blue and red, respectively.
Comparison with P. horikoshii S28E NMR structures
The solution structure of S28E from P. horikoshii has been solved independently by using the automated structure determination program AutoStructure (Aramini et al. 2003). As expected from the 64% sequence identity between the two proteins, the structures of M. thermoautotrophicum S28E and P. horikoshii S28E are virtually identical in the core region. Superposition of the β-barrel motif yields an r.m.s.d. of 0.76 Å for backbone atoms (Fig. 4 ▶). However, a major difference lies in loop L1–2, a poorly defined region in both structures. In loop L1–2, Lys 14 in M. thermoautotrophicum S28E is replaced by Gly and Met 18 by Thr in P. horikoshii S28E. The flexibility introduced by Gly in P. horikoshii S28E may account for significant conformational difference in this region. The side-chain packing in the hydrophobic core is not changed, which is not surprising, because all these residues are highly conserved among S28E proteins.
Figure 4.

Stereoview of the superposition of backbone structure of S28E from M. thermoautotrophicum (blue) and S28E from P. horikoshii (red). The backbone atoms of M. thermoautotrophicum S28E residues P6-V12, V22-I30, I38-M44, and I52-M54 were superimposed on the backbone atoms of P. horikoshii S28E residues P8-I14, V24-I31, V40-R46, and I54-I56.
Implications for S28E function
It is commonly assumed that the primary function of ribosomal proteins is to stabilize specific RNA structures and to assure correct folding of the large rRNAs. The biological role of S28E in the ribosome is not yet clearly defined, but the core region of S28E structure from M. thermoautotrophicum and P. horikoshii reveals a variant of the OB-fold that is a well-characterized RNA-binding motif. One side of the β-barrel and the C-terminal tail of S28E are colonized by 11 out of 12 basic residues as shown in Figure 5 ▶. Among them, Arg 15, Lys 26, Arg 37, and Arg 41 are strictly conserved and create a potential RNA-binding site analogous to the surface of OB-fold proteins that bind oligonucleotides. However, one significant difference between S28E proteins and typical OB-fold proteins is the absence of aromatic residues. The nucleic acid-associated OB-fold proteins, such as Aspartyl-tRNA synthetase (Cavarelli et al. 1993; Eiler et al. 1999) and ribosomal protein S17 (Jaishree et al. 1996; Broderson et al. 2002), and the ssDNA-binding protein Replication Protein A (Bochkarev et al. 1997) use aromatic residues on the binding surface to stack with the nucleotide base. Devoid of aromatic residues, S28E may bind to ribosomal RNA mainly through electrostatic interactions and hydrogen bonding. Alternatively, this site may not bind RNA, but protein, as is found in many crystal structures containing this fold. The second striking feature related to binding is the C-terminal unstructured and flexible tail, rich in charged residues. According to PROSITE analysis (Falquet et al. 2002), the C terminus of S28E proteins possesses a conserved signature sequence motif, E-[ST]-E-R-E-A-R-x-[LI] (DTIREAKEI in M. thermoautotrophicum S28E), which is predicted to form an α-helix (PHD; Rost 1996), possibly induced upon interaction with RNA.
Figure 5.
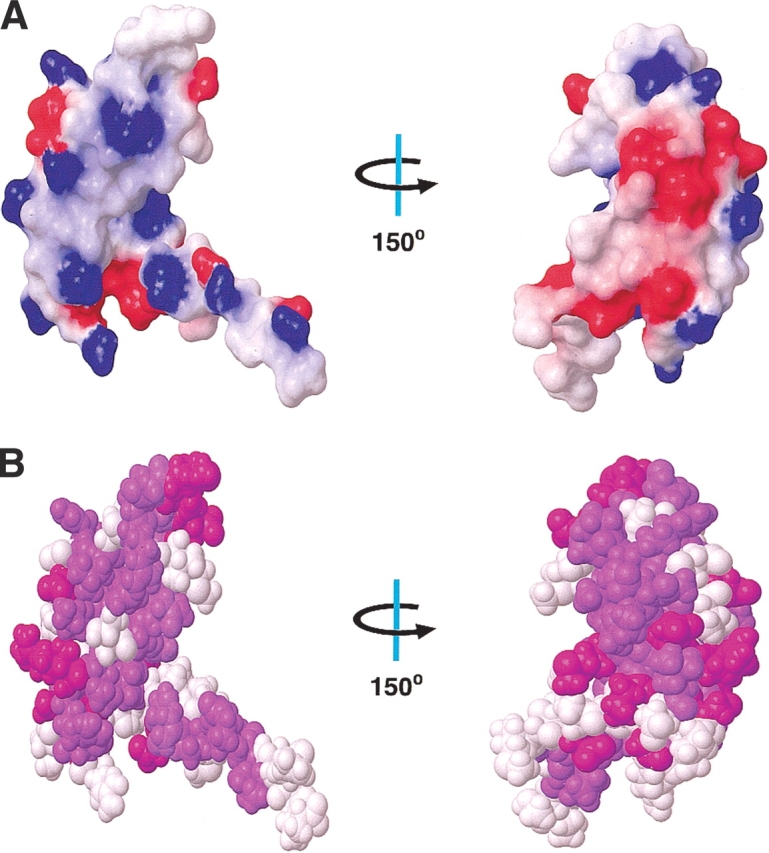
(A) Surface electrostatic potential of S28E as calculated by MOLMOL. Red and blue colors represent negative and positive electrostatic potential, respectively. The left view is in the same orientation as in Figure 2 ▶, whereas the right view is rotated by +150° along the y-axis. (B) Space-filling model of S28E with conserved residues in all S28E proteins colored by purple and conserved residues only in archaea by magenta according to alignment in Figure 1 ▶.
On the opposite face of the protein from the putative RNA-binding site is a negatively charged surface formed by residues Asp 3, Glu 8, Glu 11, Asp 31, Asp 49, and Asp 51. Residues Asp 31 and Asp 51 are well conserved in S28E proteins. Asp 3 is found only in archaea. The other two residues, Glu 8 and Glu 11, are identical among archaeal S28E proteins, whereas in eukaryotes they are replaced by Lys. The surface of P. horikoshii S28E shares the similar feature (Aramini et al. 2003). Thus, this charged surface is conserved at least within archaea (Fig. 5B ▶). The configuration of negatively charged surface among archaea likely defines the binding partners for S28E within the ribosome.
In conclusion, the solution structures of S28E from M. thermoautotrophicum and P. horikoshii provide the first high-resolution picture of this family of proteins. The structural and surface resemblance to OB-fold family of proteins and the presence of highly conserved basic residues suggest that S28E may bind to RNA. A number of structural and sequence properties of S28E provide the basis of future studies to determine the specific interaction of S28E with RNA and other binding partners.
Materials and methods
Protein purification
The gene mth256 coding for ribosome protein S28E (68 amino acids) from M. thermoautotrophicum was subcloned into the pET-15b expression vector with an N-terminal His tag. It was expressed in E. coli strain BL21(DE3) grown in M9-minimal medium supplemented with 15N ammonium chloride (1 g/l) and 13C glucose (2 g/l). The protein was purified to homogeneity using metal affinity chromatography as described previously (Yee et al. 2002). The concentration of protein samples ranged from 1.0 mM to 1.5 mM in an aqueous solution containing 25 mM sodium phosphate (pH 6.5), 450 mM NaCl, 1 mM DTT, 95% H2O/5% D2O.
NMR spectroscopy
All NMR spectra were collected at 25°C on Varian Inova 600-MHz and 750-MHz spectrometers equipped with pulsed field gradient triple-resonance probes. Chemical shifts were referenced to external DSS. Spectra were processed using the program NMRPipe (Delaglio et al. 1995) and analyzed with the program XEASY (Bartels et al. 1995) and SPARKY (Goddard and Kneller 2003). SPSCAN (Glaser and Wüthrich 1997) was used to convert NMRPipe-formatted spectra into XEASY. The backbone assignments were obtained using HNCO, CBCA(CO)NH, HNCACB, HNHA, and 15N-edited NOESY-HSQC spectra. Aliphatic side chain assignments were derived from CCC-TOCSY-NNH, HCC-TOCSY-NNH, HCCH-COSY, and HCCH-TOCSY spectra (Bax et al. 1994; Kay 1997).
Structure calculation
Distance restraints for structure calculations were derived from cross-peaks in a 15N-edited NOESY-HSQC (τm = 150 ms) and a 13C-edited NOESY-HSQC (τm = 120 ms). Slowly exchanging amide protons were monitored by dissolving the protein in D2O and acquiring a series of 15N-HSQC spectra. A 4D CC-NOESY-HMQC (D2O, τm = 125 ms) was recorded in D2O (Vuister et al. 1993). The structure calculation proceeded in three stages. In the first stage, the program CANDID/DYANA (Herrmann et al. 2002) was used for automated assignment and distance calibration of NOE intensities, structure generation calculation with torsion angle dynamics, and automatic NOE upper-distance limit violation analysis. The input for CANDID/DYANA included the chemical shift list, peak lists from a 15N-edited NOESY, and a 13C-edited NOESY and dihedral angle restraints derived from the program TALOS (Cornilescu et al. 1999). NOE peaks were picked and integrated with the program SPARKY. Seven iterative cycles of CANDID assignment and DYANA structure calculation were performed. A total of 92% of the NOE cross-peaks from a 13C-edited NOESY and 86% from a 15N-edited NOESY were assigned in cycle 7. The target function in cycle 1 and cycle 7 was 186 ± 4.55 Å2 and 5.47 ± 0.33 Å2, respectively. The r.m.s.d. drift of the mean coordinate between cycle 1 and cycle 7 was 1.25 Å for the backbone atoms in the ordered region.
In the second stage, hydrogen bond restraints were added on the basis of the structures generated in the initial stage and were restricted to the residues that were clearly in the secondary structure region as judged by NOE pattern and chemical shifts and supported by slowly exchanging amide protons. Structures were refined using default simulated annealing protocol (anneal) in the program DYANA. The 20 calculated structures with the lowest target functions were used to analyze restraint violations and assign additional NOE cross-peaks. Several rounds of calculation were required by deleting or losing consistently violated restraints derived from NOESY cross-peaks that were judged likely to be wrongly assigned, overlapped, or produced by spin diffusion. In the final cycle, the NMR-derived experimental restraints contained 1146 NOEs (266 intraresidue, 275 sequential, 103 medium-range [2 ≤ |i − j| ≤ 4] and 502 long-range [|i − j| > 4] interproton constraint), 32 distance restraints for 16 backbone hydrogen bonds, and 98 dihedral angle restraints. Two hundred structures were calculated, from which the 30 structures with the lowest target functions were selected. The average value of the DYANA target function was 0.53 ± 0.027 Å2.
In the final stage, the 30 selected structures were each subjected to molecular dynamics simulation in explicit water with the program CNS (Brunger et al. 1998). The structures were soaked in an 8 Å layer of TIP3P water molecules (Linge et al. 2003). Details of this protocol will be reported elsewhere. The 20 structures with lowest NOE energies were retained and validated by the program PROCHECK-NMR (Laskowski et al. 1996) and NESG validation software (A. Bhattacharrya and G.T. Montelione, unpubl.). Structures were visualized using the program MOLMOL (Koradi et al. 1996).
Accession numbers
The chemical shifts have been submitted to the BMRB (accession #5620), and the structure ensemble and NOE restraint file has been submitted to the PDB (accession #1NE3).
Acknowledgments
We thank J.M. Aramini and G.T. Montelione for providing atomic coordinates of P. horikoshii S28E and sharing their knowledge of S28E proteins. We thank A. Bhattacharrya and S. Goldsmith-Fishman for structure validation and annotation (www.nesg.org). S28E from M. thermoautotrophicum is target TT744 of the Northeast Structural Genomics Consortium. All spectra were performed at the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy (DOE) Office of Biological and Environmental Research, located at Pacific Northwest National Laboratory and operated by Battelle for the DOE. This work was supported by the Ontario Research and Development Challenge Fund, the NIH Protein Structure Initiative (grant P50-GM62413-02), and MOST NRDP of Korea (M1-0203-00-0020) to W.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03358203.
References
- Aramini, J.M., Huang, Y.J., Cort, J.R., Goldsmith-Fischman, S., Xiao, R., Shih, L., Ho, C.K., Liu, J., Rost, B., Honig, B., et al. 2003. Solution NMR structure of the 30S ribosomal protein S28E from Pyrococcus horikoshii. Protein Sci. (this issue). [DOI] [PMC free article] [PubMed]
- Ban, N., Nissen, P., Hansen, J., Moore, P.B., and Steitz, T.A. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289 905–920. [DOI] [PubMed] [Google Scholar]
- Bartels, C., Xia, T., Billeter, M., Günter, P., and Wüthrich, K. 1995. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6 1–10. [DOI] [PubMed] [Google Scholar]
- Bax, A., Vuister, G.W., Grzesiek, S., Delaglio, F., Wang, A.C., Tschudin, R., and Zhu, G. 1994. Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol. 239 79–105. [DOI] [PubMed] [Google Scholar]
- Bochkarev, A., Pfuetzner, R.A., Edwards, A.M., and Frappier, L. 1997. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature 385 176–181. [DOI] [PubMed] [Google Scholar]
- Broderson, D.E., Clemons Jr., W.M., Carter, A.P., Wimberly, B.T., and Ramakrishnan, V. 2002. Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: Structure of the proteins and their interactions with 16S RNA. J. Mol. Biol. 316 725–768. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system (CNS): A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cavarelli, J., Rees, B., Ruff, M., Thierry, J.C., and Moras, D. 1993. Yeast tRNAAsp recognition by its cognate class II aminoacyl-tRNA synthetase. Nature 262 181–184. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Delarbre, L., Stevenson, C.E., White, D.J., Mitchenall, L.A., Pau, R.N., and Lawson, D.M. 2001. Two crystal structures of the cytoplasmic molybdate-binding protein ModG suggest a novel cooperative binding mechanism and provide insights into ligand-binding specificity. J. Mol. Biol. 308 1063–1079. [DOI] [PubMed] [Google Scholar]
- Diederichs, K., Diez, J., Greller, G., Muller, C., Breed, J., Schnell, C., Vonrhein, C., Boos, W., and Welte, W. 2000. Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis. EMBO J. 19 5951–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiler, S., Dock-Bregeon, A.C., Moulinier, L., Thierry, J.C., and Moras, D. 1999. Synthesis of aspartyl-tRNAAsp in Escherichia coli—A snapshot of the second step. EMBO J. 18 6532–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falquet, L., Pagni, M., Bucher, P., Hulo, N., Sigrist, C.J., Hofmann, K., and Bairoch, A. 2002. The PROSITE database, its status in 2002. Nucleic Acids Res. 30 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannino, D., Frugis, G., Ticconi, C., Florio, S., Mele, G., Santini, L., Cozza, R., Bitonti, M.B., Innocenti, A., and Mariotti, D. 2000. Isolation and molecular characterisation of the gene encoding the cytoplasmic ribosomal protein S28 in Prunus persica [L.] batsch. Mol. Gen. Genet. 263 201–212. [DOI] [PubMed] [Google Scholar]
- Glaser, R. and Wüthrich, K. 1997. Spscan—software for the semi-automatic evaluation of high-resolution NMR spectra of biological macromolecules. http://www.molebio.uni-jena.de/~rwg/spscan/
- Goddard, T.D. and Kneller, D.G. 2003. Sparky—NMR assignment and integration software. http://cgl.ucsf.edu/home/sparky.
- Herrmann, T., Güntert, P., and Wüthrich, K. 2002. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319 209–227. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Jaishree, T.N., Ramakrishnan, V., and White, S.W. 1996. Solution structure of prokaryotic ribosomal protein S17 by high-resolution NMR spectroscopy. Biochemistry 35 2845–2853. [DOI] [PubMed] [Google Scholar]
- Kay, L.E. 1997. NMR methods for the study of protein structure and dynamics. Biochem. Cell. Biol. 75 1–15. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmann, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lecompte, O., Ripp, R., Thierry, J.C., Moras, D., and Poch, O. 2002. Comparative analysis of ribosomal proteins in complete genomes: An example of reductive evolution at the domain scale. Nucleic Acids Res. 30 5382–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge, J.P., Williams, M.A., Spronk, A.E.M., Bonvin, A.M.J.J., and Nilges, M. 2003. Refinement of protein structures in explicit solvent. Proteins 50 496–506. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G. 1993. OB(oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 12 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnam, V. 2002. Ribosome structure and the mechanism of translation. Cell 108 557–572. [DOI] [PubMed] [Google Scholar]
- Rost, B. 1996. PHD: Predicting one-dimensional protein structure by profile based neural networks. Methods Enzymol. 266 525–539. [DOI] [PubMed] [Google Scholar]
- Vuister, G.W., Clore, M., Gronenborn, A.M., Powers, R., Garret, D.S., Tschudin, R., and Bax, A. 1993. Increased resolution and improved spectral quality in four-dimensional 13C/13C-separated HMQC-NOESY-HMQC spectra using pulsed field gradients. J. Magn. Reson. Ser. B 101 210–213. [Google Scholar]
- Wagner, U.G., Stupperich, E., and Kratky, C. 2000. Structure of the molybdate/tungstate binding protein Mop from Sporomusa ovata. Structure 8 1127–1136. [DOI] [PubMed] [Google Scholar]
- Yee, A., Chang, X., Pineda-Lucena, A., Wu, B., Semesi, A., Le, B., Ramelot, T., Lee, G.M., Bhattacharyya, S., Gutierrez, P., et al. 2002. An NMR approach to structural proteomics. Proc. Natl. Acad. Sci. 99 1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]