

Abstract
Nitric oxide (NO) has been implicated as a contributor to the host’s innate defense against viral infections including those affecting the CNS. Reovirus infection of the CNS is a classic experimental system for understanding the pathogenesis of neurotropic viral infection. Infection with serotype 3 strains is associated with perturbations in various cellular signaling pathways including NF-κB and NO plays a regulatory role in many of these same pathways. We therefore examined whether NO production is dysregulated following reovirus serotype 3 strain Abney (T3A) infection of the mouse CNS. Nitric oxide synthase (NOS) activity was significantly higher in brain homogenates from T3A-infected animals compared to mock infected. Increased NOS activity correlated with inducible NOS (iNOS) expression in brain homogenates of T3A-infected animals. Expression of iNOS was confined to areas of viral infection and injury. T3A infection of primary neuronal and glial cultures was also associated with enhanced expression of iNOS. Immunocytochemical studies of primary glial cultures demonstrated that, in addition to its known neuronotropism, T3A was also capable of infecting immature microglial cells. T3A infection did not alter expression of either neuronal or endothelial NOS isoforms in neuronal or glial cultures or in mouse brain. The NO donor S-Nitroso- N-acetyl penicillamine (SNAP) significantly inhibited T3A growth in neuronal cultures, conversely the NOS inhibitor N-ο-Nitro-l-arginine methyl ester hydrochloride (l-NAME) augmented viral growth. Our findings provide the first evidence of reovirus-induced iNOS expression and the first demonstration that NO inhibits mammalian reovirus replication, suggesting that NO may play an antiviral role during reovirus infection.
Keywords: Reovirus, Nitric oxide, Viral encephalitis, Nitric oxide synthase, Neurons, Microglia
Introduction
Nitric oxide (NO) is generated by the oxidation of l-arginine to l-citrulline by NADPH-dependent NO synthases (NOS). Endothelial (eNOS) and neuronal (nNOS) NOS isoforms are expressed constitutively with activity dependent on calcium fluxes and subsequent calmodulin binding. In contrast, inducible NOS (iNOS) is active at basal intracellular calcium concentrations with total activity determined by levels of enzyme, substrate, and co-factor and is thus able to produce greater quantities of NO over a longer time period than either constitutive isoform (Zhang and Snyder, 1995). Excessive NO production leads to the generation of peroxynitrite potentially resulting in DNA damage, protein oxidation, protein nitration, lipid peroxidation, and inactivation of metalloenzymes which results in cell death via apoptosis or necrosis (reviewed in Murad, 1998). Consequently, dysregulation of nitric oxide production has been implicated in several neurodegenerative conditions and has become the focus of novel therapeutic strategies (see Togo et al., 2004).
Inducible NOS is expressed in the CNS following infection with several neurotropic viruses including borna disease virus (Akaike et al., 1995; Koprowski et al., 1993), rabies virus (Akaike et al., 1995; Koprowski et al., 1993), junin virus (Gomez et al., 2003), and herpes simplex virus type 1 (HSV-1) (Fujii et al., 1999; Koprowski et al., 1993; MacLean et al., 1998). The mechanism of iNOS induction during viral infection remains poorly understood although both direct (Adamson et al., 1999; Hori et al., 1999) (induced via viral replication or viral components) and indirect (induced via interferon-γ and other pro-inflammatory cytokines) processes have been suggested (see Akaike and Maeda, 2000).
Antiviral effects of NO have been described in several viral systems (for a review, seeReiss and Komatsu, 1998). Recent studies suggest NO reacts with viral proteins or host cell replicative machinery to exert antiviral activity (seeAkaike and Maeda, 2000). For example, coxsackievirus B3 replication is suppressed by NO through the inactivation of the viral cysteine protease by NO-dependent S-nitrosylation (Saura et al., 1999). iNOS expression is induced in the CNS following junin virus infection of mice and inhibition of iNOS activity in these animals results in increased mortality, but reduced astrocytosis, independent of any effect on titer, suggestive of a protective role for NO against junin virus infection (Gomez et al., 2003). NO does not consistently provide neuroprotection during neurotropic viral infection, however, and may, in certain situations, actually contribute to viral neuropathogenesis. For example, the CNS inflammatory response to borna disease virus infection may be dependent upon peroxynitrite-mediated destruction of the blood-brain barrier (Hooper et al., 2001).
Reoviruses have provided an excellent experimental system for examining virus-host interactions in general and the pathogenesis of viral infections of the CNS in particular (see Clarke and Tyler, 2003). Reoviruses belonging to serotype 3 infect and induce apoptosis in neurons (neuronotropic) (reviewed in Tyler, 1998). We wished to examine the role of NO during reovirus infection because of the significant overlap between mechanisms of reovirus-induced injury and the potential roles for NO in apoptotic mechanisms of cell death. Apoptotic mechanisms of reovirus-induced injury have been explored extensively (reviewed in Clarke and Tyler, 2003). Previous studies have demonstrated a role for the transcription factor nuclear factor kappaB (NF-κB) (Clarke et al., 2003; Connolly et al., 2000), initiator and effector caspases (Clarke et al., 2000; Kominsky et al., 2002a; Richardson-Burns et al., 2002), and mitochondrial (Clarke et al., 2004; Kominsky et al., 2002a,b) signaling events in reovirus-induced apoptosis. Each of these pathways demonstrates modulation by NO in other experimental systems. For example, NF-κB activity is dependent upon DNA binding via the p50 subunit of the transcription factor and this ability can be inhibited by NO (Matthews et al., 1996), such that the inhibition of DNA binding by NF-κB has been proposed as a mechanism of NO-induced apoptosis (Marshall and Stamler, 2002). NO-mediated S-nitrosylation (the reaction of NO with cysteine, tyrosine, or heme groups) can inhibit the activity of caspases (Mohr et al., 1997) while mitochondrial apoptotic signaling pathways can also be disrupted by NO (reviewed in Boyd and Cadenas, 2002).
We now show that infection of neonatal mice with the prototypic neurotropic reovirus strain (T3A) results in induction of iNOS expression in brain areas demonstrating reovirus antigen expression and associated virus-induced injury. Expression of iNOS correlates with increased NOS activity in whole brain homogenates while levels of nNOS and eNOS are unchanged following infection. Under in vitro conditions, T3A infection of primary neuronal and glial cultures also results in induction of iNOS expression. In addition to infecting neurons, we now show for the first time that T3A can also infect a subpopulation of microglial cells in vitro. Growth of T3A in neuronal cultures is inhibited by SNAP, an NO donor, and increased by treatment with l- NAME, an inhibitor of iNOS, suggesting iNOS expression has the potential to exert antiviral activity in vivo.
Materials and methods
Cell lines and viruses
L929 mouse fibroblasts (ATCC CL1) used for viral titer assays were maintained in 2X199 medium supplemented with 10% heat-inactivated fetal bovine serum and 4 mM l-glutamine.
Reovirus strain type 3 Abney (T3A) is a laboratory stock which has been plaque purified and passaged (twice) in L929 cells to generate working stock (Tyler et al., 1996). Virus infections in vitro were performed at a multiplicity of infection (MOI) of 100 (unless stated otherwise) to ensure that 100% of susceptible cells are infected and to maximize the synchrony of viral replication.
Primary cell culture
Pregnant Swiss Webster Hsd:nd4 mice (Harlan-Sprague Dawley, Indianapolis, IN) were anesthetized with isoflurane and euthanized on gestational day 18 to prepare primary neuronal or glial cultures. Fetuses were removed and cortices dissociated and viable cells prepared for plating as described previously from our laboratory (Richardson-Burns et al., 2002). Viable cells were plated at a density of 5 × 105 cells per well onto poly-d-lysine-coated 6-well plates (Biocoat; Becton Dickinson, Franklin Lakes, NJ) for Western blotting. For immunocytochemical studies and viral growth assays, cells were plated at a density of 1.5 × 105 cells per well on to poly-d-lysine-coated coverslips and 24-well plates (Biocoat), respectively.
Cultures enriched in cortical neurons were plated out and maintained in serum-free Neurobasal A medium supplemented with B-27 nutrient supplement (2% v/v), antibiotic/antimycotic (penicillin/streptomycin; 1000 units/ml), l-glutamine (0.6 mM) and glutamate (0.6 mM; Glutamax) from 0 to 4 days in vitro (DIV). Cortical cultures enriched in glial cell populations were prepared as described for neuronenriched cultures and maintained in Neurobasal A medium containing 10% FBS and supplemented with antibiotic/antimycotic (penicillin/streptomycin; 1000 units/ml), l-glutamine (0.6 mM), and glutamate (0.6 mM; Glutamax) from 0 to 4 days in vitro (DIV). All primary cultures were maintained at 36.5°C and 5% CO2. From DIV 5 onwards, Neurobasal A media were supplemented as described above but with 0.3 mM glutamate (DIV 5) or zero glutamate (DIV 7) as opposed to the 0.6 mM used at the time of plating. All studies were performed at DIV 9-11. All culture media and supplements were purchased from Invitrogen (Carlsbad, CA) unless stated otherwise.
Primary neuronal cultures were characterized both morphologically and immunocytochemically using mouse monoclonal antibodies directed against neuron-specific nuclear protein (NeuN; 1:250; Chemicon International Inc., Temecula, CA) or microtubule-associated protein-2 (MAP-2; 1:250; Chemicon). Primary neuronal cultures comprised 91.2% ± 3.4% neurons. Primary glial cultures comprised 75.2% ± 3.7% astrocytes and 7.4% ± 1.8% microglia, as determined by immunoreactivity with antibody directed against glial fibrillary acidic protein (GFAP; 1:200: Cymbus Biotechnology, Chandlers Ford, UK) and specific labeling with FITC-conjugated isolectin B4 (IB4-FITC; 2 μg/ml in 3% BSA/TBST; Sigma), respectively. Virus-induced cell death was assessed in primary cultures using cell type-specific markers MAP-2 or GFAP in conjunction with Hoechst 33342 (1 μg/ml; Molecular Probes, Eugene, OR). All cell counting, both for phenotypic evaluation of cultures and effect of viral infection on cell survival, was performed by the counting of at least 300 cells per coverslip and with coverslips derived from at least 3 separate batches of cultures.
Drugs
S-Nitroso-N-acetylpenicillamine (SNAP), N-ο-Nitro-l-arginine methyl ester hydrochloride (l-NAME), and its inactive analog enantiomer (d-NAME) were purchased from Sigma Aldrich (St. Louis, MO). SNAP stock solutions were prepared at sufficiently high concentrations in DMSO to prevent final DMSO concentrations from exceeding 0.1% during in vitro treatment regimens. l-NAME and d-NAME were prepared in sterile PBS.
Western blotting
Whole cell extracts were prepared from primary cultures following viral infection as we have previously described (Clarke et al., 2004; Richardson-Burns et al., 2002). Briefly, cells were detached from culture plates using a cell scraper and centrifuged (1200 rpm) for 5 min. The resulting pellet was washed once with sterile PBS before a second spin (1600 rpm for 8 min) and then stored at -80°C until further use. Pellets were resuspended in whole cell lysis buffer [1% Nonidet P40 (Octylphenolpoly(ethyleneglycolether); Amersham Biosciences, Piscataway, NJ), 0.01 M triethanolamine HCl (pH 7.8), 0.15 M NaCl, 5 mM EDTA, 1 mM PMSF (phenyl methyl sulfonyl fluoride), 0.02 mg/ml trypsin inhibitor, 0.02 mg/ml leupeptin], briefly sonicated with a microtip probe, centrifuged at 20,000 × and supernatant then mixed with 150 μl of 2 × Laemmli buffer (4% sodium dodecyl sulfate, 20% glycerol, 10% beta-mercaptoethanol, 0.004% bromophenol blue, 0.125 M Tris-HCl, pH 6.8). Lysates were boiled for 5 min and electrophoresed (Hoefer Pharmacia Biotech, San Francisco, CA) in 10% or 12% tricine/polyacrylamide gels at a constant voltage of 70 V through the resolving gel. Proteins were electroblotted onto Hybond-C nitrocellulose membranes (Amersham Biosciences) and immunoblotting performed as described previously (Poggioli et al., 2000). Whole brain lysates were prepared from Swiss Webster pups after 8 days post-infection with 1 × 103 PFU of T3A or following mock infection with sterile PBS (see In vivo studies section for details of inoculation protocol). At 8 days post-infection, brains were removed, transferred to 1 ml of sterile PBS and stored at -80°C. Vials were thawed gently to room temperature and PBS aspirated off. Brains were subsequently homogenized in 300 μl of whole cell lysis buffer using a Dounce homogenizer. Lysates were transferred to 1.5 ml eppendorf tubes and stored on ice until centrifugation at 20,000×g for 3 min and transfer of supernatant to a fresh tube with 300 μl of Laemmli buffer. Samples were boiled for 5 min in a heating block before analyzing using 8% tricine gels and transfer to Hybond-C nitrocellulose membranes as detailed above. Immunoblots were probed with antibodies directed against iNOS, nNOS, and eNOS (BD Biosciences Pharmingen, San Diego, CA; 1:1000) and actin (Calbiochem, Sunnyvale, CA; 1:10,000).
In vivo studies
The use of type 3 reovirus strains as an experimental model for virus-induced CNS injury requires the infection of newborn mice and intracranial inoculation of these animals with these strains causes lethal encephalitis between 8 and 10 days post-infection (see Tyler, 1998). Two-day old Swiss Webster pups were pooled together from multiple litters and split randomly among surrogate mothers (8-10 pups per litter). Pups were intracranially inoculated with 2.5 × 102 (NOS assay only) or 1 × 103 plaque-forming units (PFU) of T3A virus in a 10-μl volume as described previously (Richardson-Burns et al., 2002). At 8 days post-infection, mice were sacrificed and brain tissue was removed for histological studies, viral titer assay, whole tissue lysates, or NOS assay. Based on previous studies in our laboratory, 8 days post-infection has proved optimal for examination of reovirus-induced CNS injury (Oberhaus et al., 1998). All experiments were performed under IACUC-approved protocols in an AAALAC-accredited animal care facility.
Assay of nitric oxide activity
The end products of NO metabolism (nitrite and nitrate) were quantitated in tissue homogenates using the Calbiochem Colorimetric Nitric Oxide Synthase Activity Assay kit as directed by the manufacturer (Calbiochem). Briefly, pups were either mock-infected (10 μl sterile PBS) or inoculated with 2.5 × 102 PFU or 1 × 103 PFU of T3A by intracranial administration. Brains were removed at 8 days post-infection and homogenized in 1 ml PBS. Samples were centrifuged at 10,000 × g for 20 min and supernatants subsequently passed through 0.45-μm filters before centrifugation again at 2000 × g through 10-kDa molecular weight cutoff centrifugation filters. 40 μl of each filtrate was added to separate wells of a 96-well plate along with 20 μl assay buffer, 10 Al 1 mM NADPH solution, 10 μl of nitrate reductase, 10 μl of co-factor, and 50 μl each of Griess Reagent R1 and R2. Samples were analyzed at a wavelength of 540 nm using an Emax Microplate Reader (Molecular Devices, Sunnyvale, CA) alongside appropriate nitrate standards. Quantitation was performed using SOFTmax Pro software suite (Molecular Devices).
Immunochemical and histological studies
Brain tissue was fixed in 10% formalin for 20 h at room temperature. Tissue was transferred to 70% ethanol before paraffin-embedding and sectioning. Coronal brain sections (4 μm thick) were prepared and tissue injury was assessed semi-quantitatively in hematoxylin and eosin stained sections. Adjacent sections were deparaffinized in xylene and rehydrated in consecutive 100% to 75% ethanol washes. Antigen retrieval was performed using antigen-unmasking solution (Vector Laboratories Inc., Burlingame, CA) or 10 mM citrate buffer. Tissue sections were permeabilized in Neuropore (Trevigen Inc., Gaithersburg, MD) overnight at 4°C and blocked in 5% normal goat serum (NGS; in 1 × Tris-buffered saline with 0.1% TWEEN [TBST]) for 6-8 h at room temperature. Sections were incubated overnight at 4°C with rabbit polyclonal antibodies directed against iNOS (Upstate, Lake Placid, NY; 1:1000) and eNOS (BD Biosciences Pharmingen; 1:100) diluted in 3% bovine serum albumin in TBST. Following washes with TBST, sections were incubated with biotinylated secondary antibody (Vector Laboratories; 1:100) diluted in 5% NGS/TBST for 2 h at room temperature. Following further washes in TBST, sections were incubated in 0.6% H2O2 (25 min) and ABC reagent (Vector Laboratories; 1 hour) before incubating for up to 10 min in pre-warmed DAB (Trevigen Inc.). Blue counterstain (Trevigen Inc.) was applied to sections before dehydrating and mounting with Vectamount (Vector Laboratories). For dual-label fluorescence immunohistochemical staining, tissue sections were incubated with polyclonal anti-reovirus antibody (a gift kindly provided by Dr. Terence S. Dermody, Vanderbilt University; 1:100 for 1 h at room temperature), monoclonal anti-j3 reovirus antibody (4F2) (Virgin et al., 1991), polyclonal antibodies against iNOS (Upstate; 1:100), nNOS (BD Biosciences Pharmingen; 1:100), MAP-2 (Abcam, Cambridge, UK; 1:100) or monoclonal antibodies against NeuN, MAP-2 (Chemicon, Temecula, CA; 1:100) or GFAP (Cymbus, Chandler’s Ford, UK; 1:200) in 3% bovine serum albumin (BSA); in TBST overnight unless stated otherwise. Following wash steps with TBST, sections were incubated with Texas red or fluoroscein-conjugated goat anti-rabbit or mouse secondary antibodies (Vector Laboratories; 1:100) for 2 h, washing again with TBST and incubating for 20 min with 1 μg/ml Hoechst 33342 (Molecular Probes). FITC fluorophore-conjugated isolectin B4 (IB4-FITC; 2 μg/ml in 3% BSA/TBST; Sigma) was incubated overnight with sections for labeling of microglial populations. Sections were mounted using VectorShield (Vector Laboratories). Immunostaining for digital fluorescence microscopy was imaged using a Zeiss Axioplan 2 digital microscope with a Cooke sensiCam 12-bit camera.
Viral titer assays
Primary neuronal cultures were assessed for viral growth following inoculation with an MOI of 10 of T3A. Viral growth was assessed in the presence and absence of the NO donor SNAP (100-200 μM) and the NOS inhibitor l-NAME (250 μM-1 mM) at concentrations widely used in the literature (Bi and Reiss, 1995; Blond et al., 2000). Cultures were transferred to storage at -80°C immediately upon completion of the incubation period. Serial dilutions of freeze-thawed cultures were prepared in gel saline and viral titer determined by plaque assays as previously described (Debiasi et al., 1999).
Statistical analysis
One-way analysis of variance (ANOVA) was used for comparison of viral titer and cell counts with significant differences evaluated using the Tukey-Kramer multiple comparisons post hoc test. Results from nitric oxide assay experiments were evaluated using an unpaired t test. All statistical analyses were performed using Instat (GraphPad Software Inc., San Diego, CA).
Results
Elevated total NOS activity in vivo following reovirus infection
We wished to determine whether production of NO is dysregulated during reovirus infection. Following intracranial inoculation of 2-day-old mice with T3A, we evaluated NOS activity in whole brain homogenates collected at 8 days post-infection. T3A infection (250 PFU and 1000 PFU) resulted in significantly elevated NOS activity, as indicated by total nitrate and nitrite formation, in brain homogenates as compared to mock-infected lysates (Fig. 1). The 1000 PFU dose provided a robust increase in total NOS activity and was thus used for subsequent in vivo studies. The 1000 PFU dose has been widely utilized in studies of T3 reovirus-induced CNS injury and facilitates comparison of the CNS effects in the present study with that observed in other recent studies of reovirus CNS infection (Richardson-Burns and Tyler, 2004, 2005).
Fig. 1.

NOS activity is increased in whole brain homogenates following T3A infection. Two-day-old Swiss Webster mice were mock-infected or inoculated i.c. with 2.5 × 102 or 1 × or 103 brains PFU of T3A. At 8 days post-infection, brains were removed and homogenized for assessment of NOS activity. NOS activity was assessed by colorimetric determination of total nitrite and nitrate formation in whole brain homogenates using Griess Reagent. Values represent the mean of 3-4 observations with vertical bars indicating SEM. *P < 0.01.
Induction of iNOS expression in vivo following reovirus infection
Having demonstrated elevation of NOS activity in brain homogenates of T3A-infected mice, we next wished to determine the mechanism by which NOS activity was dysregulated following neurotropic reovirus infection. Whole brain homogenates were examined for changes in overall expression levels of the three major NOS isoforms: nNOS, eNOS, and iNOS. We observed substantial iNOS induction in whole brain homogenates from T3A-infected animals compared to mock infected (Fig. 2). Expression levels of nNOS and eNOS were not altered in whole brain homogenates 8 days after T3A infection, compared to mock-infected animals (Fig. 2). Whole brain homogenates were also examined at 4 and 6 days post-infection for NOS expression and similarly demonstrated no evidence of disruption to overall brain levels of nNOS or eNOS while iNOS expression was not detected at 4 days post-infection and only observed in a minority of brains at 6 days post-infection (data not shown). Following intracranial inoculation of neonatal mice with T3A (1 × 103 PFU), we observed large areas of virus-induced brain injury at 8 days post-infection. As has previously been reported, virus-induced lesions were confined predominantly to specific brain regions, including fronto-parietal and cingulate cortices, lateral septal nucleus, hippocampus (CA2/CA3), and thalamus (latero-dorso nuclei) (Oberhaus et al., 1997). Injury was most severe in the thalamus (Figs. 3C, G), cingulate cortex, and CA2/CA3 portion of the hippocampus. Expression of iNOS was evident only in areas of virus-induced injury, particularly in the thalamus (Figs. 3D, H). Dual-labeling studies were performed to examine expression of viral antigen and iNOS in mock- and T3A-infected brains (Figs. 3I-O). In mock-infected animals, we found no evidence of iNOS expression in any area associated with T3A-induced injury. In T3A-infected animals, we observed substantial co-localization of iNOS immunoreactivity and the presence of viral antigen in all areas associated with reovirus-induced CNS injury (Figs. 3J-O). iNOS expression was observed predominantly in infected cells although in rare instances we did observe rare iNOS expression in noninfected cells, but only when they were in close proximity to infected cells. Deconvolution microscopy studies showed expression of iNOS in neurons of the CA2/CA3 regions of the hippocampus.
Fig. 2.
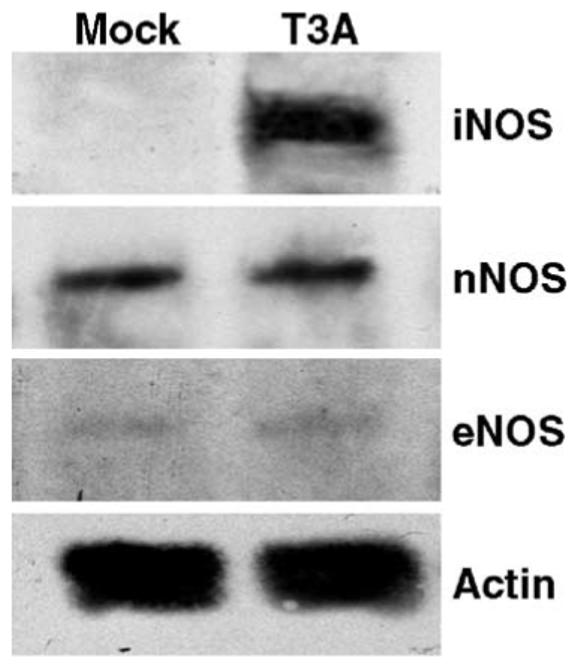
iNOS expression is induced in whole brain lysates following T3A infection. Whole brain lysates were prepared at 8 days post-infection from animals mock infected or infected i.c. with 1 × 103 PFU of T3A. Lysates were probed for NOS isoforms by Western blot. Blots of iNOS, nNOS, and eNOS protein levels are shown, each representative of n = 5 observations. Actin levels were assessed as loading control.
Fig. 3.
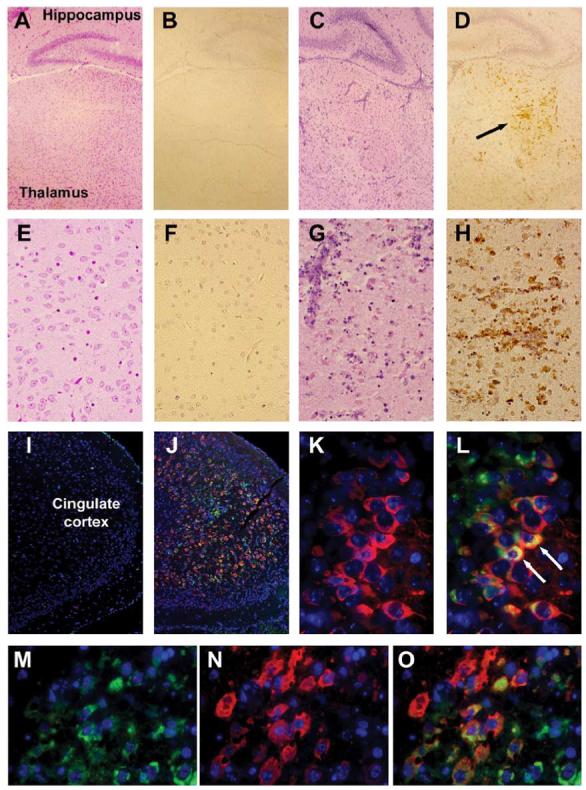
iNOS is expressed in areas of brain injury following T3A infection in vivo. Two-day-old Swiss Webster mice were mock-infected or inoculated i.c. with 1 × 103 PFU of T3A. At 8 days post-infection, brains were removed for histological examination. Hematoxylin and eosin (H&E) staining of coronal brain sections indicated areas of virus-induced injury in the brain [A and C (40×), E and G (400×)]. Injury was evident in the brain within the thalamus (C, G), cingulate cortex, and hippocampus. No injury was observed in the brains of mock-infected animals (A, E). Immunohistochemical staining was performedon adjacent brain sections with a polyclonal antibody directed against iNOS. Diaminobenzidine (DAB) was used as a marker for this staining. No evidence for iNOS expression was observed in the brain of mock-infected animals (B, F). Expression of iNOS in the thalamus [D (40×), H (400×)] and cingulate cortex and hippocampus corresponded with areas of virus-induced injury, as identified by H&E staining. Dual-label immunofluorescence was used to confirm colocalization of reovirus (identified using a reovirus α3-specific monoclonal antibody (Virgin, et al., 1991) and Texas red conjugated goat anti-mouse secondary) and iNOS (identified using an iNOS-directed polyclonal antibody and fluorescein-conjugated goat anti-rabbit secondary) (I-O). Fluorescence labeling was performed on brain tissue from mock-infected (I) and T3A-infected animals (J-O). Low-magnification images (25×) represent staining in the cingulate cortex of mock- (I) and T3A-infected (J) tissue. Higher-magnification images (630×, oil immersion) represent staining in the CA3 portion of the hippocampus (K, L) and the cingulate cortex (M-O) of T3A-infected animals.
Levels of nNOS are unchanged during T3A infection
Having observed T3A-induced iNOS expression in the brain, we next wished to determine whether T3A had any effect on nNOS expression or localization. As shown earlier, nNOS levels in whole brain homogenates were not changed during T3A infection (Fig. 2). We subsequently examined virus-induced changes in distribution of nNOS by immunohistochemistry following T3A infection in vivo. We observed low levels of nNOS expression throughout the brain of mock-infected animals (Figs. 4A, C). Following T3A infection, we found no change in the pattern of nNOS immunoreactivity (Fig. 4B). Expression remained at low levels in both areas of T3A-induced CNS injury and adjacent non-injured areas (Figs. 4B, D). We observed rare instances of colocalization of nNOS and viral antigen (Fig. 4E) never exceeding more than one or two isolated nNOS immunoreactive cells. Similar to results from in vivo studies, Western blot analysis of whole cell lysates of primary neuronal cultures revealed no effect of T3A infection (MOI = 100) on levels of nNOS expression in whole cell extracts between 0 and 48 h post-infection (Fig. 4F).
Fig. 4.
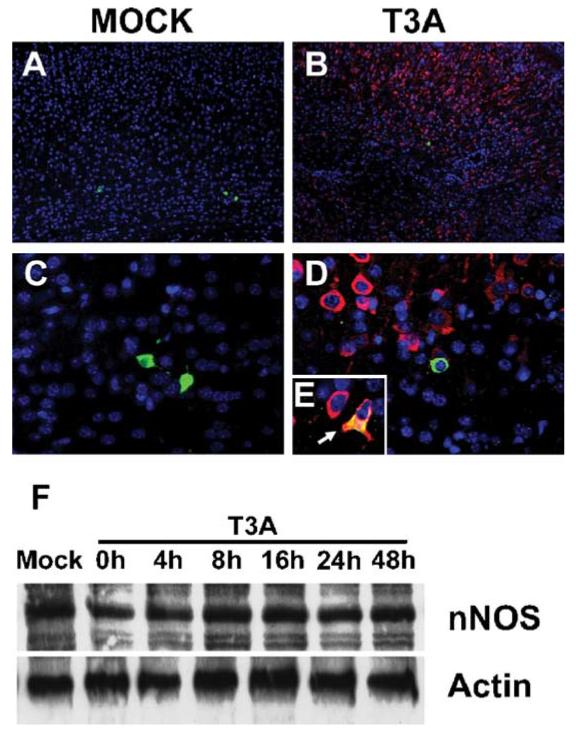
Expression of nNOS is unaltered in vivo and in vitro following T3A infection. Two-day-old Swiss Webster mice were mock infected (A, 40×; C, 400×) or inoculated i.c. with 1 × 103 PFU of T3A (B, 40×; D, 400×;E, 630× oil immersion). At 8 days post-infection, brains were removed and immunohistochemical staining was performed to examine nNOS and reovirus co-localization. Dual-label immunofluorescence was used to examine nNOS (fluorescein-conjugated anti-rabbit secondary antibody) and reovirus (Texas red conjugated anti-mouse secondary antibody against reovirus α3-specific monoclonal antibody) localization. Representative images (n = 4) demonstrate nNOS and T3Aα3 localization in cingulate cortex of mock (A, 40×; C, 400×)- and T3A (B, 40×, D, 400; E, 630 oil)-infected brains. White arrow (E) indicates evidence of nNOS and T3Aσ3 co-localization. Protein levels of nNOS were assessed in primary neuronal cultures by Western blot analysis of whole cell lysates (F). Representative blot (n = 4) demonstrates nNOS levels in primary neuronal cultures between 0 and 48 h post-infection with T3A (MOI = 100). Actin levels were assessed as loading control.
Levels of eNOS are unchanged during T3A infection
We next wished to examine whether T3A infection resulted in changes in eNOS expression within the brain. As shown earlier, eNOS levels in whole brain homogenates were unchanged following T3A infection (Fig. 2). We subsequently examined expression of eNOS in mock- and T3A-infected brains by immunohistochemistry. In mock-infected animals, we observed eNOS expression confined almost exclusively to endothelial cell populations (Figs. 5A, C). We observed no major differences in the pattern or overall level of eNOS expression in brain tissue of T3A-infected animals (Figs. 5B, D) compared to mock-infected (Figs. 5A, C). Western blot analysis of primary neuronal cultures (Fig. 5E) revealed very low levels of eNOS expression in whole cell lysates from mock-infected cultures. T3A infection (MOI = 100) had no effect on eNOS expression in whole cell extracts from primary neuronal cultures (Fig. 5E).
Fig. 5.
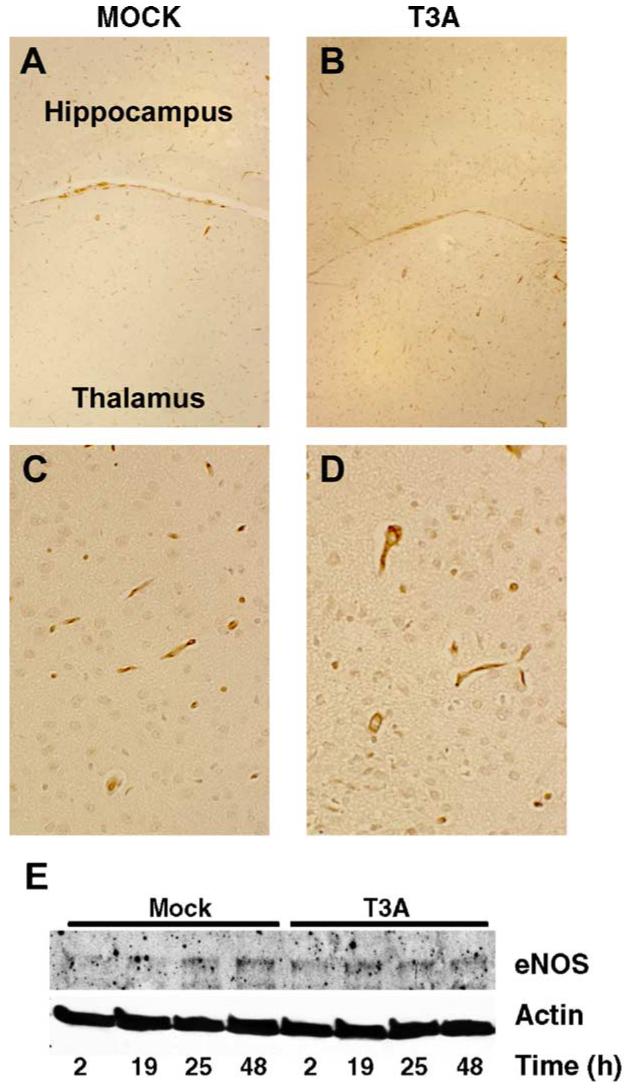
Expression of eNOS is unaltered in vivo and in vitro following T3A infection. Two-day-old Swiss Webster mice were mock infected (A, 40; C, 400×) or inoculated i.c. with 1 × 103 PFU of T3A (B, 40; D, 400 ×). At 8 days post-infection, brains were removed and DAB-based histochemical staining was performed to examine eNOS expression. Representative images (n = 4) demonstrate eNOS expression in the latero-dorsal thalamic nucleus of mock (A, 40×; C, 400)- and T3A (B, 40×; D, 400×)-infected brains. Protein levels of eNOS were assessed in primary neuronal cultures by Western blot analysis of whole cell lysates. Representative blot (n = 4) demonstrates eNOS levels in primary neuronal cultures between 2 and 48 h post-infection with T3A (MOI = 100). Actin levels were assessed as loading control.
T3A infection induces iNOS expression in primary cultures
Having demonstrated induction of iNOS expression in the brain following neurotropic reovirus infection, we wished to determine the cell populations expressing iNOS during infection by the use of primary neural cell culture models. We first examined expression of iNOS in whole cell extracts of T3A and mock-infected primary neuronal (Fig. 6A) and glial (Fig. 6B) cultures by Western blot. Infection of murine primary neuronal cultures with T3A (MOI = 100) resulted in early induction of iNOS expression within 2 h of initial infection, reaching peak levels at 8 h post-infection and maintaining a high level of expression through to 48 h post-infection (Fig. 6A). Time-course studies in glial cultures demonstrated induction of iNOS expression from 16 h post-infection with T3A (Fig. 6B). Interestingly, levels of iNOS expression consistently decreased in glial cultures between 24 and 48 h post-infection (Fig. 6B).
Fig. 6.
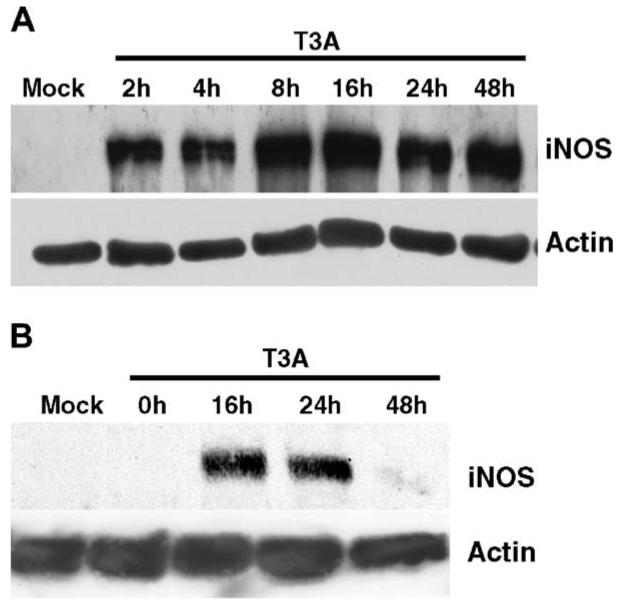
T3A infection induces iNOS expression in primary neuronal and glial cultures. Primary neuronal (A) and glial (B) cultures were examined for iNOS infection by Western blot analysis at various time points post-infection (MOI = 100). Expression of iNOS was detected in neuronal cultures as early as 2 h post-infection and reached peak levels at 8-24 h post-infection (A) and similarly peaked between 16 and 24 h post-infection in glial cultures. Representative blots (n = 4) demonstrate iNOS levels in primary neuronal and glial cultures. Actin levels were assessed as loading control.
Immunocytochemical analysis of iNOS expression was performed on primary neuronal and glial cultures (Figs. 7A-J) using dual label staining against neuronal (MAP-2), astrocyte (GFAP), and microglial (IB4) cell markers to identify iNOS-expressing cell populations following T3A infection. We found limited evidence of iNOS expression in mock-infected neuronal cultures (Figs. 7A, C). In contrast, we observed significantly higher levels of iNOS immunoreactive neurons in T3A-infected cultures (MOI = 100) at 24 h post-infection (Fig. 7C). Due to the potential for glial cell populations to express iNOS following infection, we also examined iNOS immunoreactivity in conjunction with astrocyte and microglial cell markers in glia-enriched cultures (Figs. 7D-J). We found no evidence of iNOS expression in GFAP immunoreactive cell populations (Figs. 7D-F) although iNOS expression was observed in a non-GFAP immunoreactive cell population in these cultures (Fig. 7E). We used FITC-conjugated IB4 to label microglial populations in the glial cultures (Figs. 7G-I) to determine whether these populations were expressing iNOS during infection. Dual-labeling studies of IB4 with polyclonal antibody against reovirus identified a microglial cell phenotype in our glial cultures that was susceptible to infection by T3A (Fig. 7I). The morphology of the cell population infected by T3A in our glial cultures corresponds to that of an amoeboid microglial population and we observed a high rate of infection in these cells (42.3% ± 1.2%, Fig. 7) despite their relatively low prevalence (7.4% ± 1.8%) in glial cultures.
Fig. 7.
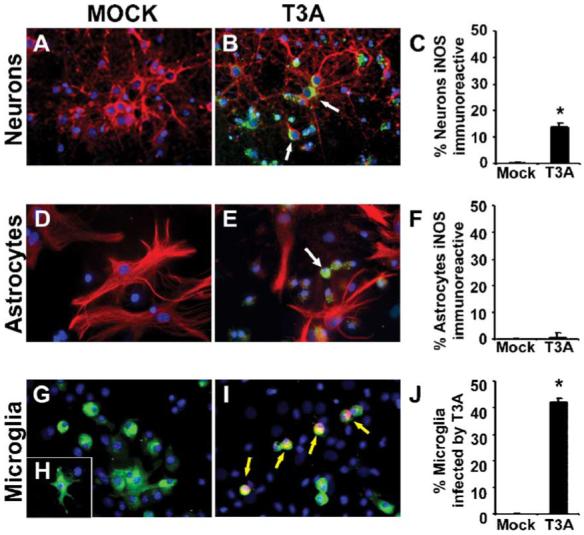
T3A infection induces iNOS expression in neurons and immature microglia. Primary neuronal (A-C) and glial (D-J) cultures were examined for expression of iNOS following infection with the neurotropic reovirus strain T3A (MOI = 100). Primary neuronal cultures comprised 91.2% ± 3.4 neurons and primary glial cultures comprised 75.2% ± 3.7 astrocytes and 7.4% ± 1.8 microglia. Representative images demonstrate dual-label immunofluorescence staining of iNOS (fluorescein-conjugated) with the neuronal marker MAP-2 (Texas red; A, B) or the astrocyte marker GFAP (Texas red; D, E) in neuronal and glial cultures, respectively. Numbers of iNOS immunoreactive neurons and astrocytes were determined by cell counting (C, F) of respective cultures. Expression of iNOS observed in primary glial cultures did not demonstrate co-localization with the astrocytic cell type marker GFAP. Dual-label studies of primary glial cultures with the microglial cell type marker IB4-FITC and the polyclonal antibody directed against reovirus identified an amoeboid microglial phenotype susceptible to T3A infection (G-I). Cell counting of T3A-infected IB4-positive cells demonstrated an infection rate of greater than 40% of microglial cells in primary glial cultures.
Modulation of NO levels alters T3A growth in primary neurons
Having demonstrated induction of iNOS expression in neurons in vivo and in vitro in primary cortical neurons following T3A infection, we wished to examine whether excess levels of NO, a consequence of iNOS expression, effected T3A growth in vitro. The effect of NO on T3A growth was assessed in primary neuronal cultures by plaque assay using the NO donor SNAP and the NOS inhibitor l- NAME (Fig. 8). SNAP dose-dependently inhibited T3A growth in neuronal cultures with significant inhibition of growth occurring at 48 h post-infection (Fig. 8A). In contrast, treatment of T3A-infected neuronal cultures with the NOS inhibitor l- NAME resulted in significantly greater viral growth at 48 h post-infection (1 mM; Fig. 8B) compared to virus alone or virus in the presence of the inactive enantiomer (d-NAME; 250 AM-1 mM). At the doses used in these studies (100-200 μM), we observed no significant effect of SNAP on neuronal survival, nor did we observe any effect of vehicle alone (0.1% DMSO) on T3A growth in primary neuronal cultures (data not shown).
Fig. 8.
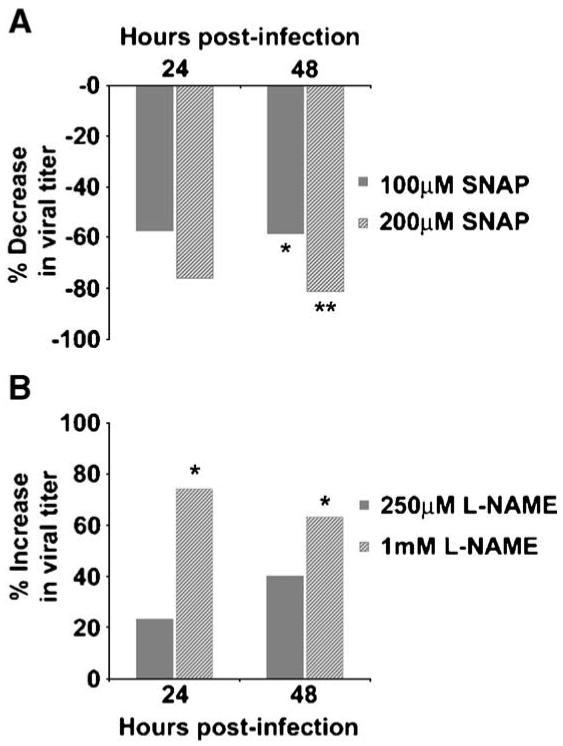
Nitric oxide modulates T3A growth in primary neuronal cultures. The effect of NO on reovirus growth was assessed in primary neuronal cultures at 0-48 h post-infection. Cultures were infected with T3A (MOI = 10) and growth was assessed by plaque assay at 24 and 48 h following infection in the presence of NO donor (A) or NOS inhibitor (B). The NO donor S-Nitroso-N-acetylpenicillamine (SNAP) was added to cultures at a final concentration of 100 or 200 μM immediately following infection (A). Repeat dosing of SNAP was required at 24 h after infection due to the short half-life of the drug. Treatment of cultures with vehicle (0.1% DMSO) had no effect on T3A growth in these studies. The NOS inhibitor l-NAME (250 μM or 1 mM) was added to cultures at the time of infection and T3A growth was assessed by plaque assay 24 and 48 h later (B). The effect of drug treatment was compared to virus infection alone at each time point and for both drug regimens to determine percent change in viral growth. Each value represents the mean of 4 observations with vertical bars indicating the SEM. *P < 0.05; **P < 0.01.
Discussion
We now show that T3A infection in vivo results in enhanced expression of iNOS in the brain in areas of viral infection and virus-induced injury. In contrast, levels of nNOS and eNOS are not significantly altered following infection. Expression of iNOS is associated with a significant increase in total NOS activity.
NO has been widely implicated as part of a host response against viral infection (for review, see Akaike and Maeda, 2000). Prior studies have demonstrated NO-mediated antiviral actions on both DNA and RNA viruses including coxsackievirus (via inactivation of the viral cysteine protease by S-nitrosylation) (Zell et al., 2003) and junin virus (via impairment of astrocytosis) (Gomez et al., 2003). The role of NO in mammalian reovirus infection has not previously been assessed. Pertile and co-workers reported that avian reovirus infection of chicken macrophages primed these cells to produce NO in response to LPS stimulation and that viral growth was inhibited in these cells following LPS stimulation (Pertile et al., 1996). Administration of a non-isoform selective NOS inhibitor, N-γ-monomethyl-l-arginine reduced the antiviral effect exhibited in LPS-stimulated chicken macrophages (Pertile et al., 1996).
In this study, we show that infection with a neurotropic reovirus strain (T3A) results in induction of iNOS expression and that iNOS localization is confined only to areas expressing viral antigen and showing evidence of virusinduced CNS injury. Induction of iNOS expression has been reported for other neurotropic viruses, including borna disease virus (Akaike et al., 1995; Koprowski et al., 1993), pseudorabies virus (Serrano et al., 2002), and herpes simplex virus type 1 (Fujii et al., 1999). Based on the colocalization of iNOS expression and viral antigen, it is not immediately clear whether the stimulus for iNOS expression is a direct result of infection, a consequence of virusinduced injury or a combination of these. Reoviridae comprise a double-stranded RNA (dsRNA) genome and recent evidence suggests accumulation of dsRNA triggers activation of protein kinase R (PKR) resulting in activation of NF-κB-mediated pathways (Auch et al., 2004). Previous studies have demonstrated a key role for the transcription factor NF-κB in reovirus-induced apoptosis (Clarke et al., 2003; Connolly et al., 2000). Importantly, NF-κB has been shown to exert transcriptional control over the gene encoding iNOS (reviewed in O’Neill and Kaltschmidt, 1997). Thus, productive T3A infection in cortical, hippocampal, and thalamic areas may trigger a PKR-NF-κB- mediated pathway to induce iNOS expression. Recent studies in our laboratory have demonstrated a biphasic pattern of reovirus-induced regulation of NF-κB activity with an initial phase of NF-κB activation followed by a phase of inactivation at later times post-infection (Clarke et al., 2003). Activation of NF-nB may provide the requisite stimulus for iNOS expression which would likely precede increased NO production. Interestingly, the DNA binding activity of NF-κB can be inhibited by NO (DelaTorre et al., 1997) and thus NF-nB-mediated iNOS expression could result in inhibitory feedback on activity of the transcription factor, possibly explaining the biphasic pattern of NF-κB activity occurring during reovirus-induced apoptosis. Enhanced iNOS expression can also occur as a result of migration of inflammatory cells, including monocytes and macrophages, into the brain of virus-infected animals (Serrano et al., 2002). However, this is unlikely to explain reovirus-induced changes in iNOS expression as inflammation is minimal during the early stages of reovirus CNS infection (reviewed in Tyler, 1998). In addition, the majority of iNOS immunoreactive cells could be phenotypically identified as neurons or microglia using cell-type specific markers. Expression of iNOS has been reported previously in neuronal populations (Minc-Golomb et al., 1996) as well as other neural cell types, including glia (Nomura and Kitamura, 1993) and oligodendrocytes (Merrill et al., 1997).
We prepared mixed glial cultures to evaluate iNOS expression in non-neuronal populations following T3A infection and observed peak levels of reovirus-induced expression between 16 and 24 h post-infection. Interestingly, reovirus-induced expression of iNOS in these cultures decreased between 24 and 48 h post-infection. Immunocytochemical studies demonstrated no evidence of iNOS expression in astrocytes (GFAP-positive cells). Similarly, evidence of T3A infection was also limited to GFAPnegative cell populations in the glial cultures. Dual-labeling studies with cell-type-specific markers demonstrated that an immature microglial cell type was vulnerable to T3A infection, providing the first evidence to suggest microglial susceptibility to reovirus infection. The reovirus-infected, IB4-positive cells were typically of an amoeboid phenotype perhaps suggesting a developmental pattern of susceptibility. In the studies described here, it should be noted that iNOS expression in our glial cultures was observed predominantly in MAP-2 or NeuN immunoreactive cells indicating neuronal localization, thus matching the classical pattern of serotype-3 reovirus infection (see Clarke and Tyler, 2003). Reovirus does not productively infect astrocytes (GFAP-positive cells) nor is there significant cell death in this population during the time course of our experiments. In addition, astrocytes do not show significant basal or reovirus-induced iNOS expression. These findings suggest that the decrease in iNOS expression between 24 and 48 h in glial cultures is due to viral effects on either neuronal or microglial cells or both. There is significant reovirus-induced CPE in both neuronal and microglial cell populations in these cultures between 24 and 48 h post-infection, suggesting that it is a loss of these cells that accounts for the decrease in iNOS expression observed by Western blot between 24 and 48 h.
Evidence for changes in expression of neuronal and endothelial NOS isoforms following neurotropic viral infection is less substantive. Experimental infection of mice with vesicular stomatitis virus (VSV) resulted in a large increase in eNOS expression in astrocytes (Barna et al., 1996) while increases in nNOS expression were reported in distinct brain regions of pseudorabies-infected rats and this was accompanied by iNOS expression in similar brain areas (Serrano et al., 2002). Our studies demonstrate no evidence of T3A-induced changes in the pattern of distribution or overall level of nNOS or eNOS expression, we can therefore conclude that increased NOS activity observed in whole brain homogenates of T3A-infected compared to mock-infected mice is due to activity of the inducible NOS isoform.
Our study provides the first evidence of an antiviral effect of NO on a mammalian reovirus. The ability of the NO donor SNAP to dose-dependently attenuate reovirus growth in primary neuronal cultures suggests susceptibility of the reovirus strain T3A to antiviral effects of NO. In addition, administration of the NOS inhibitor l-NAME significantly increased reovirus growth in our neuronal cultures providing further evidence for an antiviral effect of NO on T3A. These findings also suggest that iNOS- mediated NO production provides effective inhibition of T3A growth in neuronal cultures. The mechanism of the putative NO-mediated antiviral effect on T3A infection remains unknown; however, based on observations in other viral systems, we can speculate that reovirus replicative machinery or host components involved in protein synthesis or cell survival are likely targets. One mechanism of NO- mediated antiviral effects that has received considerable interest in recent years is that of S-nitrosylation of viral and host cell molecules (reviewed in Colasanti et al., 1999). Reactive cysteine residues provide a target for NO both on host cell and viral proteins, such as enzymes and core structural proteins. For example, NO donors inhibit the proteolytic activity of the picornavirus protease 3C while dithiothreitol is able to recover this inhibition by reducing nitrosylation (Saura et al., 1999). Treatment of model viral infections with inhibitors of NOS activity typically results in deleterious effects upon the host, resulting in increased viral replication (Kosugi et al., 2002; Saxena et al., 2001) and, on occasions, increased mortality (Tucker et al., 1996). However, the role of NO is not always straightforward and this is exemplified by the findings of Chen and Lane (Chen and Lane, 2002), who demonstrated that mouse hepatitis virusinduced mortality was decreased in iNOS-deficient mice. Decreased mortality in these studies was associated with reduced neuronal death rather than changes in viral clearance or demyelination (Chen and Lane, 2002). Together, these findings illustrate the ‘double-edged sword’ capacity of nitric oxide. Antiviral and neuroprotective roles of NO can be superseded by cytotoxicity in the event that there is substantial dysregulation of NO production. Furthering our understanding of the role of NO in reovirus infection provides a model system in which pathologic and protective roles may finally be distinguished.
In conclusion, the findings presented herein provide the first evidence that mammalian reovirus infection results in the induction of iNOS expression in primary neuronal cultures and in areas of virus-induced injury within the brain. Expression and localization of nNOS and eNOS isoforms is not altered during T3A infection, either in the CNS or in vitro in primary neuronal cultures. In addition, we have provided the first evidence of reovirus infection of an immature microglial cell type. The inhibition of T3A growth in vitro by application of an NO donor and enhancement of growth by an NOS inhibitor suggests susceptibility of reovirus to NO but other factors in vivo may serve to restrict antiviral NO effects while the possibility remains that elevated NO production may also contribute to virus-induced CNS injury. Future studies will address the mechanism of iNOS induction during neurotropic reovirus infection and assess the precise role NO plays in reovirus-induced apoptosis.
Acknowledgments
This work was supported by a Merit grant from the Department of Veterans Affairs (KLT), NIH RO1 NS050138 and the Reuler-Lewin Family Professorship of Neurology (KLT). The authors would like to acknowledge the UCHSC Histology Services for assistance with histological preparations and special thanks to Suzanne Meintzer for expert technical assistance.
References
- Adamson DC, Kopnisky KL, Dawson TM, Dawson VL. Mechanisms and structural determinants of HIV-1 coat protein, gp41-induced neurotoxicity. J. Neurosci. 1999;19:64–71. doi: 10.1523/JNEUROSCI.19-01-00064.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Maeda H. Nitric oxide and virus infection. Immunology. 2000;101:300–308. doi: 10.1046/j.1365-2567.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Weihe E, Schaefer M, Fu ZF, Zheng YM, Vogel W, Schmidt H, Koprowski H, Dietzschold B. Effect of neurotropic virus infection on neuronal and inducible nitric oxide synthase activity in rat brain. J. NeuroVirol. 1995;1:118–125. doi: 10.3109/13550289509111016. [DOI] [PubMed] [Google Scholar]
- Auch CJ, Saha RN, Sheikh FG, Liu X, Jacobs BL, Pahan K. Role of protein kinase R in double-stranded RNA-induced expression of nitric oxide synthase in human astroglia. FEBS Lett. 2004;563:223–228. doi: 10.1016/S0014-5793(04)00302-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M, Komatsu T, Reiss CS. Activation of type III nitric oxide synthase in astrocytes following a neurotropic viral infection. Virology. 1996;223:331–343. doi: 10.1006/viro.1996.0484. [DOI] [PubMed] [Google Scholar]
- Bi Z, Reiss CS. Inhibition of vesicular stomatitis virus infection by nitric oxide. J. Virol. 1995;69:2208–2213. doi: 10.1128/jvi.69.4.2208-2213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blond D, Raoul H, Le Grand R, Dormont D. Nitric oxide synthesis enhances human immunodeficiency virus replication in primary human macrophages. J. Virol. 2000;74:8904–8912. doi: 10.1128/jvi.74.19.8904-8912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd CS, Cadenas E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol. Chem. 2002;383:411–423. doi: 10.1515/BC.2002.045. [DOI] [PubMed] [Google Scholar]
- Chen BP, Lane TE. Lack of nitric oxide synthase type 2 (NOS2) results in reduced neuronal apoptosis and mortality following mouse hepatitis virus infection of the central nervous system. J. NeuroVirol. 2002;8:58–63. doi: 10.1080/135502802317247820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P, Tyler KL. Reovirus-induced apoptosis: a minireview. Apoptosis. 2003;8:141–150. doi: 10.1023/a:1022966508671. [DOI] [PubMed] [Google Scholar]
- Clarke P, Meintzer SM, Gibson S, Widmann C, Garrington TP, Johnson GL, Tyler KL. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 2000;74:8135–8139. doi: 10.1128/jvi.74.17.8135-8139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P, Meintzer SM, Moffitt LA, Tyler KL. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor-related apoptosis-inducing ligandmediated apoptosis in virus-infected cells. J. Biol. Chem. 2003;278:18092–18100. doi: 10.1074/jbc.M300265200. [DOI] [PubMed] [Google Scholar]
- Clarke P, Meintzer SM, Wang Y, Moffitt LA, Richardson-Burns SM, Johnson GL, Tyler KL. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 2004;78:13132–13138. doi: 10.1128/JVI.78.23.13132-13138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colasanti M, Persichini T, Venturini G, Ascenzi P. S-nitrosylation of viral proteins: molecular bases for antiviral effect of nitric oxide. IUBMB Life. 1999;48:25–31. doi: 10.1080/713803459. [DOI] [PubMed] [Google Scholar]
- Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. Reovirus-induced apoptosis requires activation of transcription factor NF-kappaB. J. Virol. 2000;74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiasi RL, Squier MK, Pike B, Wynes M, Dermody TS, Cohen JJ, Tyler KL. Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J. Virol. 1999;73:695–701. doi: 10.1128/jvi.73.1.695-701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelaTorre A, Schroeder RA, Kuo PC. Alteration of NF-kappa B p50 DNA binding kinetics by S-nitrosylation. Biochem. Biophys. Res. Commun. 1997;238:703–706. doi: 10.1006/bbrc.1997.7279. [DOI] [PubMed] [Google Scholar]
- Fujii S, Akaike T, Maeda H. Role of nitric oxide in pathogenesis of herpes simplex virus encephalitis in rats. Virology. 1999;256:203–212. doi: 10.1006/viro.1999.9610. [DOI] [PubMed] [Google Scholar]
- Gomez RM, Yep A, Schattner M, Berria MI. Junin virusinduced astrocytosis is impaired by iNOS inhibition. J. Med. Virol. 2003;69:145–149. doi: 10.1002/jmv.10254. [DOI] [PubMed] [Google Scholar]
- Hooper DC, Kean RB, Scott GS, Spitsin SV, Mikheeva T, Morimoto K, Bette M, Rohrenbeck AM, Dietzschold B, Weihe E. The central nervous system inflammatory response to neurotropic virus infection is peroxynitrite dependent. J. Immunol. 2001;167:3470–3477. doi: 10.4049/jimmunol.167.6.3470. [DOI] [PubMed] [Google Scholar]
- Hori K, Burd PR, Furuke K, Kutza J, Weih KA, Clouse KA. Human immunodeficiency virus-1-infected macrophages induce inducible nitric oxide synthase and nitric oxide (NO) production in astrocytes: astrocytic NO as a possible mediator of neural damage in acquired immunodeficiency syndrome. Blood. 1999;93:1843–1850. [PubMed] [Google Scholar]
- Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 2002a;9:926–933. doi: 10.1038/sj.cdd.4401045. [DOI] [PubMed] [Google Scholar]
- Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 2002b;76:11414–11424. doi: 10.1128/JVI.76.22.11414-11424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzschold B. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc. Natl. Acad. Sci. U. S. A. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi I, Kawasaki H, Arai Y, Tsutsui Y. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am. J. Pathol. 2002;161:919–928. doi: 10.1016/S0002-9440(10)64252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean A, Wei XQ, Huang FP, Al-Alem UA, Chan WL, Liew FY. Mice lacking inducible nitric-oxide synthase are more susceptible to herpes simplex virus infection despite enhanced Th1 cell responses. J. Gen. Virol. 1998;79(Pt 4):825–830. doi: 10.1099/0022-1317-79-4-825. [DOI] [PubMed] [Google Scholar]
- Marshall HE, Stamler JS. Nitrosative stress-induced apoptosis through inhibition of NF-kappa B. J. Biol. Chem. 2002;277:34223–34228. doi: 10.1074/jbc.M201638200. [DOI] [PubMed] [Google Scholar]
- Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. 1996;24:2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill JE, Murphy SP, Mitrovic B, Mackenzie-Graham A, Dopp JC, Ding M, Griscavage J, Ignarro LJ, Lowenstein CJ. Inducible nitric oxide synthase and nitric oxide production by oligodendrocytes. J. Neurosci. Res. 1997;48:372–384. [PubMed] [Google Scholar]
- Minc-Golomb D, Yadid G, Tsarfaty I, Resau JH, Schwartz JP. In vivo expression of inducible nitric oxide synthase in cerebellar neurons. J. Neurochem. 1996;66:1504–1509. doi: 10.1046/j.1471-4159.1996.66041504.x. [DOI] [PubMed] [Google Scholar]
- Mohr S, Zech B, Lapetina EG, Brune B. Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem. Biophys. Res. Commun. 1997;238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]
- Murad F. Nitric oxide signaling: would you believe that a simple free radical could be a second messenger, autacoid, paracrine substance, neurotransmitter and hormone? Recent Prog. Horm. Res. 1998;53:43–59. discussion 59-60. [PubMed] [Google Scholar]
- Nomura Y, Kitamura Y. Inducible nitric oxide synthase in glial cells. Neurosci. Res. 1993;18:103–107. doi: 10.1016/0168-0102(93)90013-g. [DOI] [PubMed] [Google Scholar]
- Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J. Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhaus SM, Dermody TS, Tyler KL. Apoptosis and the cytopathic effects of reovirus. Curr. Top Microbiol. Immunol. 1998;233:23–49. doi: 10.1007/978-3-642-72095-6_2. Reovir.ii. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Pertile TL, Karaca K, Sharma JM, Walser MM. An antiviral effect of nitric oxide: inhibition of reovirus replication. Avian Dis. 1996;40:342–348. [PubMed] [Google Scholar]
- Poggioli GJ, Keefer C, Connolly JL, Dermody TS, Tyler KL. Reovirus-induced G(2)/M cell cycle arrest requires sigma1s and occurs in the absence of apoptosis. J. Virol. 2000;74:9562–9570. doi: 10.1128/jvi.74.20.9562-9570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss CS, Komatsu T. Does nitric oxide play a critical role in viral infections? J. Virol. 1998;72:4547–4551. doi: 10.1128/jvi.72.6.4547-4551.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Tyler KL. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J. Virol. 2004;78:5466–5475. doi: 10.1128/JVI.78.10.5466-5475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Tyler KL. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp. Neurol. 2005;192:331–339. doi: 10.1016/j.expneurol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Richardson-Burns SM, Kominsky DJ, Tyler KL. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J. Neurovirol. 2002;8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- Saura M, Zaragoza C, McMillan A, Quick RA, Hohenadl C, Lowenstein JM, Lowenstein CJ. An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity. 1999;10:21–28. doi: 10.1016/S1074-7613(00)80003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena SK, Mathur A, Srivastava RC. Induction of nitric oxide synthase during Japanese encephalitis virus infection: evidence of protective role. Arch. Biochem. Biophys. 2001;391:1–7. doi: 10.1006/abbi.2001.2360. [DOI] [PubMed] [Google Scholar]
- Serrano F, Enquist LW, Card JP. Pseudorabies virus-induced expression of nitric oxide synthase isoforms. Physiol. Behav. 2002;77:557–563. doi: 10.1016/s0031-9384(02)00913-7. [DOI] [PubMed] [Google Scholar]
- Togo T, Katsuse O, Iseki E. Nitric oxide pathways in Alzheimer’s disease and other neurodegenerative dementias. Neurol. Res. 2004;26:563–566. doi: 10.1179/016164104225016236. [DOI] [PubMed] [Google Scholar]
- Tucker PC, Griffin DE, Choi S, Bui N, Wesselingh S. Inhibition of nitric oxide synthesis increases mortality in Sindbis virus encephalitis. J. Virol. 1996;70:3972–3977. doi: 10.1128/jvi.70.6.3972-3977.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL. Pathogenesis of reovirus infections of the central nervous system. Curr. Top Microbiol. Immunol. 1998;233:93–124. doi: 10.1007/978-3-642-72095-6_6. Reovir.ii. [DOI] [PubMed] [Google Scholar]
- Tyler KL, Squier MK, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J. Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H.W.t., Mann MA, Fields BN, Tyler KL. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 1991;65:6772–6781. doi: 10.1128/jvi.65.12.6772-6781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell R, Markgraf R, Schmidtke M, Gorlach M, Stelzner A, Henke A, Sigusch HH, Gluck B. Nitric oxide donors inhibit the coxsackievirus B3 proteinases 2A and 3C in vitro, virus production in cells, and signs of myocarditis in virus-infected mice. Med. Microbiol. Immunol. 2003;193(23):91–100. doi: 10.1007/s00430-003-0198-6. [DOI] [PubMed] [Google Scholar]
- Zhang J, Snyder SH. Nitric oxide in the nervous system. Annu. Rev. Pharmacol. Toxicol. 1995;35:213–233. doi: 10.1146/annurev.pa.35.040195.001241. [DOI] [PubMed] [Google Scholar]