

Abstract
Reovirus infection provides a classic experimental model system for studying the pathogenesis of viral infections of the central nervous system (CNS), with apoptosis acting as the major mechanism of cell death. The authors have examined the role of signal transducer and activator of transcription (STAT)1, a component of Janus-activated kinase (JAK)-STAT signaling, a pathway implicated in antiviral responses and pathways regulating apoptosis, following reovirus infection. Infection of primary cortical neuron cultures with reovirus serotype 3 strain Abney (T3A) resulted in phosphorylation of STAT1 at sites critical for transcriptional activity. Activated STAT1 was also detected in the brain of neonatal mice following T3A infection, with a nuclear pattern of expression in areas of virus-induced injury. Activation of STAT proteins is typically mediated by JAKs. The authors observed JAK2 phosphorylation (Tyr 1007/1008) in brain lysates from T3A-infected mice. Inhibition of JAK activity with the inhibitor AG-490 blocked reovirus-induced STAT1 activation in neuronal cultures, indicating reovirus-induced STAT activation is JAK dependent. Pretreatment of neuronal cultures with antibody raised against interferon (IFN)-α/βR2 inhibited T3A-induced STAT1 phosphorylation, whereas neither IFN-γ or IFN-γR2 antibody pretreatment had any effect on T3A-induced STAT1 phosphorylation. Mice lacking the STAT1 gene demonstrated increased susceptibility to reovirus infection, with increased mortality and higher viral titers in the brain compared to wild-type animals. The results demonstrate activation of a type I IFN-mediated, JAK-dependent STAT signaling pathway following reovirus infection and suggest that STAT1 is a key component of host defense mechanisms against reovirus infection in the brain.
Keywords: apoptosis, Janus-activated kinase, reovirus, signal transducer and activator of transcription, viral encephalitis
Introduction
Reoviruses have been utilized extensively to study the mechanisms of virus-induced pathogenesis in vivo, particularly the brain, and in several in vitro systems (Schiff et al, 2006; Tyler, 1998). Reoviruses comprise three serotypes (types 1 to 3). Viruses belonging to these serotypes possess very differing patterns of neurovirulence, with serotype 3 viruses (e.g., T3 Abney and T3 Dearing) typically infecting and inducing apoptosis in neurons (neuronotropic) while type 1 serotypes cause ependymitis and hydrocephalus and are considered non-neuronotropic (Schiff et al, 2006).
Reovirus infection of mammalian cells can lead to the induction of interferon (IFN) with the capacity to induce IFN modulated by the particular strain and host in question (see Samuel, 1998). IFNs are a multigene family of cytokines typically grouped as type I (IFN-α and IFN-β) or type II (IFN-γ), although a third type (type III) has also recently been postulated (see Takaoka and Yanai, 2006). Both type I and type II IFNs possess antiviral and immunomodulatory activities and mediate intracellular effects via the JAK-STAT (Janus-activated kinase-signal transducer and activator of transcription) pathway, although the precise mechanisms are known to differ between the two IFN types. Despite substantial evidence of IFN induction by various reovirus strains in a variety of host cell systems (see Samuel, 1998), there has been little examination of the detailed signaling events triggered following this response. Recent studies of gene expression in immortalized cell lines following infection with type 3 reovirus strains have demonstrated reovirus-induced up-regulation of the gene encoding STAT1 (O’Donnell et al, 2006; Smith et al, 2006). However, reovirus-induced effects on the JAK-STAT pathway have yet to be examined at the protein level and this is critical to understand the specific nature of host cell signaling responses to reovirusmediated IFN induction and how this may be linked to cellular survival. Furthermore, there has been no examination of IFN/JAK-STAT signaling responses in reovirus-infected neuronal populations, either in vitro or in vivo.
JAK and STAT1 signaling pathways have recently been shown to regulate inducible nitric oxide synthase (iNOS) expression and NO production in macrophages (Sareila et al, 2006) and proapoptotic mechanisms in cerebellar granule neurons (Loucks et al, 2006). Recent studies in our laboratory have demonstrated induction of iNOS expression and elevated NO production in mouse brain following reovirus infection (Goody et al, 2005). Reovirus-induced mechanisms of apoptosis have been described in vivo in the brain (Oberhaus et al, 1998; Richardson-Burns et al, 2002; Richardson-Burns and Tyler, 2005) and in vitro in primary neuronal cultures (Richardson-Burns et al, 2002; Richardson-Burns and Tyler, 2004; for a recent review, see Clarke et al, 2005). STAT1 may clearly represent a critical focal point for host cells in response to reovirus infection, with a potential role in the regulation of antiviral and proapoptotic responses. It is clear that JAK-STAT signaling following reovirus infection of the central nervous system (CNS) warrants further examination before the interplay between antiviral responses and apoptotic events in the CNS can be fully understood.
We now show that reovirus infection of primary neuronal cultures results in activation of JAK-STAT signaling cascade components. Similar results were found in brain following T3A reovirus infection of neonatal mice, demonstrating a JAK-dependent, IFN-like signaling response involving STAT1 phosphorylation and nuclear translocation. Pretreatment of neuronal cultures with antibody raised against murine IFN-α/β receptor subunit 2 (IFN-α/βR2) resulted in inhibition of T3A-induced STAT1 phosphorylation. In contrast, pretreatment with IFN-γ or IFN-γ receptor subunit 2 (IFN-γR2)-specific antibodies failed to inhibit phosphorylation induced by T3A. We wished to examine the importance of STAT1 up-regulation in the host antiviral response by the use of STAT1 gene-deficient mice. Mice lacking STAT1 suffered increased and accelerated mortality compared to wild-type controls following intracerebral inoculation (i.c.) of T3A and this effect correlated with higher viral titers in the brain. In addition, STAT1 gene-deficient mice were highly susceptible to the non-neuronotropic strain T1L with virus growth also increased in these mice compared to wild-type controls.
Results
Reovirus induces STAT1 up-regulation and activation in primary cortical neuron cultures
The role of JAK-STAT signaling following reovirus infection has not been well studied and the precise role of STAT1 in reovirus infection has yet to be evaluated either in vitro or in vivo. Tyrosine residue 701 (Y701) of STAT1 is a key site of phosphorylation and is required for STAT1’s nuclear translocation and associated STAT1-dependent transcriptional activation. We used a STAT1 Y701- phospho—specific antibody to examine STAT1 activation in primary neuronal cultures by immunocytochemistry and western blot analysis. T3A infection of primary neuronal cultures resulted in a significant increase in the number of Y701-phosphorylated STAT1 (pY701)-positive cells by 24 h post infection, compared to mock-infected controls, with elevated numbers persisting until 48 h post infection (Figure 1A-D).
Figure 1.
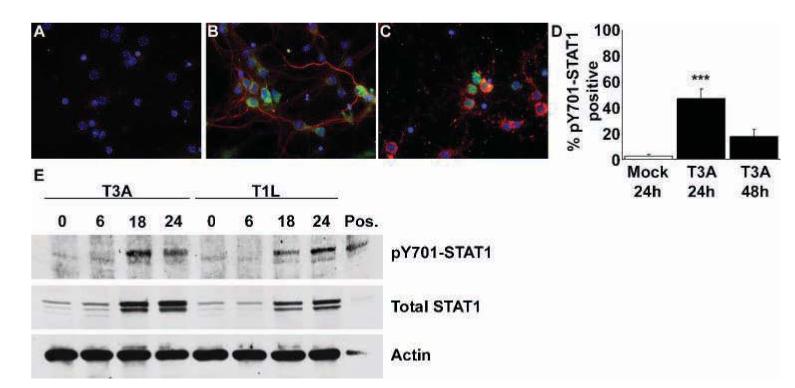
Reovirus induces STAT1 phosphorylation at Y701 in primary neuronal cultures following infection. Cortical neuron cultures were mock or reovirus infected (MOI of 100). Dual-label immunocytochemical staining was performed to identify reovirus antigen σ3 (Texas Red; Virgin et al, 1991) and pY701-STAT1 (fluorescein) immunoreactive populations. Images represent σ3 and pY701-STAT1 immunoreactivity in mock (A) and T3A-infected neuronal cultures at 24 (B) and 48 (C) h post infection, at 400× original magnification. Very few neurons from mock-infected cultures provided any demonstration of pY701-STAT1 immunoreactivity. In T3A-infected cultures significantly higher numbers of pY701-STAT1 immunoreactive neurons were observed at 24 h post infection (B, D), although these numbers diminished by 48 h post infection (C, D). Western blot analysis of primary neuronal cultures infected with T3A and T1L demonstrated increased levels of pY701-STAT1 at 18 to 24 h post infection (E), accompanied by increases in total STAT1 protein levels at the same time points. Values represent the mean of four observations, with a minimum of 300 cells counted per observation, with vertical bars indicating the SEM. ***P < .001.
Western blot analyses confirmed findings from immunocytochemical studies, with T3A inducing Y701-phosphorylation of STAT1 at 18 to 24 h post infection (Figure 1E). Phosphorylated STAT1 (pY701) levels demonstrated an identical pattern of expression to total STAT1 levels in our cultures over the initial 24 h post infection with T3A. T1L also induced up-regulation and phosphorylation of STAT1 with peak levels obtained at 24 h post infection (Figure 1E).
Reovirus induces STAT1 up-regulation and activation in the brain of reovirus-infected neonatal mice
We wished to examine whether reovirus infection in vivo resulted in similar effects upon STAT1 to those seen in vitro. We used a previously described in vivo model (Goody et al, 2005) by which 2-day-old Swiss Webster mouse pups were intracerebrally (i.c.) inoculated with T3A (1 × 103 plaque-forming units [PFU]). Total and phosphorylated STAT1 levels were evaluated in whole-brain lysates collected from these mice at 4 and 8 days post infection. We again observed phosphorylation of STAT1 at Y701, in addition to S727 (a second site of phosphorylation also reported to be critical for full transcriptional activation of STAT1 [Wen et al, 1995]) (Figure 2A), indicating full activation. STAT1 phosphorylation was observed in brain lysates from 4 and 8 days post infection but was consistently detected at much lower levels in brain lysates collected from mice at 4 days post infection. We subsequently wished to confirm that STAT1 was translocating to the nucleus to exert transcriptional activity. We prepared nuclear and cytoplasmic fractions from T3A- and mock-infected neonatal mouse brains at 8 days post infection and probed these lysates for phosphorylated STAT1. We observed both nuclear and cytoplasmic localization of Y701 and S727-phosphorylated STAT1, indicating nuclear translocation (Figure 2B). As described in vitro,we observed dramatic up-regulation of total STAT1 protein levels in brain lysates of T3A-infected mice compared to mock-infected controls at 8 days post infection (data not shown).
Figure 2.
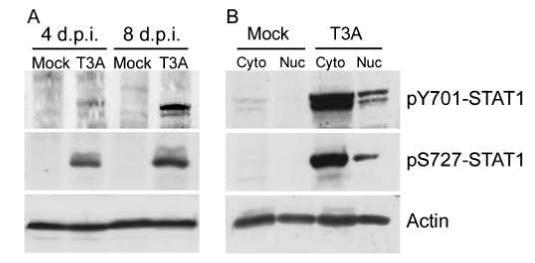
T3A induces STAT1 phosphorylation in the brain of neonatal mice following intracerebral inoculation. Two-day-old Swiss Webster mice were mock or T3A infected (1 × 103 PFU, i.c.) and sacrificed at 4 or 8 days post infection. Whole-brain lysates were prepared from each animal and samples were then probed by Western blot for STAT1 phosphorylated at either Y701 or S727 (A). Nuclear and cytoplasmic brain fractions were prepared from identically treated neonates after sacrifice at 8 days post infection. Subcellular brain fractions from mockand T3A-infected animals were probed for pY701 and pS727 STAT1 (B). Images provide representative observations from a minimum of six animals sampled per treatment.
Y701-Phosphorylated STAT1 is localized to areas of reovirus infection in the brain
We performed dual-label immunohistochemical staining of mock- and T3A-infected (1 × 103 PFU, i.c.) brain tissue, collected from mice at 8 days post infection, to examine colocalization of Y701-phosphorylated STAT1 and viral antigen. We observed no evidence of viral antigen or pY701 STAT1 staining in brain tissue from mock-infected animals (Figure 3). Immunoreactivity for reovirus σ3 protein, a marker of reovirus infection, was detected in the cingulate and frontal parietal cortices, hippocampus and thalamus (Figure 3B, D, F, and H). Immunoreactivity for pY701 STAT1 was also detected in these same brain areas with a predominantly nuclear pattern of expression. However, staining for pY701-STAT1 and σ3 antigen typically occurred in distinct, predominantly neuronal cells within the same brain regions, and only rarely was staining for pY701 STAT1 and viral antigen immunoreactivity colocalized in the same cell. Typically, activated, nuclear STAT1 was observed in cells surrounding reovirus-infected populations rather than within the infected population itself (Figure 3F, G), although there were isolated exceptions, as indicated (Figure 3D). Neuronal expression of pY701-STAT1 was confirmed in brain sections from T3A-infected animals by dual-label immunofluorescencestaining for pY701-STAT1 with the neuronal marker microtubule-associated protein (MAP)-2 (Figure 3J).
Figure 3.
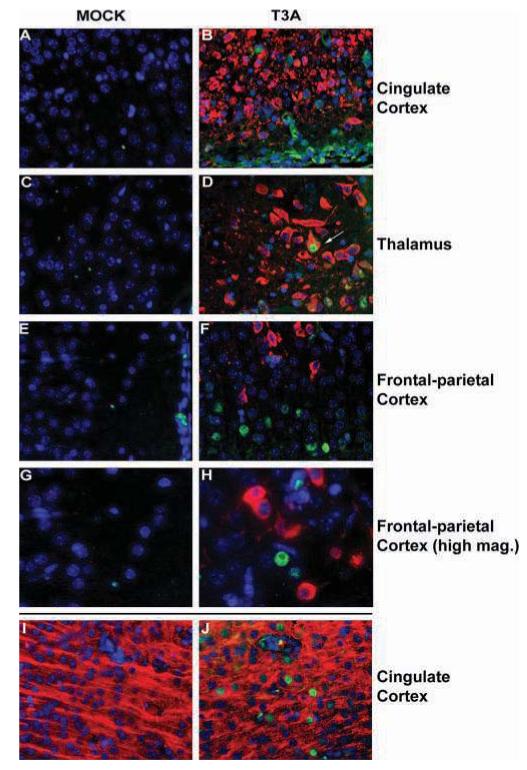
Y701-phosphorylated STAT1 is expressed in areas surrounding brain injury following T3A infection. Two-day-old Swiss Webster mice were mock or T3A infected (i.c. inoculation of 1 × 103 PFU) and sacrificed at 8 days post infection. pY701-STAT1 (fluorescein) and reovirus σ3 (Texas Red; Virgin et al, 1991) were identified by dual-label immunofluorescence (A-G). Fluorescence labeling was performed on brain tissue from mock(A, C, E, G) and T3A (B, D, F, H) infected animals. Images represent staining in the cingulate cortex (A, B), thalamus (C, D), and frontal parietal cortex (E, F at 400× and G, H at 630× [oil] original magnification). Arrow (D) indicates rare example of virus-infected cell demonstrating pY701-STAT1 immunoreactivity. Neuronal expression of activated STAT1 in T3A-infected animals was confirmed by dual label immunofluorescence of pY701-STAT1 (fluorescein) with the neuronal marker MAP-2 (Texas Red; I, J) in cingulate cortex of mock (I) and T3A- (J) infected mice.
Reovirus induces STAT1 activation via a JAK-dependent mechanism
Activation of STAT1 can be mediated via type I (IFN-α/β), or type II IFN (IFN-γ) signaling. IFN-α/β binds to the IFN-α/β receptor and triggers autophosphorylation of JAK proteins (JAK1, JAK2, and TYK2), which consequently tyrosine-phosphorylate STAT1 and STAT2, which then dimerize and form the IFN-stimulated gene factor 3 (ISGF3) complex. IFN-γ acts by binding to the IFN-γ receptor and triggering activation of JAK proteins, which, together, tyrosine-phosphorylate STAT1 to form an active homodimer. JAK-independent mechanisms have also been described that lead to STAT activation (Leaman et al, 1996; Lee et al, 2006; Wanget al, 2006), thus we wished to confirm that JAK activation was involved in STAT1 activation. We examined whole-brain lysates of mock- and T3A-infected animals for Y1007/1008-phosphorylated JAK2 (pJAK2) to further examine this. We found no evidence of JAK2 activation in brain lysates from mock-infected animals. However, we consistently observed pJAK2 in lysates of T3A-infected brains at 8 days post infection (Figure 4A). Consequently, we examined the effect of the JAK inhibitor AG-490 on reovirus-induced STAT1 up-regulation and activation in vitro. Primary neuronal cultures were treated with 50 μM AG-490 immediately prior to infection with T3A, T3D, or T1L (multiplicity of infection [MOI] = 100). Total STAT1 and pY701 STAT1 levels were assessed in wholecell lysates at 24 h post infection (Figure 4B). T3A and T3D both induced similar levels of phosphorylation at Y701 although T1L only weakly induced STAT1 phosphorylation. AG-490 (50 μM) inhibited STAT1 phosphorylation at Y701 induced by all three reovirus strains tested.
Figure 4.

Phosphorylation of STAT1 at Y701 is mediated via JAK activation. Whole-brain lysates were prepared from mock- and T3A-infected (1 × 103 PFU, i.c.) mice at 8 days post infection and probed, by Western blot, for activated JAK2 (using an antibody specific for Y1007/1008-phosphorylated JAK2) and cleaved caspase-3. Phosphorylated JAK2 could only be detected in brain lysates from T3A- and not mock-infected animals (A) and this observation correlated with the presence of the apoptotic marker cleaved caspase-3. The contribution of JAK was also examined in whole-cell extracts from mock- and reovirus-infected primary cortical neurons in the presence and absence of the JAK inhibiter AG-490 (50 μM). Treatment with AG-490 at the time of infection completely blocked activation of STAT1 and up-regulation of total STAT1 protein levels (B). Blots provide representative examples of four repeat studies in brain and neuronal culture lysates.
STAT1 is activated via an IFN-α/β-dependent mechanism
Recent studies have suggested that STAT1 can be activated independently from IFN-triggered signaling responses but still involving JAK activation (Severgnini et al, 2004; Simon et al, 1998; Tacchini et al, 2002). To confirm involvement of IFNs in JAK-mediated STAT1 activation, we examined the effect of inhibiting IFN activity on T3A-induced STAT1 activation. Similar to T3A, recombinant murine IFN-γ (50 ng/ml) induced STAT1 phosphorylation at Y701 by 24 h post infection (Figure 5A). Pretreatment with anti-murine IFN-γ antibody (10 μg/ml) for 4 h resulted in the inhibition of recombinant IFN-γ-induced STAT1 phosphorylation at Y701 (Figure 5A). In contrast, IFN-γ antibody pretreatment had no effect on T3A-induced STAT1 phosphorylation. Pretreatment of neurons with anti-murine IFN-α/βR2 antibody (10 μg/ml) for 24 h prior to infection resulted in the inhibition of T3A-induced STAT1 phosphorylation (Figure 5B). The same pretreatment regimen with anti-murine IFN-γ R2 prior to infection had no effect on T3A-induced STAT1 phosphorylation (Figure 5B).
Figure 5.
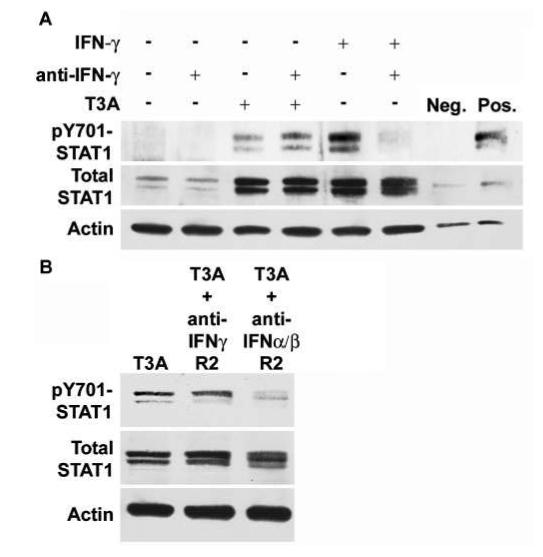
Pretreatment with interferon-specific antibodies has differing effects on T3A-induced STAT1 phosphorylation. Cortical neuron cultures were mock or T3A infected (MOI of 100) following 4 h pretreatment with antibody directed against IFN-γ (10 μg/ml) (A). Whole-cell extracts were collected for Western blot analysis at 24 h post infection and probed for Y701-phosphorylated and total STAT1 protein levels. Recombinant IFN-γ (50 ng/ml) and T3A both induced up-regulation of total STAT1 levels and phosphorylation at Y701. Recombinant IFN-γ -induced STAT1 phosphorylation was inhibited by pretreatment with IFN-γ - directed antibody, although total STAT1 protein up-regulation remained unchanged. T3A-induced STAT1 phosphorylation and total STAT1 up-regulation was not inhibited by IFN-γ antibody pretreatment. Primary cortical neuron cultures were pretreated for 24 h with 10 μg/ml of antibody raised against IFN-γ R2 or IFN-α/βR2 before infection with T3A (MOI = 100) (B). Whole-cell extracts were prepared 24 h post infection and probed for STAT1 activation. T3A-induced STAT1 phosphorylation was unaffected by pretreatment with IFN-γ R2 antibody, similar to observations with IFN-γ antibody. Pretreatment with IFN-α/βR2 resulted in inhibition of T3A-induced STAT1 phosphorylation with no major changes in total STAT1 levels. Images provide representative observations from a minimum of four replicates per treatment.
STAT1 gene-deficient neonatal mice have increased mortality following reovirus infection
We examined the importance of STAT1 in the host response to reovirus infection using STAT1 gene-deficient mice and their syngeneic controls. Newborn STAT1-deficient and wild-type pups were transferred to surrogate Swiss Webster mothers. Mice were inoculated i.c. with 1 × 102 PFU of virus at 2 days of age and monitored twice daily thereafter. Mice lacking STAT1 demonstrated accelerated mortality compared to wild-type controls following T3A infection (Figure 6A) with all inoculated gene knockout mice dying within 11 days of infection (median day of death for wild-type = 14.0; for STAT1 mutants = 9.0; P = .019). Interestingly, we also observed dramatic differences between wild-type and STAT1 gene-deficient animals following T1L infection. The LD50 for T1L in immunocompetent mice is ≥ 1 × 107 PFU/mouse (Tyler et al, 1993). There was 100% mortality in STAT1 gene-deficient animals infected i.c. with 1 × 102 PFU of T1L by 9 days post infection (median day of death = 8.0) (Figure 6A). We examined viral growth in the brain of infected animals and found significantly (approximately 10,000-fold) higher titers of both T3A (P = .049) and T1L (P = .041) in the brains of STAT1-deficient animals than wild-type controls (Figure 6B, C).
Figure 6.
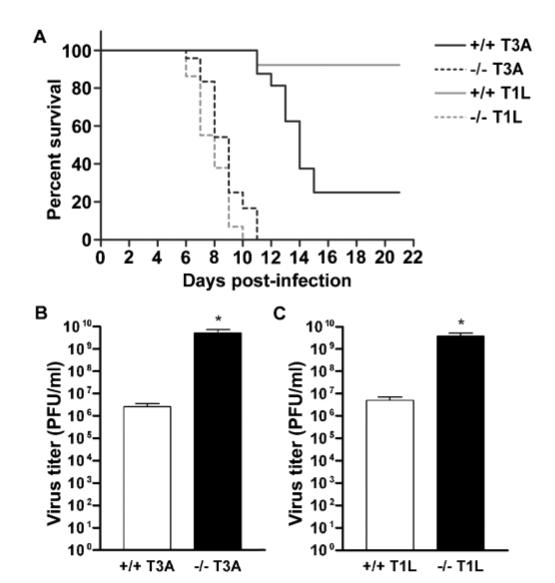
Mice lacking the STAT1 gene suffer accelerated mortality and increased viral brain titers. Newborn STAT1 gene-deficient mouse pups (dashed lines) and syngeneic controls (129SvEv) (solid lines) were transferred to Swiss Webster surrogate mothers and T3A (black) or T1L (gray) infected at 2 days of age with 1 × 102 PFU of virus (i.c.). Survival was assessed in these animals with twice daily monitoring up to 21 days post infection (A). Viral brain titers were assessed following the same viral infection paradigm in T3A- (B) and T1L- (C) infected wild-type (open columns) and STAT1 gene-deficient (closed columns) animals at 7 days post infection. Vertical bars indicate SEM from a minimum of five observations. *P < .05.
Viral spread is not altered in the brain of mice lacking STAT1 gene expression
Observations indicating significantly higher brain titers of T3A and T1L in STAT1 gene-deficient mice raise the question of whether viral spread or tropism within the brain is altered in these mice compared to syngeneic controls. We therefore chose to examine the localization of viral antigen in both wild-type and mutant animals 7 days after i.c. inoculation with 1 × 102 PFU of T3A or T1L. In T3A-infected wild-type animals, viral antigen was distributed in the expected and previously described brain areas, namely the cingulate and frontal parietal cortices, thalamus (data not shown), and hippocampus (Figure 7D, E). In T3A-infected STAT1 gene-deficient animals we again observed the same pattern of localization, although the intensity of staining was especially high in the hippocampus when considering these mice were only at 7 days post infection (Figure 7A, B). Interestingly, despite the intense staining in these areas there was little evidence of major brain pathology in these areas by this time (Figure 7C). We are currently investigating whether the absence of pathology simply reflects this earlier time point post infection at a relatively low inoculation dose or if this is a direct consequence of STAT1 deficiency on mechanisms of pathology. In T1L-infected mice we saw no dramatic difference in viral spread within the brain between wild-type and STAT1 gene-deficient mice at 7 days post infection (Figure 7G, H) and we found little evidence of CNS injury in STAT1 gene-deficient mice at this time (Figure 7I). Neuronal localization of viral antigen was evaluated by dual-label immunofluorescence staining with MAP-2. Following T3A infection, viral antigen was predominantly distributed in neuronal populations, whereas in T1L-infected animals viral antigen showed no evidence of localization within neuronal populations (data not shown).
Figure 7.
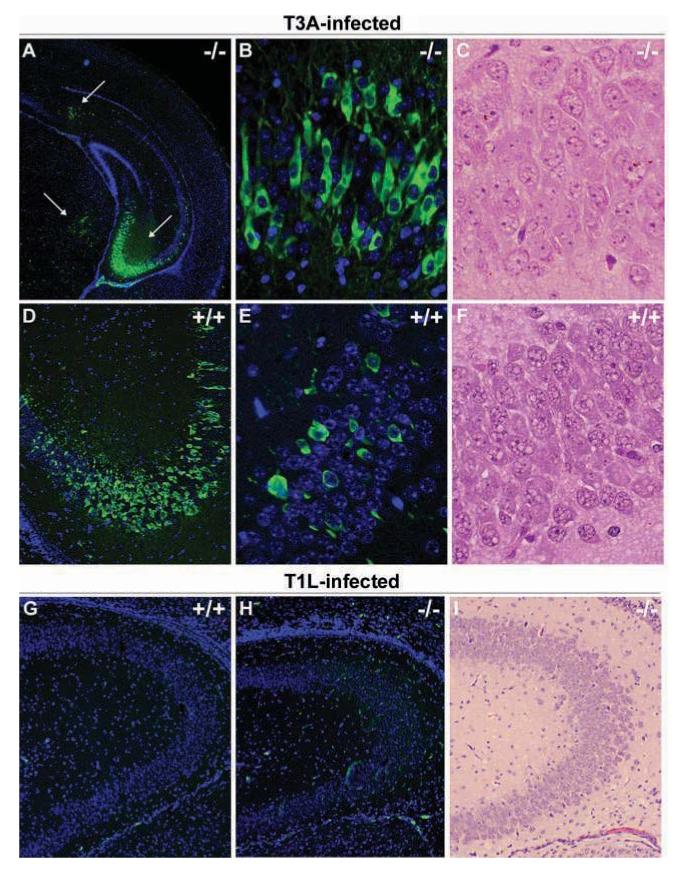
Reovirus localizes to similar brain regions in STAT1 gene-deficient mice. Newborn STAT1 gene-deficient mouse pups and syngeneic controls (129SvEv) were T3A or T1L infected at 2 days of age with 100 PFU of virus. Brain injury was evaluated in H&E-stained sections and adjacent sections stained with monoclonal antibodies specific for σ3 (clone 4F2 for T3A-infected animals) or σ 1 (clone 5C6 for T1L-infected animals) with fluorescein-conjugated anti-mouse secondary antibody used to visualize labeling (A, B, D, E, F, G). Images represent H&E (C, F, I) and dual-label immunofluorescence (A, B, D, E, G, H) staining in the hippocampus of wild-type (+/+) and STAT1 gene knockout (-/-) mice at 25× (A), 100× (D, G-I), and 400× (B, C, E, F) original magnification. Viral antigen staining was not distributed differently between +/+ and -/- animals, although the frequency of immunoreactive populations was higher in -/- animals in hippocampal areas compared to +/+ groups. Viral antigen localization was predominantly neuronal in T3A-infected animals (B, D) and non-neuronal in T1L-infected mice (G).
Discussion
Our results demonstrate that reovirus infection of primary neuronal cultures or intracerebral administration of neuronotropic strains into neonatal mice results in the dramatic up-regulation of STAT1 protein levels. Recent studies using oligonucleotide arrays have indicated that STAT1 gene expression is elevated in reovirus-infected L929 mouse fibroblasts (Smith et al, 2006) and both STAT1 and STAT2 gene expressions are up-regulated in reovirus-infected HeLa cells (O’Donnell et al, 2006); however, our study provides the first examination of STAT1 protein levels following reovirus infection and offers the only evidence of STAT1 dysregulation in neuronal populations following reovirus infection.
Phosphorylation of STAT1 at tyrosine residue 701 (Y701) and serine 727 (S727) are key events required for STAT1 dimerization, nuclear translocation, and transcription factor activity (Kovarik et al, 2001; Shuai et al, 1993). In the current studies we have demonstrated phosphorylation of STAT1 at both Y701 and S727 in the brain of neonatal mice and in primary neuronal cultures following reovirus infection. Furthermore, we have demonstrated the presence of pY701-STAT1 in nuclear fractions of infected neonatal mouse brain by Western blot and in nuclei around areas of virus-induced CNS injury by immunohistochemical staining. In addition, our in vitro and in vivo studies have both demonstrated nuclear expression of pY701-STAT1 in predominantly neuronal populations, as confirmed by colocalization of pY701-STAT1 immunoreactivity with MAP-2, a neuronal marker. An interesting feature of pY701-STAT1 localization in the T3A-infected mouse brain was the proximity of activated STAT1 to areas of viral injury and viral antigen but with a general absence of specific colocalization of pY701-STAT1 and σ3 immunoreactivities. These findings somewhat contrasted in vitro observations where phosphorylated STAT1 was consistently observed in infected populations. Our findings raise the question of whether T3A can block or switch off STAT1 phosphorylation or whether T3A spreads only to neural populations without STAT1 already activated. Protein inhibitors of activated STAT proteins (PIAS proteins) and suppressors of cytokine signaling (SOCS) are two endogenous groups of proteins capable of negative feedback on JAK-STAT signaling and we are currently investigating whether these are involved in the apparent block of STAT1 activation within reovirus-infected populations in vivo.
Activation of STAT1 is typically achieved either via IFN-α/β- or IFN-γ -mediated mechanisms and results in the phosphorylation and dimerization ofSTAT1 either with STAT2 (type I) or formation of a STAT1 homodimer (type II), followed by nuclear translocation and initiation of transcriptional activity (see Chesler and Reiss, 2002; Takaoka and Yanai, 2006). Our studies in brain lysates from T3A-infected mice found evidence of JAK2 activation (as shown by the detection of Y1007/1008-phosphorylated JAK2 in brain lysates at 8 days post infection), suggesting JAKs play a role in reovirus-induced STAT1 activation. Application of the JAK inhibitor AG-490 highlighted a critical role of JAKs in STAT1 activation, demonstrating complete inhibition of reovirus-induced STAT1 phosphorylation in neuronal cultures. Interestingly, AG-490 also blocked up-regulation of total STAT1 protein levels, perhaps reflecting a positive feedback loop through JAK-dependent pathways to regulate STAT1 production. Although STAT1 can be strongly activated by IFNs, STAT proteins can also be activated by oxidative stress (Severgnini et al, 2004; Simon et al, 1998; Tacchini et al, 2002). Our recent studies demonstrated induction of iNOS expression and elevated NO production in the mouse brain following neurotropic reovirus infection (Goody et al, 2005). Expression of iNOS and increased NO production has demonstrated regulation by a JAK-STAT-signaling mechanism with AG-490 able to inhibit IFN-γ -induced iNOS expression and NO production in macrophages via inhibition of JAK-STAT signaling (Sareila et al, 2006). Based on the potential contribution of oxidative stress to STAT1 activation, we examined the role of IFNs in activation of JAK-STAT signaling following reovirus infection. Pretreatment of neuronal cultures with antibody raised against IFN-γ inhibited Y701-phosphorylation of STAT1 induced by recombinant IFN-γ. However, the same pretreatment regimen had no effect on T3A-induced STAT1 phosphorylation at Y701, thus suggesting that T3A induces STAT1 activation in the brain and neuronal cultures via an IFN-γ -independent mechanism. A role for IFN-α/β in T3A-induced STAT1 phosphorylation was confirmed by the inhibition of T3A-induced phosphorylation of STAT1 following pretreatment with antibody raised against IFN-α/βR2. In the same experiments, pretreatment with IFN-γ R2 antibody had no effect on T3A-induced STAT1 phosphorylation, confirming that type II IFN signaling is unlikely to be involved in reovirus-induced JAK-STAT signaling. Phosphorylation of STAT2 at Y689 was also observed in primary neuronal cultures following T3A infection (data not shown), suggesting a STAT1-STAT2 heterodimer may be formed, which would further support our evidence of a type I IFN signaling response. We are currently investigating these observations more closely to confirm whether there is a link between JAK-STAT signaling and reovirus-induced iNOS expression and increased NO production.
STAT1-deficient mice, when initially described, showed increased susceptibility to infections (Durbin et al, 1996; Merazet al, 1996). However, although absence of the STAT1 gene clearly diminishes the antiviral response, not all models of viral infection have demonstrated increased lethality in mice lacking the STAT1 gene. For example, intracranial inoculation of mice with lymphocytic choriomeningitis virus (LCMV) is lethal in wild-type mice by 6 to 8 days post infection (Ousman et al, 2005). In STAT1 gene knockout mice LCMV produced lethal disease between 7 and 14 days post infection (Ousman et al, 2005). A critical role for STAT1 has also been described in vesicular stomatitis virus (VSV) infection (Chesler et al, 2004). IFN-γ treatment of a neuroblastoma cell line inhibits VSV growth via a NOS-dependent pathway and Chesler and co-workers (2004) recently demonstrated that STAT1 activation is required for this mechanism of VSV inhibition to occur (Chesler et al, 2004).
Following T3A infection, STAT1 knockout mice suffered accelerated mortality compared to wild-type animals and this was accompanied by increased viral titers in the mutant animals. Despite these observations, viral spread within the CNS was not grossly altered. Neuronal infection is not a typical feature of T1L infection, which is more commonly associated with ependymitis and hydrocephalus (Tyler, 1998). The LD50 of T1L following i.c. inoculation is approximately 107 PFU/mouse in neonatal mice (see Tyler, 1998) and it was therefore surprising to observe such rapid onset of fatal disease in T1L-infected STAT1 knockout mice with challenge doses of 1 × 102 PFU. Viral titers were significantly higher (750-fold) in the brain of these animals than syngeneic controls; however, viral tropism within the CNS was not altered with viral antigen observed predominantly in ependymal cells, as has previously been described in wild-type animals (see (Tyler, 1998)).
In conclusion, the findings presented here provide the first evidence of JAK-STAT signaling in neuronal cultures and brain tissue of neonatal mice following reovirus infection. A critical role for JAK activation and IFN-α/β activity in T3A-induced STAT1 phosphorylation is suggestive of a classical type I IFN signaling pathway. In mice lacking the STAT1 gene we observed dramatically higher mortality rates following type 3 and type 1 reovirus infection and this corresponded with significantly higher viral titers in the brain. We are currently investigating the role of oxidative stress elements in the initiation of JAK-STAT signaling pathways in the brain and the precise role STAT proteins play in reovirus-induced apoptosis.
Materials and methods
Cell lines and viruses
L929 mouse fibroblasts (ATCC CL1) were used for viral titer assays and were maintained in 2X199 medium supplemented with 10% heat-inactivated fetal bovine serum and 4 mM L-glutamine for these studies.
Reovirus strains type 3 Abney (T3A) and Dearing (T3D) and type 1 Lang (T1L) are laboratory stocks which have been plaque purified and passaged (twice) in L929 cells to generate working stocks (Tyler et al, 1996). Virus infections in vitro were performed at a multiplicity of infection (MOI) of 100 to ensure that 100% of susceptible cells were infected and to maximize the synchrony of viral replication.
Primary cell culture
Primary neuronal cultures were prepared from the cortices of embryonic day 16 Swiss Webster Hsd:nd4 mice (Harlan-Sprague Dawley, Indianapolis, IN) as described previously (Goody et al, 2005). Briefly, fetuses were removed aseptically by cesarean section, brains removed, and cortices dissected. Cortices were dissociated by trituration in Neurobasal medium supplemented with 1% fetal bovine serum (FBS) and L-glutamine (0.6 mM) and viable cells plated at a density of 1 × 106 cells per well on to poly-d-lysine-coated 6-well plates (Biocoat; Becton Dickinson, Franklin Lakes, NJ) for Western blotting. For immunocytochemical studies, cells were plated at a density of 5 × 104 cells per well on to poly-dlysine-coated coverslips (Biocoat).
Cultures were plated out and maintained in serumfree Neurobasal medium supplemented with B-27 nutrient supplement (2% v/v), antibiotic/ antimycotic (penicillin/streptomycin; 1000 units/ml), and l-glutamine (0.6 mM). All studies were performed on primary neuronal cultures at 7 to 9 days in vitro. All culture media and supplements were purchased from Invitrogen unless stated otherwise.
Cortical neurons were characterized both morphologically and immunocytochemically using a mouse monoclonal antibody directed against microtubule-associated protein-2 (MAP-2; 1:100; Abcam). Primary neuronal cultures were typically comprised of 90% to 95% neurons.
Drugs
AG-490 (Tryphostin) was purchased from Sigma Aldrich (St. Louis, MO). AG-490 has been shown to exert inhibitory activity against both JAK1 and JAK2 (Yamauchi et al, 2006). IFN-γ and murine IFN-γ -, IFN-γ R2-, and IFN-α/βR2-specific antibodies were purchased from R&D systems (Minneapolis, MN).
Subcellular fractions
Brains were dissected from mice at 4 and 8 days post infection and stored in 1 ml sterile phosphate-buffered saline (PBS) at -80°C. Samples were thawed, PBS removed, and subcellular fractions prepared, as described previously (Spencer et al, 2000). Briefly, tissue was homogenized in a dounce homogenizer with 500 μl of homogenization buffer (50 mM Tris HCl, 6 mM MgCl2, 1 mM EDTA, 10% w/v sucrose, 3 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride [PMSF], 5 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 μg/ml antipain, 1 μg/ml aprotinin, and 1 μg/ml soybean trypsin inhibitor). Samples were centrifuged at 2000 ×g for 5 min. Supernatant was removed, further centrifuged at 105,000 ×g for 30 min, and the subsequent supernatant isolated and stored at -80°C to provide the cytosolic fraction. The pellet produced following the first centrifugation step was washed twice before incubating on ice with 250 μl of homogenization buffer containing 0.5 mM NaCl for 1 h. At the end of the hour samples were centrifuged at 8000 ×g for 10 min and final supernatant transferred to a fresh Eppendorf tube and stored at -80°C to provide the nuclear fraction. Nuclear and cytoplasmic fractions were mixed with 250 μl of Laemmli buffer and boiled for 5 min before Western blot analysis.
Western blotting
Whole-cell extracts were prepared from primary neuronal cultures as described previously following viral infection (Goody et al, 2005). Whole-brain lysates were also prepared as has previously been described (Goody et al, 2005). Lysates were boiled for 5 min and electrophoresed (Hoefer Pharmacia Biotech, San Francisco, CA) in 10% or 12% tricine/polyacrylamide gels at a constant voltage of 70 V through the resolving gel. Proteins were electroblotted onto Hybond-C nitrocellulose membranes (Amersham Biosciences) and immunoblotting performed as described previously (Poggioli et al, 2000). Immunoblots were probed with antibodies directed against STAT1, phosphorylated STAT1 (Y701 and S727) (Cell Signaling Technology, Danvers, MA), and actin (Calbiochem, Sunnyvale, CA).
In vivo studies
Two-day-old Swiss Webster pups were pooled together from multiple litters and split randomly amongst surrogate mothers (8 to 10 pups per litter). Pups were intracerebrally (i.c.) inoculated with 1 × 103 plaque-forming units (PFU) of virus (T3A) in a 10-μl volume as described previously (Richardson-Burns et al, 2002). At 8 days post infection mice were sacrificed and brain tissue removed for histological studies or organ lysates. STAT1 gene-deficient mice (129S6/SvEvtm1Rds; Taconic, Hudson, NY) and syngeneic controls (129S6/SvEv) were bred, providing homozygous STAT1-deficient and wild-type offspring, respectively. At 2 days of age, pups were transferred to Swiss Webster surrogates before viral infection. Pups were inoculated i.c. with T3A or T1L (1 × 102 PFU). Mice were examined and weighed daily. All experiments performed conformed to local institutional animal care and use committee (IACUC) guidelines on the ethical use of animals.
Histological studies
Brain tissue was fixed in 10% formalin for 20 h at room temperature. Tissue was transferred to 70% ethanol before paraffin embedding and cutting of sections. Coronal brain sections (4 μm thick) were prepared and deparaffinized in xylene and rehydrated in consecutive 100% to 80% ethanol washes. Antigen retrieval was performed using 10 mM citrate buffer (pH 6.0). Tissue sections were permeabilized in Neuropore (Trevigen, Gaithersberg, MD) overnight at 4°C and blocked in 10% normal goat serum (NGS; in 1× Tris-buffered saline with 0.1 % TWEEN [TBST]) for 6 h at room temperature. Sections were incubated overnight at 4°C with rabbit polyclonal antibodies directed against Y701-phosphorylated STAT1 (Cell Signaling Technology) diluted in 3% bovine serum albumin in TBST. Following washes with TBST, sections were incubated with fluorescein-conjugated goat anti-rabbit antibody (1:100; Vector Laboratories) diluted in 10% NGS/TBST for 2 h at room temperature. For dual-label studies, tissue sections were incubated with a reovirus σ3-specific monoclonal antibody (Virgin et al, 1991) (4F2; 1:100 in 10 % NGS) or reovirus σ 1-specific monoclonal antibody (Virgin et al, 1991) (5C6; 1:100 in 10 % NGS) overnight before washing with TBST, incubating with Texas Red-conjugated goat anti-mouse secondary antibody (1:100; Vector Laboratories) for 2 h, washing again with TBST and incubating for 20 min with 1 μg/ml Hoechst 33342 (Invitrogen). Sections weremounted using VectorShield (Vector Laboratories). Immunostaining for digital immunofluorescence was visualized using a Zeiss Axioplan 2 digital deconvolution microscope with a Cooke Sensicam 12-bit camera.
Viral titer assays
Tissues prepared for determination of viral titer were immediately transferred to 1 ml of sterile PBS upon removal and stored at -80°C. Samples were freezethawed on three separate occasions before sonication. Serial dilutions of homogenized brain tissue were prepared in gel saline and viral titer determined by plaque assays as previously described (Debiasi et al, 1999).
Statistical analysis
Viral titers were assessed using a two-tailed, unpaired t test with Welch correction. Survival rates were assessed by the log-rank test. All statistical analyses were performed using Instat and Prism (GraphPad Software, San Diego, CA).
References
- Chesler DA, Dodard C, Lee GY, Levy DE, Reiss CS. Interferon-gamma-induced inhibition of neuronal vesicular stomatitis virus infection is STAT1 dependent. J Neurovirol. 2004;10:57–63. doi: 10.1080/13550280490261707. [DOI] [PubMed] [Google Scholar]
- Chesler DA, Reiss CS. The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002;13:441–454. doi: 10.1016/s1359-6101(02)00044-8. [DOI] [PubMed] [Google Scholar]
- Clarke P, Debiasi RL, Goody R, Hoyt CC, Richardson-Burns S, Tyler KL. Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation. Viral Immunol. 2005;18:89–115. doi: 10.1089/vim.2005.18.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiasi RL, Squier MK, Pike B, Wynes M, Dermody TS, Cohen JJ, Tyler KL. Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J Virol. 1999;73:695–701. doi: 10.1128/jvi.73.1.695-701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Goody RJ, Hoyt CC, Tyler KL. Reovirus infection of the CNS enhances iNOS expression in areas of virus induced injury. Exp Neurol. 2005;195:379–390. doi: 10.1016/j.expneurol.2005.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik P, Mangold M, Ramsauer K, Heidari H, Steinborn R, Zotter A, Levy DE, Muller M, Decker T. Specificity of signaling by STAT1 depends on SH2 and C-terminal domains that regulate Ser727 phosphorylation, differentially affecting specific target gene expression. EMBO J. 2001;20:91–100. doi: 10.1093/emboj/20.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman DW, Pisharody S, Flickinger TW, Commane MA, Schlessinger J, Kerr IM, Levy DE, Stark GR. Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol Cell Biol. 1996;16:369–375. doi: 10.1128/mcb.16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5:8–19. doi: 10.1158/1535-7163.MCT-05-0069. [DOI] [PubMed] [Google Scholar]
- Loucks FA, Le SS, Zimmermann AK, Ryan KR, Barth H, Aktories K, Linseman DA. Rho family GTPase inhibition reveals opposing effects of mitogen-activated protein kinase kinase/extracellular signal-regulated kinase and Janus kinase/signal transducer and activator of transcription signaling cascades on neuronal survival. J Neurochem. 2006;97:957–967. doi: 10.1111/j.1471-4159.2006.03802.x. [DOI] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Oberhaus SM, Dermody TS, Tyler KL. Apoptosis and the cytopathic effects of reovirus. Curr Top Microbiol Immunol. 1998;233:23–49. doi: 10.1007/978-3-642-72095-6_2. [DOI] [PubMed] [Google Scholar]
- O’Donnell SM, Holm GH, Pierce JM, Tian B, Watson MJ, Chari RS, Ballard DW, Brasier AR, Dermody TS. Identification of an NF-kappaB-dependent gene network in cells infected by mammalian reovirus. J Virol. 2006;80:1077–1086. doi: 10.1128/JVI.80.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, Wang J, Campbell IL. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J Virol. 2005;79:7514–7527. doi: 10.1128/JVI.79.12.7514-7527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggioli GJ, Keefer C, Connolly JL, Dermody TS, Tyler KL. Reovirus-induced G(2)/M cell cycle arrest requires sigma1s and occurs in the absence of apoptosis. J Virol. 2000;74:9562–9570. doi: 10.1128/jvi.74.20.9562-9570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Kominsky DJ, Tyler KL. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J NeuroVirol. 2002;8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- Richardson-Burns SM, Tyler KL. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J Virol. 2004;78:5466–5475. doi: 10.1128/JVI.78.10.5466-5475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Tyler KL. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp Neurol. 2005;192:331–339. doi: 10.1016/j.expneurol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Samuel CE. Reoviruses and the interferon system. Curr Top Microbiol Immunol. 1998;233 Reoviruses II:125–145. doi: 10.1007/978-3-642-72095-6_7. [DOI] [PubMed] [Google Scholar]
- Sareila O, Korhonen R, Karpanniemi O, Nieminen R, Kankaanranta H, Moilanen E. JAK inhibitors AG-490 and WHI-P154 decrease IFN-gamma-induced iNOS expression and NO production in macrophages. Mediators Inflamm. 2006;2006(2):1–7. doi: 10.1155/MI/2006/16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff LA, Nibert ML, Tyler KL. Orthoreoviruses and their replication. In: Knipe D, Howley P, editors. Fields virology. Lippincott, Williams & Wilkins; Philadelphia: 2006. pp. 1853–1915. [Google Scholar]
- Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL, Cochran BH, Simon AR. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1282–L1292. doi: 10.1152/ajplung.00349.2003. [DOI] [PubMed] [Google Scholar]
- Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–C1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. Reovirus induces and benefits from an integrated cellular stress response. J Virol. 2006;80:2019–2033. doi: 10.1128/JVI.80.4.2019-2033.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer RL, Kalman BA, Cotter CS, Deak T. Discrimination between changes in glucocorticoid receptor expression and activation in rat brain using western blot analysis. Brain Res. 2000;868:275–286. doi: 10.1016/s0006-8993(00)02341-6. [DOI] [PubMed] [Google Scholar]
- Tacchini L, Fusar-Poli D, Bernelli-Zazzera A. Activation of transcription factors by drugs inducing oxidative stress in rat liver. Biochem Pharmacol. 2002;63:139–148. doi: 10.1016/s0006-2952(01)00836-x. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Tyler KL. Pathogenesis of reovirus infections of the central nervous system. Curr Top Microbiol Immunol. 1998;233 Reoviruses II:93–124. doi: 10.1007/978-3-642-72095-6_6. [DOI] [PubMed] [Google Scholar]
- Tyler KL, Mann MA, Fields BN, Virgin HW. Protective anti-reovirus monoclonal antibodies and their effects on viral pathogenesis. J Virol. 1993;67:3446–3453. doi: 10.1128/jvi.67.6.3446-3453.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, Squier MK, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Mann MA, Fields BN, Tyler KL. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol. 1991;65:6772–6781. doi: 10.1128/jvi.65.12.6772-6781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kurosaki T, Corey SJ. Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-independent pathway. Oncogene. 2006;26:2851–2859. doi: 10.1038/sj.onc.1210092. [DOI] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Osuka K, Takayasu M, Usuda N, Nakazawa A, Nakahara N, Yoshida M, Aoshima C, Hara M, Yoshida J. Activation of JAK/STAT signalling in neurons following spinal cord injury in mice. J Neurochem. 2006;96:1060–1070. doi: 10.1111/j.1471-4159.2005.03559.x. [DOI] [PubMed] [Google Scholar]