

Abstract
Neutral lipids are an important class of hydrophobic compounds found in all cells that play critical roles from energy storage to signal transduction. Several distinct structural families make up this class, and within each family there are numbers of individual molecular species. A solvent extraction protocol has been developed to efficiently isolate neutral lipids without complete extraction of more polar phospholipids. Normal-phase HPLC was used for the separation of cholesteryl esters (CEs), monoalkylether diacylglycerols, triacylglycerols, and diacylglycerols in a single HPLC run from this extract. Furthermore, minor lipids such as ubiquinone-9 could be detected in RAW 264.7 cells. Molecular species that make up each neutral lipid class can be analyzed both qualitatively and quantitatively by on-line LC-MS and LC-MS/MS strategies. The quantitation of >20 CE molecular species revealed that challenging RAW 264.7 cells with a Toll-like receptor 4 agonist caused a >20-fold increase in the content of CEs within cells, particularly those CE molecular species that contained saturated (14:0, 16:0, and 18:1) fatty acyl groups. Longer chain CE molecular species did not change in response to the activation of these cells.
Supplementary key words: cholesteryl esters, triacylglycerol, monoalkyl diacylglycerol, ubiquinone-9, high-performance liquid chromatography, molecular species, RAW 264.7, Kdo2-lipid A
Neutral lipids constitute a family of important biomolecules central to energy storage and energy production in all cells (1). Several different families of neutral lipid compounds are present in all cells, including triacylglycerols (TAGs), diacylglycerols (DAGs), cholesteryl esters (CEs), and cholesterol itself. Superimposed on the mixture of different lipid subtypes found in cells is the observation that the glycerolipids exist as a complex mixture of molecular species that differ by fatty acyl groups, whereas the neutral CEs are a less diverse assembly of fatty acyl groups esterified to cholesterol. Methods have been developed to unambiguously determine the fatty acyl groups making up TAGs and DAGs, for example, using MS3 to deduce molecular species identity (2).
Each one of these lipids is thought to play an important role in cellular homeostasis, yet little is known about the importance of individual molecular species in such processes. It is clear that CE turnover (synthesis/hydrolysis/resynthesis) takes place during normal cellular biochemistry, such as in the CE cycle (3), and that many, if not the majority, of the glyceryl esters and CEs are stored in specialized intracellular lipid bodies (4). Understanding the importance of lipid bodies is continuing to reveal interesting information about this unique organelle with its single phospholipid boundary layer and associated proteins (5, 6), yet our level of understanding of molecular species distributions within this subcellular structure is minimal. Although not typically thought to reside in lipid bodies, DAGs serve as central intermediates in the biosynthesis of many other fatty acyl-containing lipids, including phospholipids, CEs, and TAGs, as well as signaling intermediates as products of phospholipase C hydrolysis of phospholipids (7).
CEs are a storage form of cholesterol found in lipid bodies of virtually all cells. The recycling of cholesterol and intracellular cholesterol trafficking involve the action of CE hydrolase as well as ACAT1 in most cells (8). A second enzyme, ACAT2, has been found in intestinal cells (9). The transport of CE by LDL from the liver to peripheral cells and hydrolysis by LCAT are critical events for the homeostasis of cholesterol in the intact animal. The important role played by these enzymes is also recognized as a potential pharmaceutical target for diseases such as atherosclerosis (10, 11).
There have been several approaches to quantitate molecular species of neutral lipids, exemplified by TAG at the individual molecular species level, using techniques such GC-MS, atmospheric pressure chemical ionization, and ESI (12). GC-MS with electron ionization has the advantage of providing very high chromatographic resolution of components, but at the expense of requiring thermostability for all TAGs undergoing analysis. The capillary GC column often exceeds 325°C (13), and competitive olefin formation by fatty acid elimination can occur at these temperatures (14). Electron ionization is also less than ideal because it does not provide molecular ion information, and extensive fragmentation of the TAGs takes place. The atmospheric pressure chemical ionization technique provides molecular weight information and decomposition ions corresponding to fatty acyl groups; unfortunately, the energetics of this process and the lack of outstanding chromatographic separation of all molecular species has left this approach primarily focused on the analysis of simple mixtures of TAGs, such as those found in vegetable oils like olive oil (15). ESI has excellent sensitivity for the detection of TAGs and DAGs, but only molecular weight information is generated, requiring tandem mass spectrometry to generate the additional data concerning each fatty acyl group.
Analyzing individual lipid families within mixtures of closely related compounds has led to approaches that rely on reducing the complexity of the mixture. The first step in most analytical methods has been organic solvent extraction of neutral lipids using chloroform/methanol-based systems (16). TLC methods have provided good separation of neutral lipids based on polar separations, but some lipids can be particularly prone to oxidation during TLC (17). Alternative methods include solid-phase extraction (18) and normal-phase (NP) liquid chromatography that has also been used widely for such separations (19–21). Recently, Sommer et al. (22) reported a normal-phase (NP)-HPLC-MS separation of nonpolar lipids that represented an improvement in separation and detection. Despite excellent separation, a number of factors, such as long equilibration times, extensive column washing, peak tailing, and coelution, have minimized widespread application. To that end, we report here a reliable and automatable method for the efficient separation and isolation of neutral lipid classes using NP-HPLC. Such a separation scheme, when combined with mass spectrometric strategies, can be used to identify and quantitate individual species of neutral lipids found within cellular systems.
Experimental Procedures
Materials
[13C18]oleic acid, having >99 atom% excess 13C18, was obtained from Spectra Stable Isotopes (Columbia, MD). CE reference standards were obtained from Nu-Check Prep, Inc. (Elysian, MN). Stable isotope TAG and DAG internal standards containing the labeled [1,1,2,3,3]D5 glycerol backbone substituted with various fatty acyl components, as tabulated previously (23), were obtained form Avanti Polar Lipids (Alabaster, AL). The monoalkylether diacylglycerol (MeDAG) internal standard, 1-O-pentadecanyl-2,3-(9Z-octadecenoyl)-sn-glycerol, was also obtained from Avanti Polar Lipids. Ubiquinone-9 was obtained from Fluka Chemical Corp. (Milwaukee, WI). All solvents were HPLC or Optima grade and were obtained from Sigma-Aldrich (Milwaukee, WI) and/or Fisher Scientific (Fair Lawn, NJ). Other reagents were commercially obtained and were of the highest quality available.
Synthesis of [13C18]oleate CE internal standard
To a solution of [13C18]oleic acid (0.2 mmol), cholesterol (0.2 mmol), and 20 mol% dimethylaminopyridine in 2 ml of anhydrous dichloromethane, a solution of dicyclohexyldicarbodiimide (0.2 mmol) was added drop-wise in 1 ml of anhydrous dichloromethane. The mixture was stirred overnight (∼18 h), contents were then filtered, the filtrate was collected in a clean vial, and the solvent was removed under a stream of N2. The resulting colorless syrup was purified using a Superclean (3 ml) LC-Si solid-phase extraction column (Supelco, Bellefonte, PA) with the mobile phase being 100% hexane. The combined fractions were dried under a stream of N2 to yield 60 mg (47%) of a white, free-flowing powder. The synthetic material showed one spot of expected Rf by TLC, the precursor ion and full scan spectra contained only the ammoniated parent ion, m/z 686.8, and the collision-induced decomposition mass spectrum yielded an abundant ion at m/z 369, consistent with the structure (data not shown). The synthetic [13C18]oleate CE internal standard produced a linear calibration curve against the commercial reference standard of cholesteryl oleate (r2 = 0.999) with a slope of 1.03.
Sample preparation
Cell culture, Kdo2-lipid A preparation, and cell stimulation were carried out essentially as described previously (24). Briefly, RAW cells were stimulated with Kdo2-lipid A (100 ng/ml) and incubated for 0.5, 1, 2, 4, 8, 12, and 24 h. Control samples treated with vehicle were incubated for these same times. After treatment, cells were washed twice with ice-cold Dulbecco's phosphate-buffered saline (DPBS) and scraped into vials using 2 ml of DPBS. From the DPBS suspension, 100 μl aliquots were reserved for DNA analysis and the remaining cells were lysed with 1 min of sonication. To the cell lysate was then added 75:25 (v/v) isooctane-ethyl acetate (2 ml) containing the internal standards. Internal standards were added in the following amounts: 300 pmol of [13C18]18:1-CE, 60 pmol of 15:0/18:1/18:1, and 40 pmol of each TAG and DAG d5 internal standard. The vials were again sonicated for 1 min and finally vortexed vigorously before storage as a biphasic mixture at −20°C. Stored samples were thawed and vortexed for 30 s before analysis. The phases were then separated by centrifugation (7 min, 1,000 g), and the organic layer was transferred to a clean vial. Additionally, 75:25 (v/v) isooctane-ethyl acetate (2 ml) was added, and the extraction process was repeated. The combined organic layers were dried under a stream of N2. The residue was transferred to a 300 μl conical glass vial with 2 X 100 μl of dichloromethane. The dichloromethane was removed under a stream of N2, 4.5% methyl tert-butyl ether (MTBE; 55 μl) in hexane was added, and the vials were vortexed. The prepared samples were placed immediately in the cooled autosampler to limit evaporation before injection.
DNA was quantified according to the manufacturer's instructions using the Quant-IT kit from Molecular Probes (Eugene, OR). Briefly, 10 μl of the reserved sample aliquot and 190 μl of the supplied reagent, properly diluted in the supplied buffer, were added to a black 96-well plate. Results from a fluorometer (excitation wavelength, 510 nm; emission wavelength, 527 nm) were compared with a standard curve to determine the amount of DNA. Sufficient sample was reserved from each cell aliquot to allow for triplicate DNA analysis.
In separate experiments, two aliquots of 20 X 106 untreated RAW cells each were collected in 2 ml of DPBS. One aliquot was extracted as described above using 2 X 2 ml of 75:25 (v/v) isooctane-ethyl acetate, the other using the method of Bligh and Dyer (16). At the start of each extraction, the samples were spiked with 5 μCi of [9,103H(N)]triolein (Perkin-Elmer Life and Analytical Sciences, Boston, MA). The quantity of radioactivity in these solutions was measured by scintillation counting before and after extraction, with the remainder of the extraction reserved for analyses by TLC.
Chromatographic separation
TLC separations were carried out on prewashed and activated Gel G TLC plates (Alltech, Deerfield, IL), which were developed using a mobile phase consisting of 80:20:1 (v/v/v) hexane-ethyl acetate-acetic acid. Spots were visualized by spraying with phosphomolybdic acid (20 weight% in ethanol) (Sigma-Aldrich), followed by development on a hot plate. Spots were identified by comparing their Rf values with those of standards.
For separation by NP-LC, the prepared sample solution (40 μl) was loaded onto a 250 X 4.6 mm, 5 micron, Ultremex Silica column fitted with a 2 X 4 mm silica guard cartridge, both from Phenomenex (Torrance, CA). The neutral lipid classes were eluted with a 1 ml/min gradient of MTBE in hexane as follows: 4.5% MTBE was isocratic from 0 to 10 min; from 10 to 20 min, MTBE was ramped to 45%, where it remained for 1 min; from 21 to 22 min, MTBE was returned to 4.5%, where it remained until the end of the run at 27 min. An adjustable splitter positioned just after the column directed 90% of the eluent (900 μl/min) into a fraction collector collecting 1 min/tube fractions. The remaining 10% of the eluent (100 μl/min) was then modified by the addition of 30 μl/min 10 mM ammonium acetate in 45:45:5:5 (v/v/v/v), isopropanol-acetonitrile-water-dichloromethane (hereafter referred to as the electrospray solvent) delivered by a third HPLC pump via a mixing tee on the electrospray source. This composition of the electrospray solvent system was empirically derived. The mixture was found to dissolve adequate concentrations of neutral lipids as well as ammonium acetate (10 mM) required for a stable electrospray.
Mass spectrometry
Online LC-MS and LC-MS/MS were performed on a Sciex API QTRAP hybrid quadrupole/linear ion trap mass spectrometer (Applied Biosystems, Toronto, Canada). Modified eluent was introduced into the electrospray source at 130 μl/min. The mass spectrometer was operated in positive ion mode with a spray voltage of 5,000 V. For the first 4.5 min, eluent was monitored for CE species by precursor ion scanning. Precursor of m/z 369.3 scans, from m/z 500 to 800, were collected every 3 s with a collision energy of 22 V. Full scans, from m/z 500 to 1,000, were collected every 3 s from 4.5 min until the end of the program at 27 min.
In addition to online mass spectrometry, some HPLC fractions were combined after each NP-LC-MS run to isolate a given class of lipid or other desired component(s). The solvent was removed under a stream of N2 and was diluted with electrospray solvent (200 μl). Nanospray infusion of selected samples was performed on a Sciex API4000 QTRAP hybrid quadrupole/linear ion trap mass spectrometer (Applied Biosystems) equipped with a NanoMate 100 nanoelectrospray ionization source (Advion Biosciences, Ithaca, NY). The NanoMate was operated in positive ion mode with a spray voltage of 1.7 kV, vented head space, and a gas pressure of 1.1 p.s.i. MS/MS data were collected using enhanced product ion experiments, collecting and averaging ∼20 scans from m/z 100 to 800. The mass spectrometer was operated with different collision energy for alkylether DAGs (50 V) and unknowns (70 V).
Quantitation of CE molecular species
CE analyses were carried out on isolated CE fractions. Samples were isolated as the NP-HPLC fraction between 3 and 4.5 min, prepared, and infused as described above. Identification of CE molecular species present in each sample was performed by precursor ion scanning (precursors of m/z 369.3) to determine those ion transitions to be monitored in subsequent quantitative analyses. Quantitative measurements were made by multiple reaction monitoring of all species detected. Each transition for ammoniated parent ions converted to m/z 369.3 was scanned for 200 ms, resulting in ∼10 duty cycles/min for the CE molecular species detected in RAW cells. The mass spectrometer was operated with a collision energy of 22 V. Data were collected and averaged over a 3 min period, during which overall signal intensity remained relatively stable.
Calibration curves were generated for 10 CEs. Stock solutions (10 mg/ml in dichloromethane) of reference standards were gravimetrically prepared. Then, aliquots were diluted to the appropriate concentration, which also contained 300 pmol of the stable isotope-labeled CE internal standard. Each sample was then diluted with 500 μl of the electrospray solvent. Calibration curves were generated and statistically evaluated for each species using Sigma Plot 2001 version 7.0 (SPSS, Inc., Chicago, IL).
Results
Extraction of neutral lipids
The method of Bligh and Dyer (16) is a commonly used lipid extraction procedure and is efficient for a broad spectrum of lipids, both neutral and ionized. As seen in Fig. 1B, the most abundant lipid class present in the Bligh and Dyer (16) extract of RAW cells was phospholipids, which are known to interfere with NP-HPLC chromatographic separation of less polar components by accumulating on the column or precolumn. To avoid complex washing procedures, a new extraction protocol was designed to maintain efficient neutral lipid extraction while leaving the majority of the phospholipids with the aqueous layer. After several solvent systems were assessed, 75:25 (v/v) isooctane-ethyl acetate was found to extract neutral lipid classes with recoveries comparable to those of Bligh and Dyer (16) and contained considerably lower quantities of phospholipids (Fig. 1A). When the radiolabeled TAG was added to the cellular suspensions before extraction, 93.5% was recovered by the new extraction, whereas the identical TAG was 91.8% recovered by the method of Bligh and Dyer (16). Using the new extraction procedure, the NP-HPLC was not noticeably affected by phospholipid accumulation during the analysis of over 45 consecutive samples.
Fig. 1.
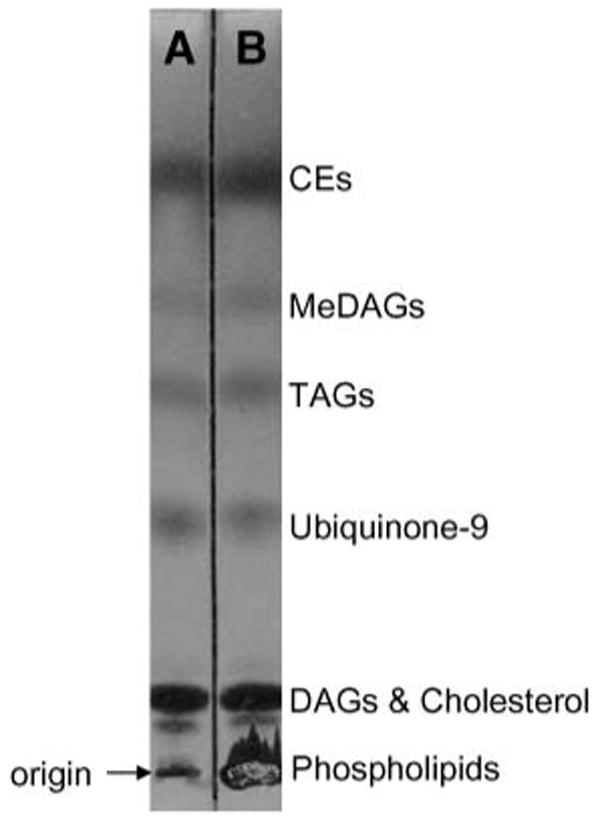
TLC separation of the components extracted from RAW cells by isooctane-ethyl acetate neutral lipid extraction (A) and the method of Bligh and Dyer (16) (B). For extraction and/or TLC conditions, see Experimental Procedures. CE, cholesteryl ester; DAG, diacylglycerol; MeDAG, monoalkylether diacylglycerol; TAG, triacylglycerol.
NP-HPLC-MS of intercellular neutral lipids
Naturally occurring complex mixtures of neutral lipids derived from RAW cells were extracted and separated by NP-HPLC. The separation of several neutral lipid classes isolated from RAW cells treated with Kdo2-lipid A was quite evident (Fig. 2A). This observed chromatography was reproducible as long as the solvents remained thoroughly degassed. The separation of these neutral lipids was extremely sensitive to slight alterations in mobile phase content and/or gradient. Retention time shifts of several minutes were observed when the isocratic portion of the gradient was changed by even 1% MTBE.
Fig. 2.
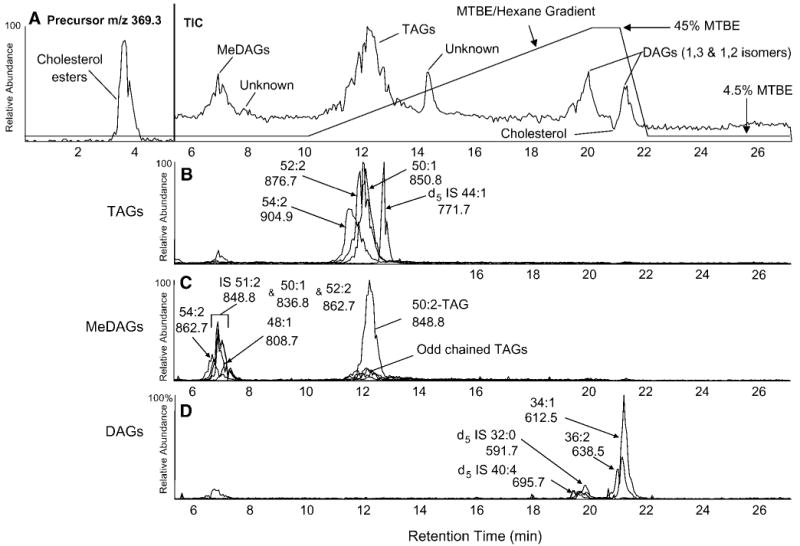
A: Normal-phase (NP) liquid chromatographic separation and mass spectrometric detection either as precursors of m/z 369.3 or as total ion current (TIC) of neutral lipids extracted from RAW cells. The HPLC gradient is indicated by the lines. MTBE, methyl tert-butyl ether. B–D: Overlaid mass chromatograms of selected ammoniated parent ions of naturally occurring MeDAG (B), TAG (C), and DAG (D) species. The identified total carbon content and the number of double bonds within the fatty acyl chains for specific mass chromatograms are labeled, as are the observed mass-to-charge ratios for the ammoniated parent ions.
The separation of CE and glyceryl ester classes was complete, with the DAG class completely separated from the TAG class (Fig. 2A). However, DAGs coeluted with free cholesterol and were split into 1,2- and 1,3-DAGs by acyl group migration, as described previously for NP separation on silica-bound columns (25). All glyceryl ester neutral lipid species formed strong ammonium ions, [M+NH4]+, when the ammonium acetate buffer was added after separation and before electrospray ionization; the elution of these neutral lipids was easily visualized in the total ion chromatogram. In initial experiments, CEs formed a barely detectable peak in the total ion chromatogram, and to better detect the CE class, the mass spectrometer was operated in precursor ion mode (precursors of m/z 369.3) during the early portion of the gradient (0–5.5 min) and the elution of the CE class became quite apparent. There was little to no separation of individual CE molecular species, likely attributable to their relatively low retention times.
There was some separation of certain TAG molecular species when [M+NH4]+ ions were extracted from the LC-MS data (Fig. 2B). The envelope of naturally occurring molecular species having 52 carbons and 2 double bonds (52:2) was clearly separated from the d5 44:1 (d5 14:0/16:1/14:0) internal standard. In fact, the elution of this single TAG molecular species (d5 14:0/16:1/14:0) likely revealed the actual elution profile of an individual molecular species. Ammoniated parent ions of naturally occurring species (e.g., 52:2, m/z 876.7) can contain >10 individual molecular species, each with a slightly different retention time, resulting in a much broader elution envelope.
The MeDAGs eluted from ∼6 to 8 min in a similar manner as TAGs, although less separation of molecular species was observed, likely because of the reduced overall retention of the MeDAG class (Fig. 2C). The abundant peak at ∼12.5 min was attributable to the signal from m/z 848.8 (internal standard 51:2, 15:0e/18:1/18:1, [M+NH4]+), which was also isobaric with TAG 50:2, because this MeDAG internal standard contained an odd carbon number radyl group at sn-1. Naturally occurring species of DAGs and DAG internal standards eluted between 19 and 22 min in a similar manner (Fig. 2D).
Mass spectra of the CE class were obtained by isolation of the HPLC fractions and subsequent infusion by nanospray. Because ammonium adduct ions of all CE molecular species can be collisionally activated to yield m/z 369.3, precursor ion scanning was used to identify the molecular species present (Fig. 3A). Each fatty acyl group was determined from the observed [M+NH4]+ ion after subtraction of the mass of the cholesterol fragment (369.3 amu) and the ammonium adduct (18 amu). The ester of 18:1 was by far the most abundant species in RAW cells, followed by 16:0 and 16:1. Several polyunsaturated species were present at low abundance. The stable isotope CE internal standard, [13C18]18:1-CE, appeared at m/z 686.8.
Fig. 3.
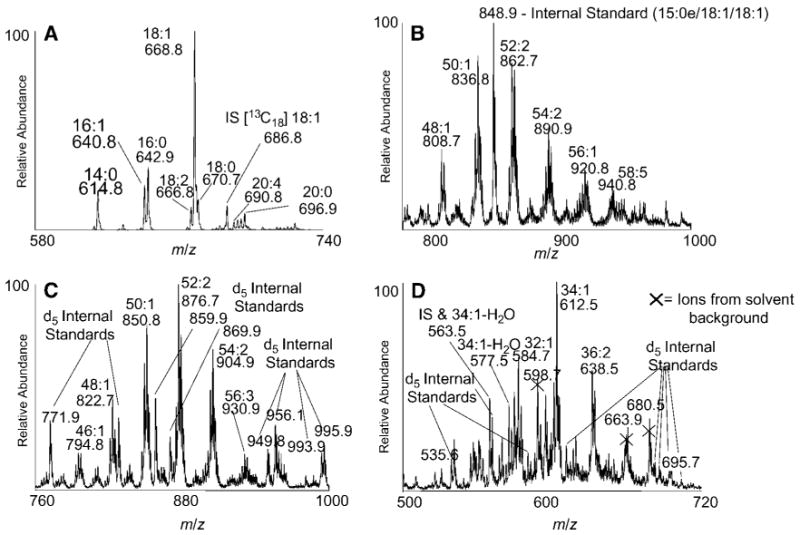
Summed positive ion mass spectral data recorded during the elution of RAW 264.7 neutral lipid classes during LC-MS analysis. Precursor ion scans for m/z 369.3 from the CE fraction (A) and total ions observed during the elution of MeDAG (B), TAG (C), and DAG (D) lipid classes. The labeled species indicate total carbon content and the number of double bonds within the fatty acyl chains as well as the observed mass-to-charge ratios of the ammoniated parent ions. The fragment ions of selected DAGs are labeled as the protonated molecular ion species minus water. Peaks from the internal standards (IS) are indicated.
The mass spectra of the neutral glycerol lipid classes were obtained by integrating full scan data for each eluting family. The elution window of the MeDAG class, ∼6–8 min (Fig. 3B), revealed a large number of ammonium ions, each corresponding to ammonium adducts of molecular species having the same total number of carbon atoms in the radyl groups and double bonds. A noticeable 6–12 amu envelope of abundant ions was observed at 24–28 amu intervals that corresponded to molecular species having the same number of total carbon atoms. The width of the envelope was the result of varying the quantity of double bonds in the variety of fatty acyl substituents present. It can be seen that as the number of total radyl carbons increased, so did the width of the envelope, indicating a greater variety of unsaturated compounds in MeDAGs containing longer chain fatty acids. A single ion envelope was observed for the internal standard (15:0e/18:1/18:1), m/z 848.8, and the naturally occurring carbon 13 isotope. The observed [M+NH4]+ fell directly between the mass envelopes of naturally occurring species containing 50 and 52 total carbons, as it contains an odd-chain ether with 51 total carbon radyl groups.
The elution of TAGs between 11 and 14 min (Fig. 3C) had a very similar envelope series character, indicating a complex mixture of molecular species. In this TAG region, numerous d5 internal standards were present, generally appearing just outside the envelopes of naturally occurring species. It was possible to quantitate the relative abundances of the parent ions using these internal standards from a separate neutral loss scan (23, 24). Less abundant odd-chain-containing TAG species were clearly present, and these would have appeared at the same mass-to-charge ratio as the major MeDAG molecular species. For example, the 50:1 MeDAGs observed at m/z 836.8 (Fig. 3B) would be directly on top of the odd-chain TAGs consisting of 49:1 molecular species (m/z 836.8 in Fig. 3C). Thus, MeDAGs and TAGs, when present in a mixture, could confound the analysis of one another; therefore, physical separation was essential for unambiguous assignment.
The elution of 1,2- and 1,3-DAGs from the NP-HPLC between 19 and 22 min (Fig. 3D) had a rather complicated mass spectrum arising not only from the mixture of molecular species but also from the complex array of [M+NH4]+ and fragment ions. There was a facile in-source fragmentation of DAG [M+NH4]+ corresponding to the loss of water. Further complicating the spectra was the presence of background ions (m/z 598.7, 663.9, and 680.5) from the HPLC column. Despite these complexities, all of the internal standards were observed, albeit at low abundances.
Identification of unknowns
During NP-LC-MS analysis of samples isolated from tissue-cultured cells, two unidentified peaks consistently appeared in the total ion current plot. The first HPLC peak, eluting between 7.5 and 8.5 min, generated essentially a single ion at m/z 798.7, clearly evident from the extracted ion chromatogram (Fig. 4B) and when this isolated fraction was infused for nanoelectrospray analysis. MS/MS of m/z 798.7 yielded a single ion of m/z 408.2, which fit for the product ion being an ammoniated monomer, [M+NH4]+, and the precursor ion (m/z 798.7) being an ammoniated dimer, [2M+NH4]+. MS2 of m/z 408.2 produced a major product ion at m/z 149.0, a rather diagnostic ion for phthalate esters (Fig. 4B, inset). Considering the monomer molecular weight, the compound was most likely dioctylphthalate and/or diethylhexylphthalate, both commonly observed plasticizer contaminants.
Fig. 4.
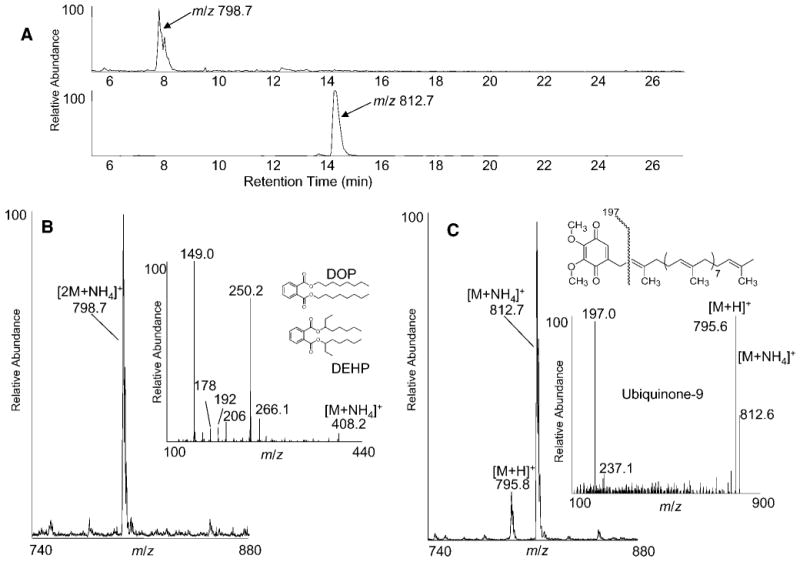
A: Mass chromatograms of two unidentified components detected in the total ion current during NP-LC-MS analysis of neutral lipids extracted from RAW cells (see Fig. 2A). B: Positive ion electrospray mass spectra integrated over the elution window of the unknown peak that eluted near 8 min. B, inset: Collision-induced dissociation spectrum of the monomer, m/z 408.2, after isolation by fraction collection from NP-LC. C: Positive ion electrospray mass spectra integrated over the elution window of the unknown peak eluting from ∼14 to 15 min. C, inset: Collision-induced dissociation spectrum of the unknown component, m/z 812.6, after isolation by fraction collection during NP-LC.
The second unidentified peak, eluting between 14 and 15 min, showed varying abundance directly proportional to other lipid components extracted from the cells. However, this component also eluted as a single compound (Fig. 4A), observed as the ammoniated adduct ion [M+NH4]+, m/z 812.7, and the protonated ion [M+H]+, m/z 795.8 (Fig. 4C). Isolation of the HPLC peak and subsequent collisional activation of m/z 812.7 followed by tandem mass spectrometry produced a single abundant fragment of m/z 197.0 (Fig. 4C). This product ion was consistent with a previously published collision-induced decomposition spectrum of ubiquinone-9 (26). The identity of this species was confirmed by coinjection of authentic ubiquinone-9, which exactly coeluted with this HPLC peak. The structure and suggested origin of the product ion are indicated in the inset to Fig. 4C.
CE quantitation
Calibration curves for several CE reference standards using [13C18]18:1-CE as the internal standard revealed a general trend of increasing responses with increases of both chain length and total number of double bonds (Fig. 5). For CEs of 18 fatty acyl carbon atoms or less, response factors appeared to be entirely dependent on the total number of double bonds in the fatty acyl group. For species having 20 carbons or more, the response increased with the total carbon number as well as unsaturation. This behavior has been reported previously, although much different mass spectrometric strategies were used (27, 28). To accommodate the changing response factor relative to the internal standard, molecular species of CE were quantified using the standard curve closest in both chain length and unsaturation. The limit of quantification was determined by the lowest point on the standard curve, which was 3 pmol. There was no attempt to test the absolute detection level of CEs by this method. The majority of the CE species monitored fell within the concentration range of the standard curves (Fig. 5).
Fig. 5.

Calibration curves for 10 CE reference standards (RS). Tabulated values indicate the slope and r2 value for each regression line using the isotope-labeled [13C18]18:1-CE as internal standard (IS). Linear least-fit square regression lines were forced through the origin by applying the constraint y0 = 0 during regression generation.
Using these methods, it was possible to monitor changes in the absolute quantity of individual CE species in RAW cells during Toll-like receptor 4 activation. For this study, Kdo2-lipid A was added to RAW cells for various lengths of time, and untreated samples were also incubated for the same times as control experiments. After incubation, neutral lipids were extracted and separated by NP-HPLC-MS. The HPLC fraction containing the CEs was isolated, and individual molecular species were subsequently quantified by nanoelectrospray and multiple reaction monitoring analysis for 23 different CEs. The profile of CE molecular species in untreated RAW cells revealed 18:1 as the most abundant species, with this single species accounting for ∼40% of the total CEs. The next most abundant species, 16:0, 16:1, and 14:0, together accounted for 33% of total CEs. Several polyunsaturated species were present at lower abundances (Fig. 6A).
Fig. 6.
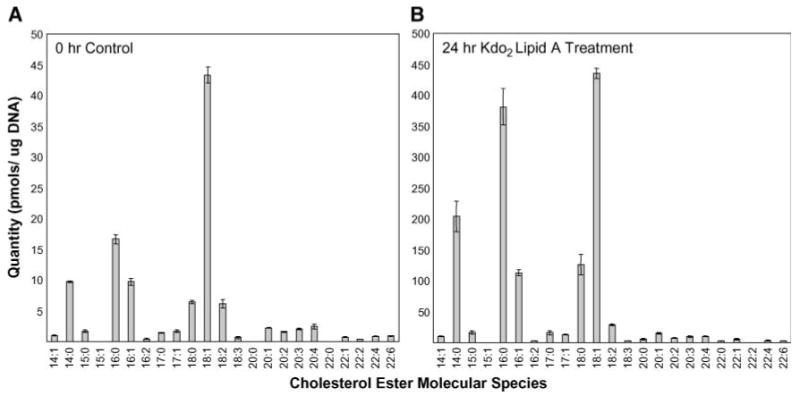
Abundance of individual CE molecular species extracted from control untreated cells (A) and RAW 264.7 cells exposed to Kdo2-lipid A (100 ng/ml) for 24 h (B). Molecular species are indicated by the total number of carbons and the number of double bonds in the fatty acyl chain. Values are means ± SEM of triplicate experiments.
Significant changes in some CE molecular species were observed in RAW cells exposed to Kdo2-lipid A (Fig. 6B). Not only did the overall amount of CEs increase by ∼13-fold, but the profile of individual molecular species changed significantly. The most abundant species remained 18:1; however, after stimulation, this single molecular species accounted for only 30% of the overall CEs. The unsaturated species 14:0, 16:0, and 18:0 were now the next most abundant species, together accounting for >50% of the total CE content.
Discussion
Neutral lipids constitute a family of important biomolecules central to energy storage and energy generation in virtually all cells (1). Although the study of this class of lipids has been carried out since the development of facile chromatographic strategies such as thin-layer chromatography, it is only relatively recently that techniques have been available to analyze all neutral lipids at the level of their individual components or molecular species. Many of the different neutral lipids, such as CEs, TAGs, and DAGs, exist in cells not as a single entity but rather as a mixture of hundreds, if not thousands, of different individual molecular species that differ by fatty acyl identity and position of esterification on the glycerol backbone (for the glycerol esters). There have been recent studies using various mass spectrometric and chromatographic techniques that report the identification and quantitation of molecular species of neutral lipids (22, 27, 29), but typically these reports have focused on a single family of neutral lipids rather than the entire group that is readily extracted by immiscible organic solvents. Individual molecular species of CEs have been reported using an LC-MS/MS approach (27, 28). We also recently reported on an MS3 method to identify individual molecular species that occur within natural complex mixtures (2). However, in many cases, it is of interest to get a picture of all of the changes of individual molecular species of neutral lipids in a study, especially those focusing on global changes that occur within a cell when a particular biochemical pathway is perturbed. Interest in being able to follow alterations in the quantity of individual molecular species has been driven by expectations that such information would reveal a level of biochemical control that is currently unexplored.
Many methods had been reported for the isolation of neutral lipids, including organic solvent extraction and solid-phase extraction techniques. It was found in this study that excellent recovery of CEs and the glycerol lipids could be obtained using the organic solvent system isooctane-ethyl acetate (75:25). The advantage of this technique compared with other organic extraction protocols, which typically involve chloroform, was that this less polar immiscible solvent inefficiently extracted the more polar phospholipids and sphingolipids while extracting >90% of the less polar neutral lipids such as TAGs. The isooctane-ethyl acetate extraction has the additional practical advantage of recovery of the top layer of the separated phases, eliminating the need to puncture the insoluble protein interface, as in the method of Bligh and Dyer (16). This extraction approach greatly simplified the subsequent purification of the extracted neutral lipid by the NP-HPLC silica phase column, in that it was not necessary to carry out extensive cleaning of the HPLC column to remove the polar lipids that would be extracted by the more traditional chloroform-methanol base extraction solvent systems. This strategy improved the sample-to-sample reproducibility and minimized the amount of time required to regenerate the column for the next analytical sample. The HPLC gradient used achieved sufficient narrow peak shape as well as reasonable column capacity to ensure complete separation of each of the neutral lipid classes. The mobile phase used (4.5% MTBE in isocratic hexane for 10 min, then gradient-programmed to 45% MTBE in 10 min) could achieve separation of the target neutral lipids in ∼25 min.
The detection of the elution of neutral lipids from the HPLC column by mass spectrometry required the presence of a counter ion, such as an alkali metal ion or an ammonium ion, that would facilitate electrospray ionization and charge the neutral lipid during the electrospray process. Following the report of Sommer et al. (22), a postcolumn solvent modification scheme was implemented to introduce the necessary ionic solvent to the highly nonpolar separation solvent containing the neutral lipids. This resulted in excellent sensitivity for the detection of all lipid classes. However, because the silica was the stationary phase, the DAGs eluted as two components after acyl group migration of the biologically derived DAGs. This is a well-known phenomenon attributed to the acidic properties of silica in promoting the formation of 1,3-DAGs from 1,2-DAGs. Although not attempted in these studies, it would be possible to mitigate this acyl group migration through chemical derivatization of the DAGs after immediate extraction. This approach has been used by Gross and coworkers (30) in their studies to improve the ionization efficiency of DAGs through the formation of a betaine ester. Because the resulting mass spectra of the eluting DAGs were complicated by the presence of not only [M+NH4]+ ions but also ions corresponding to the loss of water, such a derivatization approach would appear to be essential for the subsequent quantitative analysis of the DAG species.
The identification of the MeDAGs in RAW 264.7 cells as a completely separable class of neutral lipids from the TAGs revealed the importance of the separation of neutral lipid classes to facilitate identification. The MeDAGs are a class of neutral lipids that were first described in 1961 by Eichberg, Gilbertson, and Karnovsky (31), but little is known about their biosynthesis and metabolism. Recent work has suggested that they are efficiently stored in lipid bodies and perhaps efficiently remodeled (32). The molecular weight differences between the MeDAGs and TAGs are attributable to the structural difference of an alkylether at sn-1 relative to a fatty acyl ester at sn-1 of the glycerol backbone. The difference in mass between these structural entities is only 14 Da, which is attributable to the presence of two additional hydrogen atoms and one less oxygen atom in the ether moiety of the MeDAGs. However, without very high-resolution mass spectrometric analysis, this mass difference of 14 Da could appear to be the result of the presence of an odd-chain fatty acyl group esterified to the glycerol backbone in a TAG molecular species. Thus, if one did not completely separate these two families of closely related molecules, the resulting mass spectrum in this region between m/z 700 and 1,000 would be a complex mixture of overlapping envelopes of the two individual families, in which the even radyl carbon-numbered MeDAGs would appear between the envelopes of the even fatty acyl-numbered TAGs formed by those molecular species having a certain number of carbon atoms and varying degrees of unsaturation. These envelopes differ by typically 24–28 mass units. Furthermore, the MeDAGs would fall directly on top of the odd fatty acyl carbon-numbered TAGs if they were present. Using this chromatographic separation approach, it was possible to identify the MeDAGs and their molecular species distribution completely separate from the analysis of the TAG molecular species. Indeed, there was evidence for odd-chain TAG molecular species; however, they were of substantially lower abundance than what would be assigned to them if there was no separation of MeDAGs from TAGs.
This approach of using NP-LC-MS to analyze neutral lipids also permitted the identification of unanticipated neutral lipids and contaminants with very lipophilic properties. The identification of ubiquinone-9 was unanticipated, but this method clearly could be modified to quantitate the level of ubiquinones in cell extracts. The use of this NP-LC-MS approach for the quantitative analysis of neutral lipids was exemplified by the analysis of the molecular species of CEs that were initially extracted from the RAW cells followed by separation by NP chromatography and subsequent quantitation of the CE HPLC peak using stable isotope dilution mass spectrometry. A single internal standard, [13C18]18:2-CE, and 10 different molecular species of CE were available for reference standards to establish calibration curves. With these standard curves, it was possible to estimate the quantity of 13 additional CE molecular species for which reference standards were not available.
Approximately 20 major as well as minor molecular species of CE were quantitated in RAW 264.7 cells. The most abundant CE molecular species corresponded to 18:1-CE and 16:0-CE. In part, this may be attributable to the low abundance of polyunsaturated fatty acids, which become depleted during the tissue culturing of cells. The CE molecular species present in peritoneal macrophages isolated from the mouse were much more enriched in polyunsaturated fatty acids (data not shown) than cells carried in culture reported here. It was of interest that the saturated and monounsaturated molecular species of CE did increase during culture for this shorter fatty acyl chain (14:0-CE to 18:0-CE) relative to the 20:0-CE and 22:0-CE molecular species even without agonist treatment. There was very little change in the polyunsaturated CE molecular species of RAW cells carried in culture.
CEs are the storage form of cholesterol found primarily in lipid bodies of virtually all cells, including the RAW 264.7 cell. The recycling of cholesterol and intracellular cholesterol trafficking involve the action of CE hydrolase as well as ACAT1 in most cells (3). Toll-like receptor 4 is known to play an important role in changing macrophages into foam cells, which is thought to be a critical event in atherosclerosis (33). The accumulation of lipids within these macrophages is regulated by a number of enzymes, including fatty acid binding proteins and ACAT1. Previous studies have shown that in RAW 264.7 cells, lipopolysaccharides (Toll-like receptor 4 agonist) can lead to an increase in fatty acid binding protein mRNA expression (34) and the accumulation of total CEs. No study has previously reported the effect of Toll-like receptor 4 activation on individual molecular species of CEs and the magnitude of the increase of saturated and monounsaturated CEs. Oxidized LDL has been reported to cause a significant accumulation of CEs in THP-1 macrophages, as measured by the incorporation of [3H]oleate into CEs separated by TLC (35). However, there has been no report of individual molecular species of CE changing during transformation of the THP cell into more foam-like cells.
In summary, this robust method of neutral lipid analysis permitted both qualitative and quantitative assessment of changes in neutral lipids using mass spectrometric techniques. It is clear from the time course studies that the activation of Toll-like receptor 4 by Kdo2-lipid A caused a significant increase in certain molecular species of CEs of RAW 264.7 cells. This might reveal a particular effect of Toll-like receptor 4 on the activity of ACAT1 and fatty acid binding proteins such as FABP4 on specific molecular species of CEs in mounting the cellular response to this agonist by driving certain fatty acyl biochemistry into the storage of the fatty acyl group as a CE in lipid bodies within the RAW 264.7 cell.
Acknowledgments
This work was supported, in part, by a Lipid MAPS Large-Scale Collaborative Grant from the National Institutes of Health (Grant GM-069338).
Abbreviations
- CE
cholesteryl ester
- DAG
diacylglycerol
- DPBS
Dulbecco's phosphate-buffered saline
- MeDAG
monoalkylether diacylglycerol
- MTBE
methyl tert-butyl ether
- NP
normal-phase
- TAG
triacylglycerol
References
- 1.Jaworski K, Sarkadi-Nagy E, Duncan RE, Ahmadian M, Sul HS. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1–G4. doi: 10.1152/ajpgi.00554.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McAnoy AM, Wu CC, Murphy RC. Direct qualitative analysis of triacylglycerols by electrospray mass spectrometry using a linear ion trap. J Am Soc Mass Spectrom. 2005;16:1498–1509. doi: 10.1016/j.jasms.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 3.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 4.Murphy DJ. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res. 2001;40:325–438. doi: 10.1016/s0163-7827(01)00013-3. [DOI] [PubMed] [Google Scholar]
- 5.Martin S, Parton RG. Caveolin, cholesterol, and lipid bodies. Semin Cell Dev Biol. 2005;16:163–174. doi: 10.1016/j.semcdb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Weller PF, Bozza PT, Yu W, Dvorak AM. Cytoplasmic lipid bodies in eosinophils: central roles in eicosanoid generation. Int Arch Allergy Immunol. 1999;118:450–452. doi: 10.1159/000024161. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32:27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Jonas A. Lecithin cholesterol acyltransferase. Biochim Biophys Acta. 2000;1529:245–256. doi: 10.1016/s1388-1981(00)00153-0. [DOI] [PubMed] [Google Scholar]
- 9.Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr Opin Lipidol. 2001;12:289–296. doi: 10.1097/00041433-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Leon C, Hill JS, Wasan KM. Potential role of acylcoenzyme A:cholesterol transferase (ACAT) inhibitors as hypolipidemic and antiatherosclerosis drugs. Pharm Res. 2005;22:1578–1588. doi: 10.1007/s11095-005-6306-0. [DOI] [PubMed] [Google Scholar]
- 11.Rudel LL, Lee RG, Parini P. ACAT2 is a target for treatment of coronary heart disease associated with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2005;25:1112–1118. doi: 10.1161/01.ATV.0000166548.65753.1e. [DOI] [PubMed] [Google Scholar]
- 12.Murphy RC, Fitzgerald M, Barkley RM. Neutral lipidomics and mass spectrometry. In: Griffiths W, editor. Metabolomics, Metabonomics, and Metabolic Profiling. RSC Publishing; London: 2008. pp. 161–194. [Google Scholar]
- 13.Geeraert E, Sandra P. Capillary GC of triglycerides in fats and oils using a high temperature phenylmethylsilicone stationary phase. II. The analysis of chocolate fats. J Am Oil Chem Soc. 1987;64:100–105. [Google Scholar]
- 14.Smith GG, Wetzel WH. The effect of molecular size and structure on the pyrolysis of esters. Org Biomol Chem. 1957;79:875–879. [Google Scholar]
- 15.Holcapek M, Jandera P, Zderadicka P, Hruba L. Characterization of triacylglycerol and diacylglycerol composition of plant oils using high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J Chromatogr A. 2003;1010:195–215. doi: 10.1016/s0021-9673(03)01030-6. [DOI] [PubMed] [Google Scholar]
- 16.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 17.Wren JJ, Szczepanowksa D. Chromatography of lipids in presence of an antioxidant, 4-methyl-2-,6-di-tert-butylphenol. J Chromatogr A. 1964;14:387–404. [Google Scholar]
- 18.Kim HY, Salem N., Jr Separation of lipid classes by solid phase extraction. J Lipid Res. 1990;31:2285–2289. [PubMed] [Google Scholar]
- 19.Hamilton JG, Comai K. Separation of neutral lipid, free fatty acid and phospholipid classes by normal phase HPLC. Lipids. 1988;23:1150–1153. doi: 10.1007/BF02535282. [DOI] [PubMed] [Google Scholar]
- 20.Ingalls ST, Kriaris MS, Xu Y, DeWulf DW, Tserng KY, Hoppel CL. Method for isolation of non-esterified fatty acids and several other classes of plasma lipids by column chromatography on silica gel. J Chromatogr. 1993;619:9–19. doi: 10.1016/0378-4347(93)80441-6. [DOI] [PubMed] [Google Scholar]
- 21.Hoving EB. Chromatographic methods in the analysis of cholesterol and related lipids. J Chromatogr B Biomed Appl. 1995;671:341–362. doi: 10.1016/0378-4347(95)00223-6. [DOI] [PubMed] [Google Scholar]
- 22.Sommer U, Herscovitz H, Welty FK, Costello CE. LC-MS-based method for the qualitative and quantitative analysis of complex lipid mixtures. J Lipid Res. 2006;47:804–814. doi: 10.1194/jlr.M500506-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Krank J, Murphy RC, Barkley RM, Duchoslav E, McAnoy AM. Qualitative analysis and quantitative assessment of changes in neutral glycerol lipid molecular species within cells. Methods Enzymol. 2007;432:1–20. doi: 10.1016/S0076-6879(07)32001-6. [DOI] [PubMed] [Google Scholar]
- 24.Murphy RC, McAnoy AM, Krank J, Duchoslav E, Barkley RM. Detection and abundance of diacylglycerol and triacylglycerol molecular species in cells using neutral loss mass spectrometry. Anal Biochem. 2007;366:59–70. doi: 10.1016/j.ab.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blank ML, Snyder FL. Chromatographic analysis of ether-linked glycerolipids, including platelet-activating factor and related cell mediators. In: Shibamoto T, editor. Lipid Chromatographic Analysis. CRC Press; New York: 1994. pp. 219–316. [Google Scholar]
- 26.Teshima K, Kondo T. Analytical method for ubiquinone-9 and ubiquinone-10 in rat tissues by liquid chromatography/turbo ion spray tandem mass spectrometry with 1-alkylamine as an additive to the mobile phase. Anal Biochem. 2005;338:12–19. doi: 10.1016/j.ab.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Liebisch G, Binder M, Schifferer R, Langmann T, Schulz B, Schmitz G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS) Biochim Biophys Acta. 2006;1761:121–128. doi: 10.1016/j.bbalip.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Cullen P, Fobker M, Tegelkamp K, Meyer K, Kannenberg F, Cignarella A, Benninghoven A, Assmann G. An improved method for quantification of cholesterol and cholesteryl esters in human monocyte-derived macrophages by high performance liquid chromatography with identification of unassigned cholesteryl ester species by means of secondary ion mass spectrometry. J Lipid Res. 1997;38:401–409. [PubMed] [Google Scholar]
- 29.Han X, Gross RW. Quantitative analysis and molecular species fingerprinting of triacylglyceride molecular species directly from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem. 2001;295:88–100. doi: 10.1006/abio.2001.5178. [DOI] [PubMed] [Google Scholar]
- 30.Li YL, Su X, Stahl PD, Gross ML. Quantification of diacylglycerol molecular species in biological samples by electrospray ionization mass spectrometry after one-step derivatization. Anal Chem. 2007;79:1569–1574. doi: 10.1021/ac0615910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eichberg J, Gilbertson JR, Karnovsky ML. Neutral plasmalogens analogous to the neutral triglycerides. J Biol Chem. 1961;236:PC15–PC16. [PubMed] [Google Scholar]
- 32.Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RG, Liu P, Chapman KD. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007;48:837–847. doi: 10.1194/jlr.M600413-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Groeneweg M, Kanters E, Vergouwe MN, Duerink H, Kraal G, Hofker MH, de Winther MP. Lipopolysaccharide-induced gene expression in murine macrophages is enhanced by prior exposure to OxLDL. J Lipid Res. 2006;47:2259–2267. doi: 10.1194/jlr.M600181-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Kazemi MR, McDonald CM, Shigenaga JK, Grunfeld C, Feingold KR. Adipocyte fatty acid-binding protein expression and lipid accumulation are increased during activation of murine macrophages by Toll-like receptor agonists. Arterioscler Thromb Vasc Biol. 2005;25:1220–1224. doi: 10.1161/01.ATV.0000159163.52632.1b. [DOI] [PubMed] [Google Scholar]
- 35.Batt KV, Avella M, Moore EH, Jackson B, Suckling KE, Botham KM. Differential effects of low-density lipoprotein and chylomicron remnants on lipid accumulation in human macrophages. Exp Biol Med (Maywood) 2004;229:528–537. doi: 10.1177/153537020422900611. [DOI] [PubMed] [Google Scholar]