

Abstract
Human fibroblasts, capable of expressing a kinase-dead form of ATR (ATRkd), can be sensitized to the cytotoxic effects of methyl methanesulfonate (MMS) by the PARP inhibitor 4-amino-1,8-naphthalimide (4-AN). The combination of MMS + 4-AN results in accumulation of cells in S-phase of the cell cycle and activation of Chk1. Inhibition of ATR activity by expression of ATRkd suppresses the S-phase accumulation and partially reverses the Chk1 phosphorylation. The results confirm involvement of an ATR-mediated damage response pathway in the MMS + 4-AN-induced S-phase cell cycle checkpoint in human fibroblasts. Consistent with this hypothesis, the inhibitors caffeine and UCN-01 also abrogate the ATR- and Chk1-mediated delay in progression through S-phase. In the absence of ATR-mediated signaling, MMS + 4-AN exposure results in a G2/M arrest, rather than an S-phase checkpoint. Thus, whereas ATR mediates the S-phase response, it is not critical for arrest of cells in G2/M.
Keywords: PARP-1, PARP inhibitor, Base excision repair, Methyl methanesulfonate, Cell cycle, Cell signaling, Chk1, ATR, γH2AX
1. Introduction
The monofunctional DNA methylating agent methyl methanesulfonate (MMS) reacts directly with cellular DNA forming a number of methylated base adducts. Repair of such single base lesions is initiated by a damage-specific monofunctional glycosylase, N-methylpurine-DNA glycosylase. In the most simple ‘single-nucleotide’ base excision repair (BER) pathway, removal of the damaged base is followed by strand cleavage on the 5′ side of the sugar by apurinic/apyrimidinic endonuclease. Next, gap filling and cleavage on the 3′ side are conducted by the DNA synthesis and deoxyribose phosphate lyase activities, respectively, of DNA polymerase β, and finally there is sealing of the nick by a DNA ligase [1]. It has been proposed that the MMS hypersensitivity phenotype of mouse fibroblasts deficient in DNA polymerase β reflects accumulation of cytotoxic repair intermediates, such as a 5′-deoxyribose phosphate group at the margin of a nick, following removal of methylated bases from DNA [2].
Poly(ADP-ribose) polymerase (PARP)-1 is an abundant nuclear enzyme, and the first described member of a family of at least eighteen poly(ADP-ribosyl)ating enzymes [3]. PARP-1 is involved in damage surveillance and can detect and bind to nicks and strand breaks in cellular DNA, including those formed during the BER process. Binding to damaged DNA results in rapid enzymatic activation of PARP-1 leading to poly(ADP-ribosyl)ation of numerous nuclear proteins, including itself, using NAD+ as substrate. As a consequence of this automodification, PARP-1 loses its affinity for DNA, and is released from its DNA binding site [4,5]. Photoaffinity labeling studies of the interaction of BER proteins with DNA repair intermediates revealed the level of DNA probe cross-linked to PARP-1 was greater than that of any other BER protein [6]. Also, labeling was specific and was found to be strongest with DNA representing the 5′-sugar phosphate intermediate of BER implicated in MMS-induced cytotoxicity [6].
Inhibition of PARP activity in mouse fibroblasts by a PARP inhibitor such as 4-amino-1,8-naphthalimide (4-AN) results in tremendous sensitization to MMS [6,7]. In the presence of a chemical inhibitor, PARP-1 can still detect and bind to strand breaks in DNA, but now the inactivated protein will not be automodified and may remain bound to damaged DNA for a prolonged period of time. Such modification of DNA may entirely prevent access of repair proteins [8] leading to persistence of single-strand breaks or degeneration into double strand breaks [9], and result in the observed extreme sensitization. Previous studies have demonstrated that mouse fibroblasts treated with MMS + 4-AN undergo a caffeine-sensitive S-phase arrest that requires the presence of PARP-1 protein and leads to cell death by apoptosis [10,11]. The S-phase replication checkpoint acts to delay the firing of late origins of replication when active replication forks are stalled in response to unrepaired strand breaks and protein-DNA lesions [12]. Since we demonstrated that the S-phase arrest in mouse cells involves activation (phosphorylation) of Chk1, and Chk1 is an essential downstream effector kinase regulated by ATR, it was logical to propose that ATR might play a key role in the observed DNA damage checkpoint. However a role for ATR was not certain since both ATR and ATM kinases are inhibited by caffeine.
In the present study, we have extended our initial observations in mouse fibroblasts [10] by utilizing human cells expressing an inducible dominant negative kinase-dead form of ATR (ATRkd) [13]. With exposure to the tetracycline derivative, doxycycline (dox), the cells overproduce a protein containing a D2457A substitution that inactivates endogenous ATR kinase activity. It has been shown that expression of ATRkd results in hypersensitivity to several DNA damaging agents, including MMS, and that ATR is a critical component of multiple damage response pathways [13]. By making use of these ATRkd cells, we were able to examine the specific role of ATR in the cellular response following exposure to a sub-lethal concentration of MMS combined with a PARP inhibitor. Analysis of cell signaling in treated cells revealed that an ATR- and Chk1-dependent signaling pathway is involved in the S-phase checkpoint response.
2. Materials and methods
2.1. Cell cultures
GM847 SV40-transformed human fibroblasts expressing either tetracycline-inducible ATR-wild-type (ATRwt cells) or ATR-kinase-dead (ATRkd cells) expression vectors have been characterized previously and were obtained from Fred Hutchinson Cancer Research Center [13]. Both cell lines were routinely grown at 37 °C in a 10% CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glutamine (Invitrogen, Carlsbad, CA) and 10% fetal bovine serum (FBS; HyClone, Logan, UT). Expression of either wild-type or kinase-dead ATR protein was achieved by treatment of the appropriate cell line for 48 h with dox (Sigma-Aldrich, St. Louis, MO) at a concentration of 1 μg/ml. All cells were routinely tested and found to be free of mycoplasma contamination.
2.2. Cytotoxicity studies
Cytotoxicity was determined by growth inhibition assays as described previously [2]. ATRwt or ATRkd human fibroblasts were seeded at a density of 20,000 cells/well in six-well dishes and either treated or not with dox prior to exposure to DNA damaging agents. Cells were exposed to MMS (Sigma-Aldrich) for 1 h then washed with Hanks’ balanced salt solution (HyClone) and fresh medium was added. Dishes were incubated for approximately 10 days in a 10% CO2 incubator until untreated control cells were approximately 80% confluent. Cells (triplicate wells for each drug concentration) were counted by a cell lysis procedure [14] and results were expressed as the number of cells in drug-treated wells relative to control wells (% control growth). In many studies, cells were treated with MMS in the presence of the previously described PARP inhibitors 4-AN (10 μM) or 1,5-dihydroxyisoquinoline (DIQ; 250 μM) [10,15,16] obtained from Sigma-Aldrich, and the incubation with inhibitor was continued for a total of 48 h. Stock solutions of the inhibitors were prepared in dimethyl sulfoxide, and were diluted to the appropriate concentration in medium just before use. In some studies, cells were treated with caffeine (1 mM) or 7-hydroxystaurosporine (UCN-01; 100 nM) as well as 4-AN during the 1 h MMS exposure and for the following 23 h. Concentrated stock solutions of caffeine (Sigma) were made by dissolving it directly in medium at the time of the experiment. Stock solutions of UCN-01, an agent proprietary to Kyowa Pharmaceuticals, Tokyo, Japan and obtained from the Cancer Therapy Evaluation Program, NCI, were made in dimethyl sulfoxide and stored at -20 °C. The IC90 for MMS is the concentration that results in 90% growth inhibition.
2.3. Flow cytometric cell cycle analysis
Cell cycle and DNA synthesis, as assessed by staining with propidium iodide (PI) and incorporation of bromodeoxyuridine (BrdUrd) respectively, were analyzed simultaneously, as described previously [2]. ATRkd cells were seeded in 100 mm dishes at a density equivalent to that used in the cytotoxicity experiments and either treated or not with dox to induce ATRkd protein expression. The cells were treated for 1 h with MMS (0.5 mM) ± 4-AN (10 μM) and incubation with 4-AN was continued for up to 32 h. In some studies, cells were cotreated with 1 mM caffeine or 100 nM UCN-01 in addition to 4-AN during and for 23 h following the 1 h MMS. At specified times (1-32 h) after the beginning of exposure to MMS, 10 μM BrdUrd (Sigma) was added to the dishes for 2 h to pulse label the cells. Cells were washed with phosphate buffered saline (PBS), harvested by trypsinization and then washed a second time with PBS. The cell pellet obtained after centrifugation was resuspended in 100 μl cold PBS and the cells dropped slowly into 70% ethanol and allowed to fix at 4 °C overnight. The samples were washed, then suspended in 2N HCl containing 0.5% Triton X-100 and incubated for 30 min at room temperature to denature DNA. The cell samples were pelleted, resuspended in 0.1 M sodium borate, pH 8.5, to neutralize the sample and then washed with PBS. Cells were then incubated at 4 °C overnight with 20 μl anti-BrdUrd-FITC-conjugated antibody (BD Biosciences, San Jose, CA) in PBS containing 0.5% Tween 20 and 1% bovine serum albumin and 5 μl of 10 mg/ml RNAse (Sigma) stock solution. The following day, the cells were pelleted, washed with PBS and resuspended in 1 ml PBS containing 5 μg/ml PI (Sigma). The samples were analyzed by flow cytometry using Cell Quest software (BD Biosciences) and cell cycle histograms of PI fluorescence (DNA content) versus BrdUrd incorporation were generated. Cell cycle populations are designated G0/G1 (2N DNA content with no BrdUrd incorporation), S (variable DNA content with BrdUrd incorporation) and G2/M (4N DNA content without BrdUrd incorporation). Data represent the mean of at least two independent experiments. For most samples, data was also analyzed using ModFit LT software (Verity Software House, Inc., Topsham, ME).
2.4. Cell lysate preparation and Western blotting
ATRkd fibroblasts were seeded in 145 mm dishes at a cell density of 6.9 × 106 cells/dish. They were either induced or not with dox prior to treatment with MMS (0.5 mM) or 4-AN (10 μM) or a combination of the two agents as described previously. In some studies, cells were treated with 1 mM caffeine or 100 nM UCN-01 as single agents, or in addition to 4-AN during and following the 1 h MMS. After 16 h, treated and control untreated cells were harvested by scraping and washed with PBS. Cell lysates were prepared in a lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM NaF, 0.1 mM sodium orthovanadate, 0.2% Triton X-100, 0.3% Nonidet P-40) containing protease inhibitors, 0.1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin and 5 μg/ml leupeptin as described previously [17]. Cells in lysis buffer were incubated on ice for 30 min, then centrifuged at 14,000 rpm for 30 min at 4 °C and the supernatant fraction was transferred to another tube. The protein concentration in the extract was determined using the Bio-Rad protein assay, with bovine serum albumin as standard.
Equal amounts (100 μg) of total protein were loaded on gels and separated by 4-12% SDS-PAGE. The proteins were then transferred onto a nitrocellulose membrane in a transblot apparatus for 2 h at 150 mA, or overnight at 25 V. The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20 (TBS-T) and then first probed with anti-Chk1 polyclonal antibody (1:1000 dilution; Cell Signaling, Beverly, MA). Goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) (1:1000 dilution; Cell Signaling) was used as the secondary antibody and immobilized HRP activity was detected using the Phototope-HRP (Cell Signaling) chemiluminescence detection kit for Western blotting. The blot was stripped by incubating with buffer containing 62.5 mM Tris-HCl, pH 6.8, 100 mM β-mercaptoethanol and 1% SDS for 30 min at 50 °C, or with commercially available stripping buffer (Pierce, Rockford, IL) for 10 min at room temperature followed by 5 min at 37 °C. The blots were then washed twice for 20 min with TBS-T at room temperature. Detection of phospho-Chk1 was with rabbit polyclonal anti-phospho-Chk1 (serine345) antibody (1:1000; Cell Signaling). The membrane was stripped again and, as a loading control, probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using anti-GAPDH mouse monoclonal antibody (1:1000, Alpha Diagnostic International, Inc., San Antonio, TX); the secondary antibody was goat anti-mouse IgG-HRP conjugate (1:5000; Bio-Rad, Hercules, CA).
2.5. Flow cytometric analysis of phosphorylated H2AX
ATRkd cells (2 × 106) were seeded in 100 mm dishes and then treated or not with dox to induce ATRkd protein expression. Cells were either γ irradiated (1-8 Gy) and returned to the incubator, or treated for 1 h with MMS (0.5 mM) in the presence or absence of 4-AN (10 μM, 24 h) as described above. In some experiments, MMS + 4-AN-treated cells were additionally treated with caffeine (1 mM, 24 h). Cells were harvested by trypsinization 50 min after irradiation or 24 h after MMS, centrifuged and washed with PBS, and then analyzed by flow cytometry using the H2AX Phosphorylation Kit (Millipore, Billerica, MA) following the instructions of the manufacturer. Cells were first resuspended in 1× Fixation Solution, washed with PBS, and permeabilized. The anti-phospho-histone H2AX (serine139) monoclonal antibody conjugated to FITC (Millipore) was added and incubated on ice 20 min in the dark, with periodic gentle agitation to mix the cells. To aid in washing away excess FITC-labeled antibody, 1× Wash Solution was added to each tube. Cells were then pelleted by centrifugation and the supernatant was aspirated. The labeled cell pellets were resuspended in 500 μl PBS. Samples were read on a FACS flow cytometer and analyzed using Cell Quest software.
3. Results
3.1. MMS-induced cytotoxicity in human fibroblasts
We first confirmed that exposure of ATRkd cells to dox resulted in induction of ATRkd protein. Cells treated with dox for 48 h were shown to over-express kinase-dead protein detectable by Western blotting with an anti-ATR antibody (Fig. 1A). As demonstrated previously [13], dox induction resulted in hypersensitivity to MMS (Fig. 1B and C). This sensitizing effect was not seen in cells transfected with tetracycline-inducible wild-type ATR protein (ATRwt; Fig. 1D). In the absence of dox induction, and consistent with data obtained previously in mouse fibroblasts, both of these human cell lines were sensitized to MMS by the PARP inhibitor 4-AN (Fig. 1), though the effect was not as extreme as that seen in mouse cells. To rule out the possibility of an effect specific to 4-AN, another PARP inhibitor, DIQ, was tested and found to have a similar MMS-sensitizing effect in ATRkd cells (2.8- and 2.7-fold sensitization, respectively, for 4-AN and DIQ; Fig. 1B and C). A third PARP inhibitor, EB-47, also sensitized ATR cells to MMS (data not shown) demonstrating that the sensitization is neither PARP inhibitor-specific nor cell line-specific. Expression of ATRkd (Fig. 1B and C), but not ATRwt (Fig. 1D), further sensitized the cells to the MMS + PARP inhibitor combination. The results suggest that ATR plays a role in the damage response pathway following MMS-induced and MMS + PARP inhibitor-induced DNA damage.
Fig. 1.

Expression of ATRkd protein sensitizes cells to MMS, and MMS combined with a PARP inhibitor. (A) Detection of ATRkd by Western blotting 48 h after exposure to dox (1 μg/ml). (B) and (C) Effect of dox induction (closed symbols) on sensitivity of ATRkd cells to MMS alone (circles) and MMS combined with the PARP inhibitors (squares) 4-AN (B, 10 μM for 48 h) and DIQ (C, 250 μM for 48 h). (D) Effect of ATRwt induction on sensitivity to MMS alone, or MMS + 4-AN. Immunoblotting and growth inhibition experiments were carried out as described in Section 2. Survival data represent the mean ± S.E.M. of at least three independent experiments or are from one or two representative experiments where the values are the mean of triplicate determinations.
Signaling protein inhibitors produced similar effects in both ATRkd and ATRwt cell lines. Caffeine is known to inhibit the activity of both ATM and ATR upstream checkpoint kinases [18]. UCN-01 is a specific and potent inhibitor of Chk1 at the concentration (100 nM) utilized here; Chk2 is 100-fold more resistant [19]. Similar to the results obtained following dox induction, UCN-01 was found to sensitize cells to MMS as well as MMS + 4-AN. Interestingly, caffeine was able to sensitize both cell types to MMS alone, but not to the combination of MMS + 4-AN (data not shown).
3.2. Cell cycle arrest following exposure to MMS and 4-AN
Using flow cytometric analysis of BrdUrd incorporation and PI staining, we next investigated the effects of MMS and 4-AN as single agents, and in combination, on DNA synthesis and cell cycle progression. ATRkd cells, without dox induction, were treated for 1 h with MMS (0.5 mM) in the absence or presence of 4-AN (10 μM continuously) and analyzed at times up to 32 h for their ability to incorporate BrdUrd. The occurrence of BrdUrd positive (S-phase cells) is dependent on the proportion of cells undergoing DNA synthesis during the 2 h labeling period. This is around 30% in the slowly growing control population (Fig. 2A). Non-toxic exposures of cells to 4-AN alone had no effect on BrdUrd incorporation or cell cycle distribution compared to control, untreated cells (data not shown). Treatment with MMS alone resulted in a transient decrease in BrdUrd incorporation (DNA synthesis) that was rapidly reversed (Fig. 2A). When the two agents were combined, there was some inhibition of DNA synthesis at 4 and 6 h. More significantly, by 16-20 h after treatment, greater than 60% of the cells incorporated BrdUrd and were accumulated in S-phase of the cell cycle (Fig. 2A). Combination of MMS with DIQ resulted in a similar accumulation of BrdUrd-incorporating cells at 20 h, while there were no changes when DIQ was administered alone (Fig. 2B).
Fig. 2.
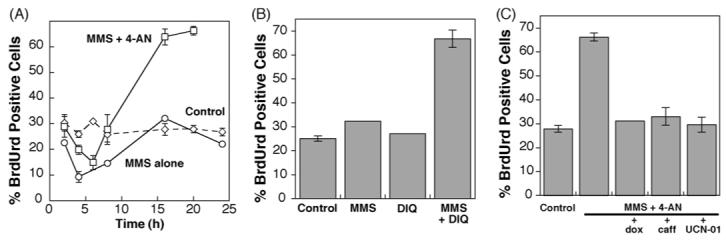
Accumulation of BrdUrd positive (S-phase) cells following exposure to MMS combined with a PARP inhibitor. (A) Control, untreated cells, and cells treated with MMS alone (0.5 mM for 1 h) or a combination of MMS and 4-AN (10 μM continuously), were pulse labeled with BrdUrd at the times indicated after initiation of MMS exposure, and harvested for flow cytometric analysis as described in Section 2. Plotted is the time course of % of cells staining positive for BrdUrd incorporation. (B) % BrdUrd positive cells 20 h after exposure to MMS alone, or to the PARP inhibitor DIQ (250 μM continuously) ± MMS (0.5 mM for 1 h). (C) Effect of pre-treatment with dox for 48 h to induce expression of ATRkd, or co-treatment with caffeine (1 mM) or UCN-01 (100 nM), on accumulation of BrdUrd positive cells 20 h following initiation of MMS combined with 4-AN exposure. Data represent the mean ± S.E.M. of at least three independent experiments or are the mean of two independent experiments.
3.3. Inhibition of cell signaling pathways in cells treated with MMS and 4-AN
As described above, exposure of human fibroblasts to MMS + 4-AN results in accumulation of cells in S-phase of the cell cycle 16-20 h after treatment (Fig. 2A). When cells were pre-treated with dox to inhibit ATR activity, this S-phase delay observed (at 16-20 h) was abrogated (Fig. 2C). In the next experiments, cells were treated with either caffeine or UCN-01 in addition to the MMS + 4-AN combination. Both agents were equally as effective as dox induction in preventing the MMS + 4-AN-mediated accumulation of S-phase cells at 16-20 h (Fig. 2C). Since ATR signaling will be inhibited under each of these treatment conditions, the results confirm that an ATR-mediated signaling pathway is involved in the MMS + 4-AN-induced S-phase cell cycle checkpoint.
A significant effect between 16 and 24 h after MMS + 4-AN treatment of dox-induced cells is the steady increase in the G2/M population (Fig. 3A). Therefore, in the absence of ATR-mediated signaling, MMS + 4-AN exposure results in a G2/M cell cycle arrest, without an earlier accumulation of S-phase cells. The result suggests that, whereas ATR mediates the S-phase delay, it is not essential for the G2/M arrest. Increased populations of G2/M cells were also observed at 20 h in the presence of caffeine or UCN-01 (Fig. 3B), but smaller proportions of cells were arrested in G2/M at 24 h than under conditions where ATR activity alone is inhibited (i.e., +dox). In cells treated with MMS + 4-AN, and no additional inhibitor, the major population of cells (66%) was accumulated in S-phase of the cell cycle at 20 h (Fig. 3B). However by 24 h, some cells had escaped the S-phase checkpoint and were now arrested in G2/M. Now more equal proportions of cells were accumulated in S- and G2/M-phases (42 and 55%, respectively; Fig. 3C) and by 32 h after treatment, 66% cells are arrested in G2/M (data not shown).
Fig. 3.

Accumulation of G2/M cells following exposure to MMS combined with 4-AN. ATRkd cells, pre-treated or not with dox for 48 h to induce expression of ATRkd, were treated with MMS (0.5 mM for 1 h) and 4-AN (10 μM, continuously). Cell cycle analysis was conducted as described in Section 2. (A) Plotted is the time course of % cells in G2/M. In some experiments, cells were co-treated with caffeine (1 mM) or UCN-01 (100 nM) as indicated, and then the incubation with 4-AN, caffeine or UCN-01 was continued for a total of (B) 20 h or (C) 24 h. Plotted are stack columns showing the proportions of G0/G1, S and G2/M phase cells following the various treatments. Data represent the mean of at least two independent experiments.
3.4. Activation of Chk1 following combined exposure to MMS and 4-AN
Chk1 activation has been observed previously in mouse fibroblasts undergoing an S-phase delay following treatment with MMS + 4-AN [10]. Here, in human fibroblasts, we find that the accumulation of S-phase cells following exposure to MMS + 4-AN is suppressed by UCN-01 (Fig. 2C). These results implicate Chk1 activation in the MMS + 4-AN-induced S-phase checkpoint observed in human fibroblasts. Since S-phase cells were maximally induced by exposure to MMS + 4-AN at 16-20 h (Fig. 2A), we wished to confirm Chk1 activation by examining cells at 16 h after a number of cell treatments.
The level of activated phosphorylated Chk1 (P-Chk1) was detectable, but present at a similar low level in control, untreated cells and in cells treated with either 4-AN or MMS alone (Fig. 4, lanes 1-3). When cells were treated with the combination of MMS + 4-AN, the amount of P-Chk1 was significantly increased, and in addition, a band corresponding to hyperphosphorylated Chk1 protein was also observed (lane 4). Caffeine exposure alone (lane 8), or dox induction (lane 10), decreased the amount of P-Chk1 even below that observed in control cells. Both of these treatments partially reversed the increase in P-Chk1 observed with MMS + 4-AN exposure (lanes 5 and 7). The effect was most pronounced in dox-induced cells where ATR activity is inhibited, but P-Chk1 still remained at a slightly higher level than in control cells. The results confirm that MMS + 4-AN exposure results in activation of both ATR and Chk1 kinases. Treatment with UCN-01 alone increased the level of P-Chk1 above background (lane 9). This has been observed by others and may represent an attempt to activate the essential kinase Chk1 in the presence of a Chk1 inhibitor [20]. When UCN-01 was combined with MMS + 4-AN, the level of P-Chk1 was greater than in cells treated with MMS + 4-AN and no inhibitor (lane 6). However, since UCN-01 occupies the ATP-binding pocket of Chk1, it is known that Chk1 activity will be suppressed even though the protein is present in a super-phosphorylated form [21]. The level of unphosphorylated Chk1 protein remained constant after all the cell treatments (Fig. 4).
Fig. 4.

Analysis of cell signaling proteins in ATRkd cell extracts. Immunoblot analysis of P-Chk1 and Chk1 in control, untreated cells, and 16 h after treatment with MMS (0.5 mM for 1 h) or inhibitors alone, and MMS combined with 4-AN (10 μM) plus caffeine (1 mM), UCN-01 (100 nM) or dox (1 μg/ml) induction. Cells were treated and lysates were prepared as described in Section 2; equal amounts of protein (100 μg) were resolved by 4-12% SDS-PAGE. The blots were also probed for GAPDH as a loading control.
3.5. Phosphorylation of H2AX following exposure to MMS and 4-AN
Phosphorylated H2AX (γH2AX) is formed in response to double stand breaks. Foci of γH2AX have been localized at sites of ionizing radiation (IR)-induced double strand breaks [22], and also in response to replication arrest following treatment of cells with UV or hydroxyurea [23]. Since replication is stalled following exposure to MMS + 4-AN, we looked for formation of γH2AX under these treatment conditions. First, as a positive control for the flow cytometric assay, IR-exposed cells were analyzed (Fig. 5A). As expected, the intensity of γH2AX fluorescence 50 min following irradiation, as determined by the shift in position of FITC-γH2AX histogram, correlated with the dose of IR. Cells treated with MMS + 4-AN had a level of γH2AX fluorescence at 24 h as least as high as that seen following exposure to 4 Gy irradiation (Fig. 5B). This fluorescence intensity was apparent between 16 and 24 h following treatment. In cells treated with MMS alone, there was a small increase in the fluorescence intensity of a sub-population of the cells (Fig. 5B). No shift was seen when cells were treated with 4-AN as a single agent (data not shown). The results suggest that double strand breaks are formed in cells exposed to the MMS + 4-AN combination.
Fig. 5.

Flow cytometric analysis of γH2AX. (A) ATRkd cells were exposed to γ irradiation (1-8 Gy) and the formation of γH2AX was analyzed at 50 min as described in Section 2. Shown is the FITC fluorescence histogram for control, untreated cells (green), and cells treated with 1 Gy (red), 4 Gy (blue) or 8 Gy IR (orange). (B) Cells were treated with MMS (0.5 mM for 1 h) in the presence or absence of 4-AN (10 μM, continuously) and analyzed for γH2AX after 24 h. Control, untreated cells (green), MMS alone (red), MMS + 4-AN (blue). (C) Cells, either induced or not with dox, were treated with MMS + 4-AN in the presence or absence of caffeine (1 mM) and analyzed after 24 h. Control, untreated cells (green), MMS + 4-AN (blue), MMS + 4-AN + dox (orange), MMS + 4-AN + caffeine (yellow).
Different protein kinases have been implicated in the phosphorylation of H2AX depending upon the mechanism of induction of double strand breaks [23,24]. In cells treated with either dox to inhibit ATR activity, or with the ATM and ATR inhibitor, caffeine, there was no significant change in γH2AX fluorescence compared with control, untreated cells (data not shown). Inhibition of ATR activity did not decrease the γH2AX fluorescence intensity following MMS + 4-AN-treatment (Fig. 5C). This result suggests that, whereas the S-phase checkpoint is mediated by ATR, the phosphorylation of H2AX observed following MMS + 4-AN exposure is ATR-independent. When cells were co-treated with caffeine, there was a significant decrease in the γH2AX fluorescence intensity of MMS + 4-AN-treated cells (Fig. 5C). Since we know that ATR is not involved, this result suggests that the H2AX phosphorylation observed following treatment with MMS + 4-AN is mediated by ATM.
4. Discussion
Previous studies have implicated Chk1-dependent signaling in the S-phase arrest of mouse fibroblasts following exposure to the combination of MMS and the PARP inhibitor 4-AN [10]. Since the kinase Chk1 is regulated by ATR, known to be involved in the DNA replication checkpoint, we proposed that ATR might also play a role. ATR-deficient mice and cell lines are non-viable [25,26], but we were able to follow up on our initial observations in mouse cells by using ATRkd human fibroblasts expressing an inducible dominant negative kinase-dead allele of ATR [13].
Three PARP inhibitors, 4-AN, DIQ (Fig. 1) and EB-47 (data not shown) were found to moderately sensitize the human fibroblasts to MMS-induced cytotoxicity. Previous results indicated that inhibition of PARP activity by 4-AN results in a more dramatic (40-fold) sensitization of wild-type mouse fibroblasts to the DNA methylator MMS. PARP inhibition has been shown to result in delayed resolution of stalled replication forks [27] and formation of γH2AX, an indicator of DNA double strand breaks [28,29]. Here we demonstrate that treatment of human fibroblasts with MMS combined with 4-AN results in significant phosphorylation of H2AX at 24 h (Fig. 5B). Replication fork collapse will generate double strand breaks that are preferentially repaired by homologous recombination (HR) [9,30,31] and mouse cells with BRCA1 and BRCA2 dysfunction have been shown to be hypersensitive to PARP inhibitors as a result of deficiency in double strand break repair by HR [28,29]. It is possible that the mouse fibroblasts used in previous studies have some unknown deficiency in the HR pathway resulting in the enhanced sensitization compared with human fibroblasts.
Following DNA damage and stalling of replication, checkpoints have been identified in cells that lead to a delay in progression through the cell cycle. Defects in checkpoint regulation can lead to accumulation of DNA damage, enhanced sensitivity to DNA damaging agents, and decreased genomic stability. Previously it was shown that expression of ATRkd, therefore inhibition of ATR activity, results in sensitization of human fibroblasts to MMS [13]. Consistent with the idea that ATR has a role in the response to MMS-induced damage, we find that caffeine and UCN-01 similarly sensitized the human fibroblasts to MMS-induced cytotoxicity. Now we show that ATR is also involved in the damage response induced by MMS combined with a PARP inhibitor. ATRkd induction, i.e., inhibition of ATR activity, sensitizes cells to MMS + 4-AN, MMS + DIQ (Fig. 1) and MMS + EB-47 (data not shown). Sensitization may be associated with a previously reported lesser efficiency of HR in the dox-induced S-phase checkpoint checkpoint-deficient ATRkd cells [32].
The combination of MMS + 4-AN results in an accumulation of S-phase cells at 16-20 h after treatment (Fig. 2A). This may be due to a slowing of progression of cells through S-phase of the cell cycle as a result of stalling of active replication forks by intermediates of BER bound to inactivated PARP-1 protein. An increased length of S-phase has been reported previously in cells following treatment with a methylating agent and a PARP inhibitor [33], and a similar accumulation of S-phase cells was observed earlier in MMS + 4-AN-treated mouse fibroblasts [10]. Induction of ATRkd protein prevented the MMS + 4-AN-induced accumulation of S-phase cells (Fig. 2C). This observation confirms our earlier suggestion that ATR-mediated signaling is responsible for the S-phase cell cycle checkpoint. Exposure of cells to MMS + 4-AN results in ATR-mediated phosphorylation of Chk1 at 16 h (Fig. 4), and suppression of the S-phase checkpoint by induction of ATRkd is associated with abrogation of Chk1 phosphorylation. These results suggest that Chk1-mediated signaling is involved in the S-phase accumulation. Consistent with this hypothesis, treatment of cells with either caffeine or UCN-01 was just as effective as dox in abrogating the S-phase checkpoint (Fig. 2C).
Our results are reminiscent of those obtained in ATRkd cells using a topoisomerase 1 inhibitor. Topoisomerase 1-DNA cleavage complexes can be reversibly or irreversibly trapped by camptothecins, such as topotecan, resulting in slowing of DNA replication and activation of an S-phase checkpoint [34]. Like the combination of MMS + 4-AN, camptothecins are selectively toxic during S-phase of the cell cycle. Expression of ATRkd in human fibroblasts sensitized them to topotecan-induced cytotoxicity and also reversed the observed increase in the S-phase population [35]. In addition, induction of ATRkd abrogated phosphorylation of Chk1 after topotecan exposure [35]. Results with inhibitors show that the MMS + 4-AN-induced (Fig. 3C) and the topotecan-induced checkpoints [36] are mediated by caffeine- and UCN-01-sensitive pathways. Therefore, for both types of damage, the ATR-mediated S-phase checkpoint is regulated by Chk1 activation.
At 24 h and later following MMS + 4-AN exposure, there is arrest of cells in G2/M (compare Fig. 3C with B). This observation is in agreement with previously published results in cells treated with MMS combined with the PARP inhibitor 3-aminobenzamide [37]. The late arrest of cells in G2/M, observed subsequent to the ATR-dependent S-phase checkpoint, may be a result of formation of double strand breaks associated with collapsed replication forks [9,31]. Indeed γH2AX, which is formed at sites of double strand breaks, was detected 16-24 h after MMS + 4-AN exposure (Fig. 5B). It is likely that ATR-mediated signaling is not involved in the MMS + 4-AN-induced G2/M arrest since it is most apparent in ATR-inhibited cells (Fig. 3). Similarly, since γH2AX is observed in dox-induced as well as non-induced ATRkd cells (Fig. 5C), the results show that the phosphorylation of H2AX occurring as a result of MMS + 4-AN exposure does not require ATR.
There is a lesser accumulation of cells in G2/M in the presence of caffeine or UCN-01 than following dox induction (Fig. 3B and C). The difference in the ability of cells to arrest in G2/M phase under conditions where ATR is inhibited (dox induction) compared with exposure to the less specific inhibitor, caffeine, suggests that caffeine is exerting an effect in addition to inhibition of ATR activity. Since caffeine is a known inhibitor of ATM, as well as ATR, the results suggest involvement of ATM-mediated signaling. Interestingly, it has been reported that ATM is activated following inhibition of PARP activity [9]. Both ATM- and DNA-dependent protein kinase (DNA-PK) have been shown to phosphorylate H2AX after exposure to IR [24], whereas γH2AX formation in response to replication blocks following treatment with UV or hydroxyurea is ATR-dependent [23]. The data are consistent with the idea that γH2AX formation, and the G2/M arrest observed following exposure to MMS + 4-AN, is ATM-dependent.
Taken together, the results suggest that exposure to MMS + 4-AN induces activation of an ATR- and Chk1-mediated S-phase checkpoint leading to formation of double strand breaks and an ATM-mediated G2/M cell cycle arrest. The results with UCN-01 suggest that Chk1 may be activated by upstream kinase(s) in addition to ATR, therefore, even though the G2/M arrest is not mediated by ATR, Chk1 activation may still be involved in this checkpoint. Future studies will further investigate the signaling pathway(s) involved in the observed G2/M arrest.
Acknowledgements
We thank Dr. William A. Beard for help with figure preparation, Zachary Weiner for technical assistance, and Jennifer Myers for editorial assistance. We also thank Christina Markunas and Daniel Shaughnessy for help with gamma irradiation. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
REFERENCES
- [1].Srivastava DK, Vande Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- [2].Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair (Amst.) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- [3].Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- [4].Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- [5].D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- [6].Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- [7].Horton JK, Baker A, Vande Berg BJ, Sobol RW, Wilson SH. Involvement of DNA polymerase β in protection against the cytotoxicity of oxidative damage. DNA Repair (Amst.) 2002;1:317–333. doi: 10.1016/s1568-7864(02)00008-3. [DOI] [PubMed] [Google Scholar]
- [8].Satoh MS, Lindahl T. Role of poly (ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- [9].Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34:1685–1691. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest following DNA methylating agent exposure. J. Biol. Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- [11].Horton JK, Stefanick DF, Wilson SH. Involvement of poly(ADP-ribose) polymerase activity in regulating Chk1-dependent apoptotic cell death. DNA Repair (Amst.) 2005;4:1111–1120. doi: 10.1016/j.dnarep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- [12].Zhang Y-W, Hunter T, Abraham RT. Turning the replication checkpoint on and off. Cell Cycle. 2006;5:125–128. doi: 10.4161/cc.5.2.2308. [DOI] [PubMed] [Google Scholar]
- [13].Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Butler WB. Preparing nuclei from cells in monolayer cultures suitable for counting and for following synchronized cells through the cell cycle. Anal. Biochem. 1984;141:70–73. doi: 10.1016/0003-2697(84)90426-3. [DOI] [PubMed] [Google Scholar]
- [15].Banasik M, Komura H, Shimoyama M, Ueda K. Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J. Biol. Chem. 1992;267:1569–1575. [PubMed] [Google Scholar]
- [16].Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J. Biol. Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- [18].Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- [19].Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000;60:2108–2112. [PubMed] [Google Scholar]
- [20].Liu X, Guo Y, Li Y, Jiang Y, Chubb S, Azuma A, Huang P, Matsuda A, Hittelman W, Plunkett W. Molecular basis for G2 arrest induced by 2′-C-cyano-2′-deoxy-1-β-d-arabino-pentofuranosylcytosine and consequences of checkpoint abrogation. Cancer Res. 2005;65:6874–6881. doi: 10.1158/0008-5472.CAN-05-0288. [DOI] [PubMed] [Google Scholar]
- [21].Zhao B, Bower MJ, McDevitt PJ, Zhao H, Davis ST, Johanson KO, Green SM, Concha NO, Zhou BB. Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem. 2002;277:46609–46615. doi: 10.1074/jbc.M201233200. [DOI] [PubMed] [Google Scholar]
- [22].Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- [23].Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- [24].Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- [25].de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- [26].Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- [27].Yang Y-G, Cortes U, Patnaik S, Jasin M, Wang Z-Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 2004;23:3872–3882. doi: 10.1038/sj.onc.1207491. [DOI] [PubMed] [Google Scholar]
- [28].Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- [29].Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- [30].Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol. 2001;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- [31].Haince J-F, Rouleau M, Hendzel MJ, Masson J-Y, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol. Med. 2005;11:456–463. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- [32].Wang H, Wang H, Powell SN, Iliakis G, Wang Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004;64:7139–7143. doi: 10.1158/0008-5472.CAN-04-1289. [DOI] [PubMed] [Google Scholar]
- [33].Jacobson EL, Meadows R, Measel J. Cell cycle perturbations following DNA damage in the presence of ADP-ribosylation inhibitors. Carcinogenesis. 1985;6:711–714. doi: 10.1093/carcin/6.5.711. [DOI] [PubMed] [Google Scholar]
- [34].Wang JL, Wang X, Wang H, Iliakis G, Wang Y. CHK1-regulated S-phase checkpoint response reduces camptothecin cytotoxicity. Cell Cycle. 2002;1:267–272. [PubMed] [Google Scholar]
- [35].Cliby WA, Lewis KA, Lilly KK, Kaufmann SH. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 2002;277:1599–1606. doi: 10.1074/jbc.M106287200. [DOI] [PubMed] [Google Scholar]
- [36].Wang H, Wang X, Zhou XY, Chen DJ, Li GC, Iliakis G, Wang Y. Ku affects the ataxia and Rad 3-related/CHK1-dependent S phase checkpoint response after camptothecin treatment. Cancer Res. 2002;62:2483–2487. [PubMed] [Google Scholar]
- [37].Boorstein RJ, Pardee AB. Factors modifying 3-aminobenzamide cytotoxicity in normal and repair-deficient human fibroblasts. J. Cell. Physiol. 1984;120:335–344. doi: 10.1002/jcp.1041200312. [DOI] [PubMed] [Google Scholar]