

Abstract
The cyclin-dependent kinase (CDK) inhibitor p16INK4a and the MDM2 ubiquitin ligase inhibitor p19ARF are critical to the regulation of cell cycle progression. Their loss by deletion, mutation or epigenetic silencing is a common molecular alteration in many human cancers. To investigate the role of p16INK4a/p19ARF deficiency in CNS tumor pathogenesis, pregnant mice bearing p16-/-/p19-/-, p16+/-/p19+/-, and p16+/+/p19+/+ embryos were exposed transplacentally on gestation day 14 to a single dose of the potent carcinogen, ethylnitrosourea (ENU). p16+/-/p19+/- male mice treated with ENU developed meningial proliferative lesions with a high incidence (5/10). The incidence was lower in other ENU-treated animals of both sexes and none occurred in saline-treated control animals. The lesions ranged from widespread meningeal proliferation and plaque-like thickening by neoplastic spindle cells consistent with meningiomatosis to a larger discrete mass consistent with a meningioma. Ultrastructural analysis revealed the presence of intercellular junctions between cells, supporting a meningothelial histogenesis. Spontaneous meningiomas occur rarely in wild-type mice but are a common neoplasm afflicting humans, accounting for between 13 and 26% of primary intracranial neoplasms. This ENU inducible meningeal lesion in p16+/-/p19+/- mice may provide additional insight into the pathogenesis of meningeal neoplasia and aid the development of therapeutics.
Keywords: Animal model, central nervous system, meningioma, meningiomatosis, mouse, tumor suppressor gene deficiency, ethylnitrosourea
Introduction
Meningiomas account for between 13% and 26% of primary intracranial neoplasms in humans in the United States and constitute a significant cause of morbidity and mortality, particularly among older individuals and women (Louis, 2000). Although most meningiomas have a benign clinical course and many are amenable to surgical resection, approximately 10% display more aggressive clinical features such as brain invasion and a tendency to recur after surgery and, for these neoplasms, treatment options are more limited (Louis, 2000; Lusis and Gutmann, 2004). Although significant progress has been made in understanding meningioma pathogenesis, research into this important topic has been hindered by the small number of suitable animal models (McCutcheon et al., 2000; Kalamarides et al., 2002; Baia et al., 2006).
The cyclin-dependent kinase (CDK) inhibitor p16INK4a is a critical regulator of cell cycle progression through its inhibition of CDK 4 and CDK 6, thereby preventing the cell from passing the G1/S checkpoint. Functional loss of this protein by deletion, mutation or epigenetic silencing is one of the most commonly observed molecular alterations in human cancer and has been documented in a subset of human CNS tumors, including glioblastomas, ependymomas, and atypical and anaplastic meningiomas (Ruas and Peters, 1998; Bostrom et al., 2001; Korshunov et al., 2003; Rousseau et al., 2003; Sharpless, 2005). p19ARF (called p14ARF in humans) is an inhibitor of the ubiquitin ligase MDM2, which is responsible for ubiquitinating, and thereby inducing the degradation of the p53 tumor suppressor protein. Functional loss of p19ARF effectively allows the cell to bypass the G2/M checkpoint, and has been found in many types of human cancer (Serrano, 2000; Sharpless, 2005). Combined loss of p16INK4a and p19ARF has been consistently recognized in several highly aggressive human neoplasms, including aggressive meningiomas, and has been proposed as an important step on the road to malignancy (Sharpless, 2005; Kim and Sharpless, 2006). The p16INK4a and p19ARF genes partially overlap in the mammalian genome, sharing exons 2 and 3. Curiously, the reading frames of the two proteins are different and so there is no sequence similarity between them (Serrano, 2000).
Recent studies have demonstrated the importance and utility of p16INK4a deficient mouse models for studying cancer (Ruas and Peters, 1998; Serrano, 2000; Sharpless et al., 2001). The p16INK4a-deficient or Ink4a/Arf null (B6.129-Cdkn2atm1Rdp) mouse carries a targeted deletion of exons 2 and 3 of the INK4a/ARF locus which eliminates both p16INK4a and p19ARF . On a mixed genetic background, these mice displayed spontaneous development of various tumors by up to 20 weeks of age and loss of this locus is associated with an increased incidence of several carcinogen-induced tumors (Serrano et al., 1996; Kamijo et al., 1999; Sharpless et al., 2001). Thus, p16INK4a/p19ARF deficient mice are both a powerful new model for screening potential carcinogens and also an important tool for performing mechanistic studies of cancer development.
In the present report, we investigated the effect of p16INK4a/p19ARF deficiency on the development of CNS tumors by exposing p16-/-/p19-/-, p16+/-/p19+/-, and p16+/+/p19+/+ pups in utero to the well known carcinogen, ethylnitrosourea (ENU), at gestation day 14.
Materials and Methods
Animals and Animal Care
Male and female genetically altered mice were purchased from Taconic Farms (Germantown, NY). The p16-/-/p19-/- mouse (commercial name: Ink4a/Arf null; strain nomenclature: B6.129-Cdkn2atm1Rdp) was derived from the DePinho mouse (Serrano et al., 1996) and carries a targeted deletion of exons 2 and 3 of the INK4a/ARF locus which eliminates both p16INK4a and p19ARF. In total, there were 34 p16-/-/p19-/- females (129/Sv × C57BL/6 × SJL)-(K)1INK4a carrying p16-/-/p19-/- pups, 6 C57BL/6 females carrying p16+/-/p19+/- pups, and 10 C57BL/6 females carrying p16+/+/p19+/+ pups. Mice were 6-13 weeks old at receipt, and their body weight range was 16.0-23.7 g. A subset of the female mice was purchased from Taconic Farms pregnant; however, due to small litter sizes from many animals, additional male and nonpregnant female mice were purchased and breedings were performed on-site in order to obtain a sufficient number of animals of the p16-/-/p19-/- genotype.
Mice were fed Purina Mouse Diet 5015 (Granville Milling Creedmoor, NC) or NTP 2000 diet (Madison, WI) and tap water (City of Durham) ad libitum. Environmental conditions included ambient temperature ranging from 17-28°C, relative humidity ranging from 12-77% and a 12-hour light-dark cycle. Animals were housed in polycarbonate cages with absorbent hardwood bedding (Beta Chips, Northeastern Products Corp., Warrensburg, NY). Animal care and use was in accordance with the United States Public Health Service policy on humane care and use of laboratory animals and the Guide for the Care and Use of Laboratory Animals (DHHS NIH No. 86-23, 1996).
Chemical and Treatment
N-ethyl-N-nitrosourea [ethylnitrosourea (ENU)] was used for tumor induction. ENU Isopac (CAS No. 759-73-9) was obtained from Sigma Chemical Co. (St. Louis, MO). The ENU was prepared in sterile saline at a dose level of 25 mg/kg body weight and a dosing volume of 10 mL/kg body weight. Sterile saline was used as a negative control. The dosing solutions were administered to the animals intraperitoneally on gestation day 14.
Clinical Observation
All mice were observed once (weekends) or twice (week-days) daily for mortality and morbidity. Clinical observations of mice were recorded weekly from weaning until study termination.
Genotyping
Genotyping of newborn mice was performed using an approximately 2 mm tail fragment and a commercially available DNA extraction and amplification kit (Sigma Extract N Amp Tissue PCR Kit, Sigma-Aldrich, Inc.). Procedures were performed according to the manufacturer’s instructions. Primer sequences 5′-GTG ATC CCT CTA CTT TTT CTT CTG ACT T-3′ and 5′-CGG AAC GCA AAT ATC GCA C-3′ were employed to amplify the wild-type allele and 5′-GTG ATC CCT CTA CTT TTT CTT CTG ACT T-3′ and 5′-GAG ACT AGT GAG ACG TGC TAC TTC CA-3′ were employed to amplify the modified allele. After amplication the DNA samples were examined using 3% agarose gel containing ethidium bromide. Amplified DNA fragment sizes were 278 bp for the wild-type allele and 313 bp for the genetically modified allele.
Pathology
At the time of scheduled terminal sacrifice or when the mice were clinically moribund, they were humanely euthanatized via carbon dioxide asphyxiation and a full necropsy was performed. Due to significantly higher morbidity and mortality for the homozygous knockout mice, the age at terminal sacrifice varied from 14-19 weeks for the p16-/-/p19-/-mice to 30-39 weeks for the p16+/-/p19+/- and p16+/+/p19+/+ mice. Animals that were found dead were generally not necropsied due to advanced autolysis. At necropsy, the brain, liver, lung, kidney, spleen, pancreas, urinary bladder, mammary gland, skin, and any gross lesions were collected and immersion fixed in 10% neutral-buffered formalin. All tissues were then processed routinely and embedded in paraffin wax, sectioned at 5 μm, and stained with hematoxylin and eosin (HE) for microscopic examination. Select cases of the brain were further characterized using the periodic acid-Schiff (PAS) stain reaction for polysaccharides, alcian blue stain for myxoid connective tissue, and Masson’s trichrome stain for collagen fibers.
Immunohistochemistry
Immunohistochemistry was performed for vimentin and cytokeratin using commercially available antibodies and standard protocols. The primary antibodies used were a monoclonal mouse anti-vimentin (DAKO EPOS™ Anti-vimentin/HRP, Clone Vim 3B4, Carpinteria, CA) prediluted, with a 1 hour incubation at room temperature (RT) and rabbit polyclonal pan-cytokeratin (DAKO, Carpinteria, CA) at 1:500, with a 30 minute incubation at RT. Antigen retrieval pretreatments were pronase digestion for 20 minutes at RT for vimentin and incubation in 0.1 M Sodium Citrate solution, pH 6.0 for approximately 5 minutes in a pressurized steamer (Decloaking Chamber™ Biocare Medical, Concord, CA) for cytokeratin; 3,3′-diaminobenzidine was used as the chromagen and hematoxylin was the counterstain for both antigens.
Ultrastructural Analysis
For electron microscopic examination, a formalin-fixed and paraffin-embedded sample from 1 animal was deparaf-finized and processed for electron microscopy. The deparaffinized section was immersed in 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.4) before postfixing in 0.1% osmium tetroxide in 0.1M phosphate buffer (pH 7.4). Then, the sample was routinely processed and embedded in Poly/Bed 812 epoxy resin (Polysciences, Inc, Warrington, PA). Ultrathin sections were stained with uranyl acetate and lead citrate, and selected target areas were examined by transmission electron microscopy using a Tecnai 12, Model BioTWIN transmission electron microscope.
Statistical Analysis
Fisher’s exact test was used to compare tumor incidences. One-sided p-values were used for comparing the ENU and saline groups.
Results
Mortality
Mortality was significantly increased in ENU-treated homozygous p16-/-/p19-/- animals as compared to saline-treated animals (log rank test p < 0.001 for females, p = 0.011 for males). In ENU-treated male p16+/-/p19+/- mice, mortality was 70% (7/10), while mortality was 40% (12/30) in p16-/-/p19-/- males. The ENU-treated animals had a higher incidence of neoplasia in several organ systems, but particularly the lung and the meninges (see Table 1 for meningeal neoplasia incidence). Since the heterozygous animals were on study for 30-39 weeks, while homozygous negative animals were only on the study for approximately 14-19 weeks, the increased mortality of the heterozygous animals likely reflects the increased neoplasm development with time.
TABLE 1.
Incidence of meningiomatosis and meningioma in p16/p19-deficient mice after in utero exposure to ENUa
| Treatment Genotype | Saline |
ENU |
||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| p16-/-/p19-/- | 0/19b (0%) | 0/23 (0%) | 2/26 (7.7%)d | 0/17 (0%) |
| p16+/-/p19+/- | 0/5 (0%) | 0/4 (0%) | 5/10 (50%)c,d | 1/8 (12.5%)d |
| p16+/+/p19+/+ | 0/8 (0%) | 0/8 (0%) | 0/14 (0%) | 0/10 (0%) |
p16-/-/p19-/- = homozygous knockout, p16+/-/p19+/- = heterozygous knockout, p16+/+/p19+/+ = wild-type, ENU = Ethylnitrosourea.
In utero exposure to a single dose of ENU or saline on gestation day 14.
The number of animals having the lesion as a function of the total number of animals with this genotype examined histopathologically.
One of these animals had meningiomatosis and a meningioma.
Within the genotype, the values are not statistically significant using a Fisher’s exact test.
Gross and Microscopic Observations
Clinically and at necropsy, a subset of ENU-treated animals (10.3%; 9/87) had a dome shaped head with excess cerebrospinal fluid (CSF) that was apparent as soon as the skull was opened. The location of the excess fluid accumulation was superficial, and no evidence of ventricular dilation (hydrocephalus) was present in the vast majority of animals with this gross lesion. No grossly recognizable meningeal or brain alterations were noted at necropsy.
The gross finding of a dome shaped head correlated microscopically with widespread, typically plaque-like, leptomeningeal thickening by small streaming bundles or whorling perivascular aggregates of spindle shaped neoplastic cells (Figures 1 and 2). These meningial lesions were induced in 50% (5/10) of the p16INK4a/p19ARF deficient heterozygous males after in utero exposure to ethylnitrosourea at gestation day 14. All affected animals had widespread meningeal thickening ranging from minimal to marked severity, and, in addition, 1 animal also had an expansile meningioma. Incidences were lower in all other ENU-treated groups, as summarized in Table 1.
Figure 1.
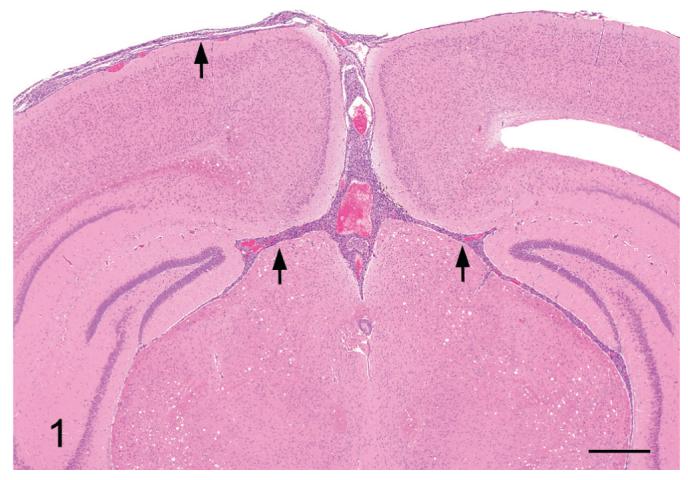
A section of brain with meningiomatosis from a p16+/-/ p19+/- mouse exposed in utero to ENU. Note the moderate meningiomatosis involving the leptomeninges of the dorsum, the dorsal cerebral fissure and the dorsal meninges of the brainstem (arrows). H&E Bar = 1 mm.
Figure 2.
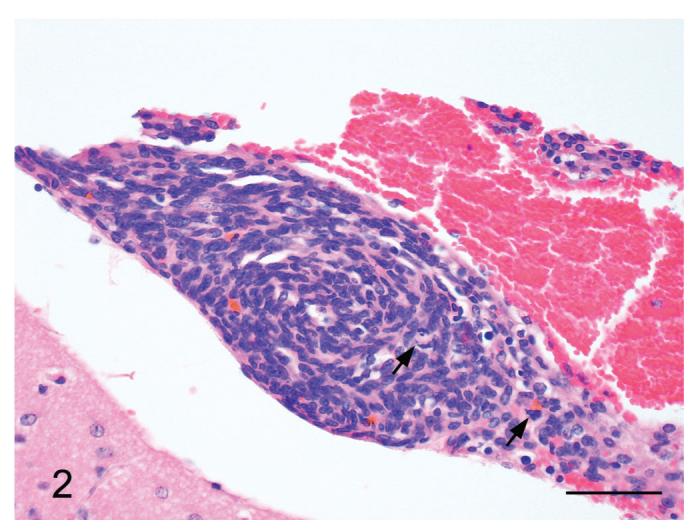
Meningiomatosis from a p16+/-/p19+/- mouse exposed in utero to ENU. Note the small meningeal whorls of spindle shaped neoplastic cells with occasional interspersed mitotic figures (arrows). H&E Bar = 50 μm.
The spindle shaped cells had round to elongate nuclei, scant to small amounts of pale eosinophilic cytoplasm and illdefined cellular margins. Rarely, the cells displayed more epithelioid differentiation, characterized by increased amounts of eosinophilic cytoplasm and plump nuclei, a morphology that is characteristic for meningothelial cells (Figure 3). In several areas, small clusters of cells with small hyperchromatic nuclei and scant cytoplasm were present (lymphocyte-like) (Figure 3) and occasionally, individual cells displayed mild karyomegaly, with nuclei approximately twice as large as the adjacent cells. The mitotic rate was variable, with occasional mitotic figures in the diffuse lesions and an increased number (1-3 mitotic figures/HPF) in the mass lesion. Clusters of cells multifocally penetrated the superficial brain parenchyma along the peri-vascular (Virchow-Robbins) spaces, particularly in the thicker plaques, but frank neuroparenchymal invasion was not present. In less severely affected animals, the leptomeningeal thickening averaged only 5-20 cells thick but still extended to cover large areas of the brain (ranging from approximately 20-60% in the sections examined). In one animal, a mass composed of interweaving bundles of cells invaded and markedly expanded the leptomeninges of the choroid plexus stroma of the fourth ventricle (Figures 4 and 5). Substantial brain compression was not a feature in any of the affected animals.
Figure 3.
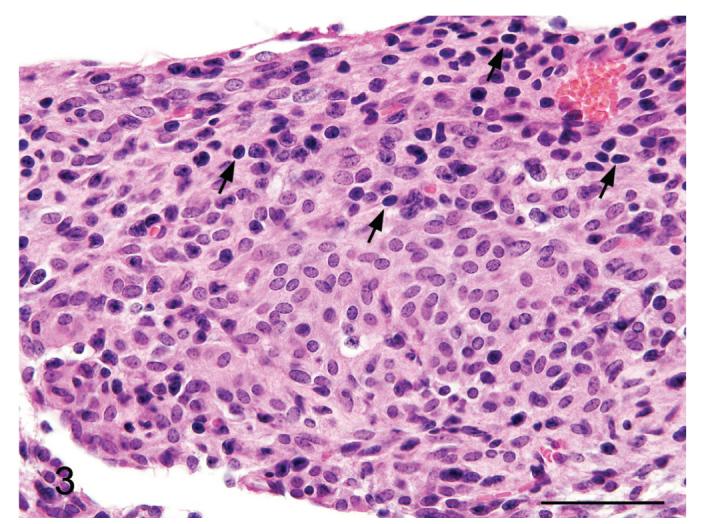
Meningiomatosis from a p16+/-/p19+/- mouse exposed in utero to ENU. In several areas of the meninges the cytomorphology was more epithelioid, with bland nuclei and more abundant eosinophilic cytoplasm, which is a typical feature of meningeal neoplasms. Also note the scattered lymphocyte-like cells (arrows); their presence is a feature of aggressive meningiomas in humans. H&E. Bar = 100 μm.
Figure 4.
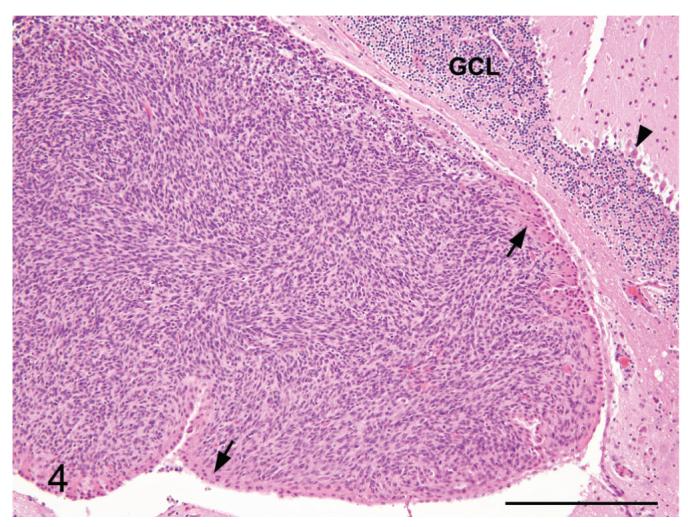
Meningioma from a p16+/-/p19+/- mouse exposed in utero to ENU. A meningioma is expanding the stroma of the choroid plexus of the 4th ventricle. Note the overlying choroid plexus epithelium (arrows), and the Purkinje cell layer (arrowhead) and the granule cell layer (GCL) of the cerebellum for orientation. H&E. Bar = 250 μm.
Figure 5.
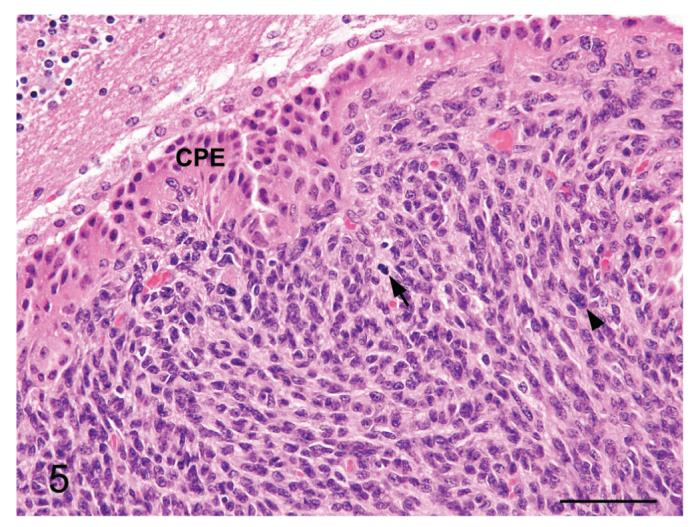
Meningioma from a p16+/-/p19+/- mouse exposed in utero to ENU. Higher magnification image of Figure 4. Note the occasional mitotic figure (arrow), karyomegalic cell (arrowhead) and the choroid plexus epithelium (CPE). H&E. Bar = 100 μm.
Several special histochemical and immunohistochemical stains were performed to further characterize the meningeal lesions. Masson’s trichrome stain highlighted a small amount of collagen interspersed amongst the spindle cells (Figure 6). Neither cells nor matrix stained with alcian blue or periodic acid Schiff (PAS). Immunohistochemistry for vimentin and cytokeratin was also performed. All the lesions displayed strong positive staining for vimentin (Figure 7), although it was typically multifocal. None of the samples was positive for cytokeratin (Figure 8).
Figure 6.
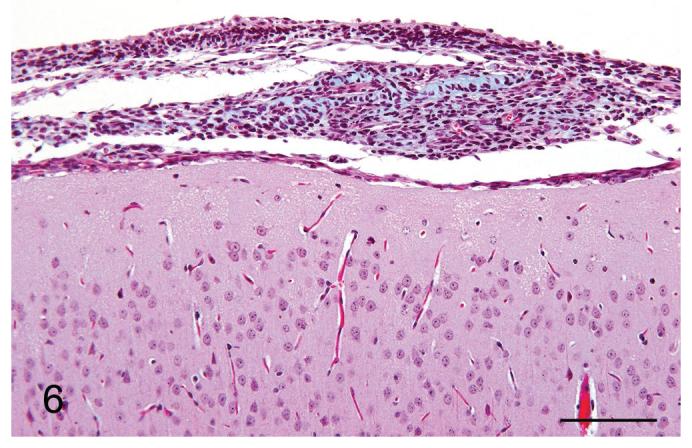
Meningiomatosis from a p16+/-/p19+/- mouse exposed in utero to ENU. A small amount of collagen was present in the meningeal plaque, as evidenced by the light blue staining. Masson’s trichrome stain. Bar = 100 μm.
Figure 7.
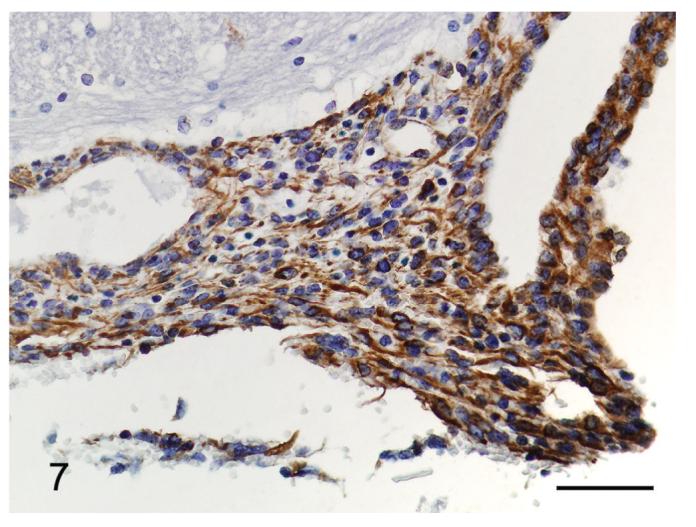
Meningiomatosis from a p16+/-/p19+/- mouse exposed in utero to ENU. The neoplastic cells are strongly positive for vimentin. Vimentin immunohistochemistry. Bar = 50 μm.
Figure 8.
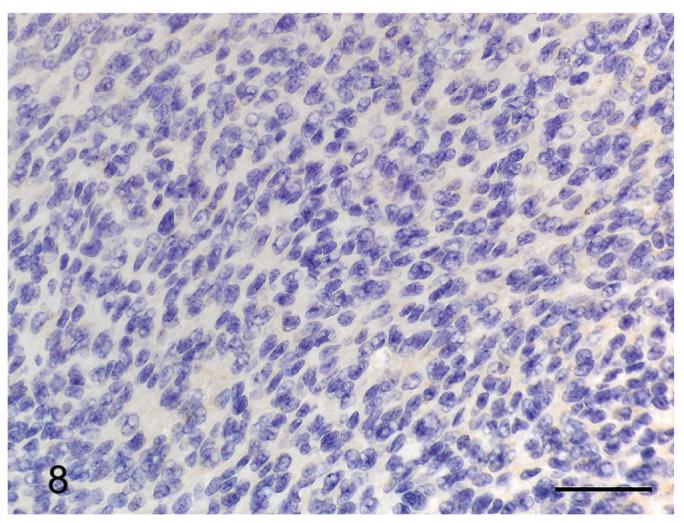
Meningioma from a p16+/-/p19+/- mouse exposed in utero to ENU. The neoplastic cells are diffusely negative for cytokeratin. Pan-cytokeratin immunohistochemistry. Bar = 50 μm.
Although preservation was less than ideal due to the availability of only paraffin embedded, formalin fixed tissue, transmission electron microscopy (TEM) performed on one animal revealed the presence of small numbers of intercellular (desmosome-like) junctions between neoplastic cells, supporting a meningothelial histogenesis (Figure 9).
Figure 9.
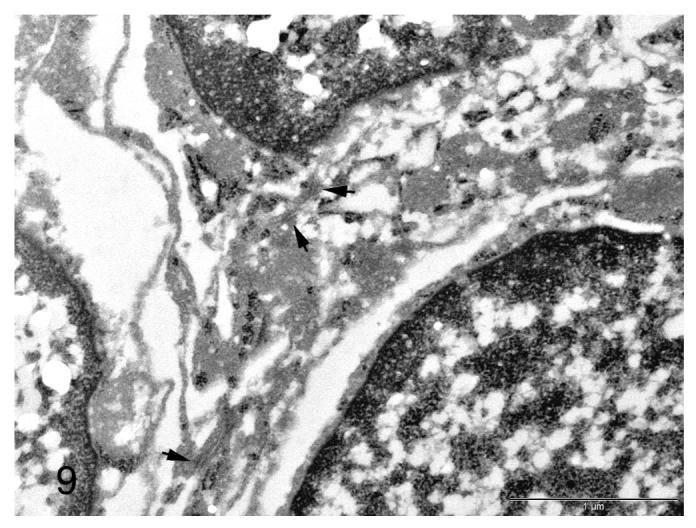
Electron micrograph of a meningiomatosis lesion from a p16+/-/p19+/- mouse exposed in utero to ENU. Small numbers of intercellular junctions are present between neoplastic cells (arrows). Bar = 1 μm.
In addition to the meningeal proliferative lesions, a variety of epithelial and mesenchymal neoplasms were observed in other organ systems of the ENU-treated mice of all genotypes and both sexes. These included a high incidence of pulmonary alveolar-bronchiolar adenomas and lower incidences of hepatocellular adenomas, undifferentiated sarcomas, and histiocytic sarcomas. The high mortality in the ENU-treated groups in this study is attributed to the high incidence of neoplastic lesions present in a variety of organ systems. In the subset of ENU-treated animals with the meningeal lesions, single or multiple pulmonary alveolar-bronchiolar adenomas were present in 7/8 (87.5%) animals. However no other neoplasms, aside from the meningeal lesions, were apparent in these animals on the study, effectively excluding the possibility that the meningeal lesions represent distant metastases of a neoplasm from another organ system.
Discussion
In the present study, male p16+/-/p19+/- mice exposed in utero to a single dose of ethylnitrosourea (ENU) on gestation day 14 showed a high incidence (50%) of meningial proliferative lesions, which we have designated meningiomatosis for the widespread, plaque-like lesions, and meningioma for the single discrete, expansile mass. A lower incidence was present in homozygous knockout (KO) male mice (2/26) and heterozygous KO female mice (1/8) that were treated with ENU. None of the saline-treated control animals or the wild-type, ENU-treated animals developed meningeal lesions. Although the incidences are not statistically significant (see Table 1), this is likely due to the small number of animals in some groups on the study. The p16/p19 deficient mouse has been used extensively in the National Toxicology Program and the meningeal lesion described in this report has not been seen as a lesion in control or treated mice (0/648) 27-40 weeks of age (National Toxicology Program a-d, 2005-2007). This included 120 untreated control male and female p16/p19-deficient mice. Therefore, the possibility that the results are due to chance is considered minimal.
The lower incidence of these lesions in ENU-treated, homozygous KO animals may be related to their decreased life span; the date of their terminal sacrifice was much earlier due to their overall poor clinical condition. No other central nervous system tumors occurred in these animals, contrary to expectations based on reports of the preferential induction of CNS parenchymal tumors in rats by in utero exposure to ENU (Zook and Simmens, 2005) and the loss of the p16INK4a and p19ARF tumor suppressor proteins in several human CNS parenchymal neoplasms, such as glioblastoma multiforme and ependymoma (Ruas and Peters, 1998; Rousseau et al., 2003).
Most animals with a histologically apparent meningeal lesion also had the gross finding of a dome-shaped head, the cause of which was not conclusively determined. It may have resulted directly from the proliferative lesions or possibly have been secondary to interference of cerebrospinal fluid resorption at the arachnoid villi by these meningeal lesions. The proliferative lesions were variable in severity but were similar in cellular morphology and in the lesion’s widespread occurrence, which typically involved between 20-60% of the leptomeninges of the brain in the sections examined. The widespread, plaque-like lesions were diagnosed as meningiomatosis, which is a rare neoplastic condition described in human neuropathology characterized by neoplastic cells having widespread to diffuse infiltration of the leptomeninges in the absence of a large circumscribed mass (Russell, 1989; Avellino et al., 1994; Wakabayashi et al., 1997).
In one animal there was a discrete mass that expanded the choroid plexus stroma and had more malignant features, such as a higher mitotic rate, mild anisokaryosis and the presence of small cells with scant cytoplasm and hyperchromatic (lymphocyte-like) nuclei (Perry et al., 1997, 1999; Louis, 2000). This animal also had extensive meningiomatosis throughout the remaining leptomeninges and the mass was interpreted as evidence of progression of the meningiomatosis lesions to a more aggressive phenotype.
The cytomorphology of the lesions was predominantly spindle shaped with rare areas displaying more epithelioid differentiation, which is a common feature of meningiomas in other species, particularly humans. The large variety of patterns described in the literature for benign meningiomas (e.g., transitional, meningothelial, psammomatous, etc.) was not present in this study. Immunohistochemical staining for vimentin and cytokeratin was positive and negative, respectively. Positive staining for cytokeratin has frequently been reported in human meningiomas and the staining is typically multifocal, with the positive staining most commonly in areas with an epithelioid morphology. Epithelioid neoplastic cells were present only rarely in this study, possibly accounting for the lack of cytokeratin expression. Although the preservation was less than ideal due to formalin fixation and paraffin embedding, the transmission electron microscopy performed on one sample revealed several intercellular junctional complexes between neoplastic cells, supporting a meningothelial histogenesis (Louis, 2000).
Meningiomas in humans and other species are typically benign, slow-growing tumors that presumably arise from the arachnoidal cap cells of the leptomeninges. Approximately 5-10% of human meningiomas have more aggressive histologic features including increased numbers of mitoses and increased cellular atypia and for these neoplasms the recurrence rate is significantly higher (Louis, 2000). The histomorphology of the meningeal lesions in these mice more closely resembles the aggressive human variants of meningeal neoplasia, the atypical and anaplastic meningiomas and meningiomatosis, than their more common benign counterparts. Given these features and the reported loss of p16 and p19 in atypical and anaplastic meningiomas in humans (Bostrom, 2001; Korshunov, 2003), this mouse model may provide particular insight into these forms of meningeal neoplasia, thereby complementing the existing models. Current models described in the literature employ various techniques to model the human disease such as intracranial or subcutaneous injections of human meningioma cells into immunodeficient mice (McCutcheon, 2000; Malham, 2001), the study of meningioma cells in cell culture (Baia, 2006), or injection of recombinant adenovirus into the CSF to ‘knock-out’ the tumor suppressor NF2 within the leptomeninges (Kalamarides, 2002).
The p16INK4a gene, located in the 9p21 chromosomal region in humans, encodes a protein that binds to cyclin dependent kinase (CDK) 4 and CDK6, inhibiting the formation of the CDK4 or 6/cyclin D complexes required for kinase activity and the phosphorylation of Rb, and thereby preventing entry into the S phase of the cell cycle. Thus, p16INK4a acts as a potent negative regulator of the cell cycle (Ruas and Peters, 1998; Kim and Sharpless, 2006).
The p19ARF gene (referred to as p14ARF in humans) is located at the same chromosomal locus as p16INK4a and these 2 genes share exons 2 and 3, although they have different open reading frames and so have no protein sequence similarity (a rare event in metazoans). p19ARF is also a potent tumor suppressor protein, which, through binding to and inhibiting the ubiquitin ligase MDM2, stabilizes the p53 tumor suppressor protein and allows it to accumulate and block cell cycle progression at the G2/M checkpoint (Serrano, 2000; Sharpless, 2005).
The INK4A/ARF locus at chromosome 9p21 in humans is among the most frequent sites of genetic loss in human cancer and the deletion of this locus has been documented with high frequency in a variety of human malignancies (Sharpless, 2005). In addition, the importance of the loss of these proteins in carcinogenesis is strongly supported by studies in transgenic p16INK4a/p19ARF deficient mice. These mice develop a variety of spontaneous tumors with a high frequency, including soft tissue sarcomas, histiocytic sarcomas, lymphoma, and various carcinomas beginning by approximately 20 weeks of age. They also display a dramatic susceptibility to neoplasia upon exposure to various carcinogens (Serrano et al., 1996; Sharpless et al., 2004) and are being evaluated for use in toxicological studies as an efficient and sensitive means to measure carcinogenic potential in vivo (National Toxicology Program a-d, 2005-2007). It has been proposed that combined deficiency of these proteins in human neoplasms represents an important step on the road to malignancy (Sharpless, 2005).
Several recent reports have identified the loss of p16INK4a and/or p19ARF in human meningiomas, particularly in the 2 more aggressive variants, atypical and anaplastic/malignant meningiomas (WHO Grade II and III, respectively) (Bostrom et al., 2001; Simon et al., 2001; Korshunov et al., 2003; Perry et al., 2004). The single more advanced meningeal lesion in this study had more malignant features such as a higher mitotic rate and invasion of the choroid plexus stroma. This supports our contention that despite the differences in morphology between the lesions in this study and benign meningiomas in humans, this may be a valuable model of human-meningeal neoplasms and will potentially be more useful in the study of the aggressive forms, such as the atypical and anaplastic meningiomas.
The highest incidence of meningeal lesions in our study occurred in the ENU-treated p16+/-/p19+/- animals. This difference may be due to the increased time that p16+/-/p19+/- animals were on the study compared with the p16-/-/p19-/- animals, which quickly became moribund and were sacrificed much earlier, at 14-19 weeks, as a result. This shortened lifespan potentially did not allow sufficient time for significant meningeal lesion development. The p16-/-/p19-/- animals exhibited a variety of neoplasms in multiple organ systems, to which their shortened life span is attributed. The incidence of meningeal lesions was much higher in male mice compared to female mice, which is contrary to the incidence of spontaneous meningiomas in humans. A pronounced female bias occurs in humans, and it has been proposed that sex hormones such as estrogen and progesterone play a role in meningioma pathogenesis (Claus et al., 2005). The reason for such a marked sex difference in the mice in this study was not apparent.
In conclusion, p16+/-/p19+/- mice exposed transplacentally to a single, nonlethal dose of ethylnitrosourea (ENU) developed meningiomatosis with a high incidence and a meningioma in one mouse. Functional loss of p16INK4a/p19ARF has been strongly associated with the malignant progression of meningiomas in humans and this animal model may provide valuable insight into meningioma pathogenesis, as well as aiding in the development and evaluation of much needed therapeutic agents.
Acknowledgments
The authors thank Dr. Peter B. Little, from Charles River Laboratories, Pathology Associates, Durham, NC, and Drs. John Peckham and Gordon Flake of the Laboratory of Experimental Pathology of the NIEHS for their critical review of the manuscript and insightful comments. We also thank Ms. Natasha Clayton of the NIEHS for her excellent technical assistance, Ms. Mary Ellen Sutphin and colleagues from the Experimental Pathology Laboratories, Inc. (EPL), RTP, NC for support in retrieving specimens and data from the NTP archives, Ms. Maureen Puccini, also of EPL, for assistance with digital photography, Dr. Glenda Moser from Integrated Laboratory Systems, RTP, NC for the conduct of the study, and the late Dr. Joel Mahler of the NIEHS for the original pathology evaluation.
This work was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Abbreviations
- CDK
cyclin-dependent kinase
- CNS
central nervous system
- ENU
N-ethyl-N-nitrosourea (ethylnitrosourea)
References
- Avellino AM, Hair LS, Symmans WF, Gold AP, Carmel P, Powers JM. Meningeal meningiomatosis in a child: case report. Clin Neuropathol. 1994;13:82–7. [PubMed] [Google Scholar]
- Baia GS, Slocum AL, Hyer JD, Misra A, Sehati N, Vandenberg SR, Feuerstein BG, Deen DF, McDermott MW, Lal A. A genetic strategy to overcome the senescence of primary meningioma cell cultures. J Neurooncol. 2006;78:113–21. doi: 10.1007/s11060-005-9076-y. [DOI] [PubMed] [Google Scholar]
- Bostrom J, Meyer-Puttlitz B, Wolter M, Blaschke B, Weber RG, Lichter P, Ichimura K, Collins VP, Reifenberger G. Alterations of the tumor suppressor genes CDKN2A (p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2C (p18(INK4c)) in atypical and anaplastic meningiomas. Am J Pathol. 2001;159:661–9. doi: 10.1016/S0002-9440(10)61737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus EB, Bondy ML, Schildkraut JM, Wiemels JL, Wrensch M, Black PM. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57:1088–95. doi: 10.1227/01.neu.0000188281.91351.b9. [DOI] [PubMed] [Google Scholar]
- Kalamarides M, Niwa-Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A, Thomas G, Gutmann DH, Giovannini M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16:1060–5. doi: 10.1101/gad.226302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–22. [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Korshunov A, Shishkina L, Golanov A. Immunohistochemical analysis of p16INK4a, p14ARF, p18INK4c, p21CIP1, p27KIP1 and p73 expression in 271 meningiomas correlation with tumor grade and clinical outcome. Int J Cancer. 2003;104:728–34. doi: 10.1002/ijc.11013. [DOI] [PubMed] [Google Scholar]
- Louis DN, Scheithauer BW, Budka H, von Deimling A, Kepes JJ. Meningiomas. In: Kleihues PC, editor. World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of the Nervous System. IARC Press; Lyon: 2000. pp. 176–84. W. K. [Google Scholar]
- Lusis E, Gutmann DH. Meningioma: an update. Curr Opin Neurol. 2004;17:687–92. doi: 10.1097/00019052-200412000-00008. [DOI] [PubMed] [Google Scholar]
- Malham GM, Thomsen RJ, Synek BJ, Baguley BC. Establishment of primary human meningiomas as subcutaneous xenografts in mice. Br J Neurosurg. 2001;15:328–34. doi: 10.1080/02688690120072487. [DOI] [PubMed] [Google Scholar]
- McCutcheon IE, Friend KE, Gerdes TM, Zhang BM, Wildrick DM, Fuller GN. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg. 2000;92:306–14. doi: 10.3171/jns.2000.92.2.0306. [DOI] [PubMed] [Google Scholar]
- NTP Technical report GMM #1. NIEHS; Research Triangle Park, NC: 2005. National Toxicology Program (a) Toxicity studies of aspartame in FVB/N-TgN (v-HA-ras) Led (Tg.AC) hemizygous mice and carcinogenicity studies of aspartame in B6.129-Trp53tm1Brd (N5) haploinsufficient mice and 9-month study in Cdkn2a deficient mice. NIH publication # 03-4459. [Google Scholar]
- NTP Technical report GMM #13. NIEHS; Research Triangle Park, NC: 2007. National Toxicology Program (b) Toxicology and carcinogenesis study of glycidol in genetically modified haploinsufficient p16/p19 mice. NIH publication #06-5962. (In press) [Google Scholar]
- NTP Technical report GMM #8. NIEHS; Research Triangle Park, NC: 2007. National Toxicology Program (c) Toxicology and carcinogenesis study of benzene in genetically modified haploinsufficient p16/p19 mice. NIH publication #06-4425. (In press) [Google Scholar]
- NTP Technical report GMM #12. NIEHS; Research Triangle Park, NC: 2007. National Toxicology Program (d) Toxicology and carcinogenesis study of phenolphthalein in genetically modified haploinsufficient p16/p19 mice. NIH publication #06-5961.(In press) [Google Scholar]
- Perry A, Gutmann DH, Reifenberger G. Molecular pathogenesis of meningiomas. J Neurooncol. 2004;70:183–202. doi: 10.1007/s11060-004-2749-0. [DOI] [PubMed] [Google Scholar]
- Perry A, Scheithauer BW, Stafford SL, Lohse CM, Wollan PC. “Malignancy” in meningiomas: a clinicopathologic study of 116 patients, with grading implications. Cancer. 1999;85:2046–56. doi: 10.1002/(sici)1097-0142(19990501)85:9<2046::aid-cncr23>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Perry A, Stafford SL, Scheithauer BW, Suman VJ, Lohse CM. Meningioma grading: an analysis of histologic parameters. Am J Surg Pathol. 1997;21:1455–65. doi: 10.1097/00000478-199712000-00008. [DOI] [PubMed] [Google Scholar]
- Rousseau E, Ruchoux MM, Scaravilli F, Chapon F, Vinchon M, De Smet C, Godfraind C, Vikkula M. CDKN2A, CDKN2B and p14ARF are frequently and differentially methylated in ependymal tumours. Neuropathol Appl Neurobiol. 2003;29:574–83. doi: 10.1046/j.0305-1846.2003.00505.x. [DOI] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–77. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Russell DS, Rubinstein LJ. Pathology of Tumours of the Nervous System. Williams and Wilkins; Baltimore: 1989. Tumours of the Meninges and Related Tissues; p. 512514. [Google Scholar]
- Serrano M. The INK4a/ARF locus in murine tumorigenesis. Carcinogenesis. 2000;21:865–9. doi: 10.1093/carcin/21.5.865. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–85. doi: 10.1038/sj.onc.1207074. [DOI] [PubMed] [Google Scholar]
- Simon M, Park TW, Koster G, Mahlberg R, Hackenbroch M, Bostrom J, Loning T, Schramm J. Alterations of INK4a(p16-p14ARF)/INK4b(p15) expression and telomerase activation in meningioma progression. J Neurooncol. 2001;55:149–58. doi: 10.1023/a:1013863630293. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Kawasaki K, Ono K, Nishiyama K, Tanaka R, Takahashi H. Primary leptomeningeal meningiomatosis with widespread whorl formation. Brain Tumor Pathol. 1997;14:139–43. doi: 10.1007/BF02478883. [DOI] [PubMed] [Google Scholar]
- Zook BC, Simmens SJ. Neurogenic tumors in rats induced by ethylnitrosourea. Exp Toxicol Pathol. 2005;57:7–14. doi: 10.1016/j.etp.2005.04.001. [DOI] [PubMed] [Google Scholar]