

Abstract
When Saccharomyces cerevisiae cells are starved of inorganic phosphate, the Pho80-Pho85 cyclin–cyclin-dependent kinase (CDK) is inactivated by the Pho81 CDK inhibitor (CKI). The regulation of Pho80-Pho85 is distinct from previously characterized mechanisms of CDK regulation: the Pho81 CKI is constitutively associated with Pho80-Pho85, and a small-molecule ligand, inositol heptakisphosphate (IP7), is required for kinase inactivation. We investigated the molecular basis of the IP7- and Pho81-dependent Pho80-Pho85 inactivation using electrophoretic mobility shift assays, enzyme kinetics and fluorescence spectroscopy. We found that IP7 interacts noncovalently with Pho80-Pho85-Pho81 and induces additional interactions between Pho81 and Pho80-Pho85 that prevent substrates from accessing the kinase active site. Using synthetic peptides corresponding to Pho81, we define regions of Pho81 responsible for constitutive Pho80-Pho85 binding and IP7-regulated interaction and inhibition. These findings expand our understanding of the mechanisms of cyclin-CDK regulation and of the biochemical mechanisms of IP7 action.
In conditions of nutrient limitation, cells maintain homeostasis by altering their metabolomic, proteomic and genomic profiles1,2. Many nutrient-starvation response systems have been intensively studied in S. cerevisiae2, and the cellular response to inorganic phosphate (Pi) starvation (the PHO system) is one of the most well characterized3.
When yeast cells are starved of Pi, the Pho80-Pho85-Pho81 cyclin-CDK-CKI system is inactivated4, which leads to the dephosphorylation5 and nuclear localization of the transcription factor Pho4 (refs. 6,7) and subsequent transcription of PHO genes8. Previous studies of Pho80-Pho85-Pho81 regulation have indicated that Pho81, the CKI, is constitutively associated with the cyclin-CDK (ref. 4), but CDK activity is only inhibited in Pi starvation conditions4. For other CKIs, CDK-CKI protein-protein interactions are sufficient to inactivate the CDK (ref. 9). Because Pho81 is constitutively associated with Pho80-Pho85, we hypothesized that there might be an additional factor or modification that is required for Pi starvation–induced Pho80-Pho85 inactivation.
To identify the additional factor required for the regulation of cyclin-CDK, we biochemically isolated a Pho81-dependent Pho80-Pho85 inhibitor from Pi-starved cell extract and identified it as an isomer of IP7—either 1, 2 or 3 (Fig. 1a)10, which are all myo-D-inositol (4; Fig. 1a) derivatives bearing one pyrophosphate and five phosphates. IP7 is found in all eukaryotic cells11 and is produced through the action of a conserved set of inositol multiphosphate kinases from the precursor myo-D-inositol 1,4,5-trisphosphate (5; Fig. 1a)11,12, which can be sequentially phosphorylated to generate inositol poly-phosphates such as myo-inositol 1,2,3,4,5,6-hexakisphosphate (IP6, 6)11. Depending on the location of these pyrophosphates, several different IP7 isomers are possible11,12; at least two distinct isomers of IP7—5-IP7 (1) and 4-IP7 or 6-IP7 (4/6-IP7; 2 or 3, respectively)—are found in natural systems (Fig. 1a)12–14.
Figure 1.

Reversible regulation of Pho80-Pho85-Pho81-MD by IP7. (a) Structures of myo-D-inositol derivatives used in this study (P and PP denote phosphate and pyrophosphate, respectively). The absolute configuration of 4/6-IP7 is not completely defined (either 4-IP7 or 6-IP7, which are mirror images of each other). (b) SDS-PAGE of Pho80-Pho85-Pho81 (8 μM each) preincubated with 40 μM 4/6-[β-32P]IP7 (5 μCi μmol−1). Coomassie staining of the gel (left); autoradiogram of the gel (right). (c,d) Time-dependent (c) and Pho80-Pho85 concentration-dependent (d) IC50 changes. For c, following 15 (●), 30 (○), 60 (▲) and 120 (△) min of incubation with 4/6-IP7, the kinase activity of Pho80-Pho85-Pho81-MD (1 nM Pho80-Ph85, 450 nM Pho81-MD) was measured by the addition of buffer containing ATP and Pho4 (1 mM and 1 μM, respectively). Average and s.d. (n = 3) are shown. For d, the IC50 value for the inhibition by 4/6-IP7 (30 min incubation) was measured at different Pho80-Pho85 concentrations (40 (●), 2 (○) and 0.4 nM (▲); data represent mean values ± s.d.; n = 3).
Studies of loss-of-function mutants have revealed that IP7 is involved in diverse cellular processes including recombination15, vacuole function16, gene expression, endo- and exocytosis17,18, osmotic stress16, telomere length control19,20, nutrient sensing and kinase regulation10,14. However, for many of these processes the identities of relevant IP7 targets are unknown. In one of the best studied examples, 5-IP7 regulates Dictyostelium discoideum chemotaxis by competing with phosphatidylinositol phosphates (PIPs) for binding to pleckstrin homology domains21. Additionally, 5-IP7 and 4/6-IP7 can nonenzy-matically pyrophosphorylate certain yeast and mammalian proteins in vitro22,23.
In S. cerevisiae, there are two enzymes that are known to synthesize IP7 from IP6 and ATP (Fig. 1a). The first of these kinases (which are generally known as IP6Ks) identified in yeast, based on its homology to mammalian IP6Ks, was Kcs1 (ref. 24). Kcs1 is thought to produce 5-IP7 (Fig. 1a)13. More recently, another IP6K in yeast was described14—Vip1, which synthesizes a distinct isomer of IP7 tentatively assigned to have a pyrophosphate moiety at its 4 or 6 position (Fig. 1a) and whose absolute configuration is still undefined14. We demonstrated that 4/6-IP7 directly inactivates recombinant Pho80-Pho85 in vitro in a manner dependent on the Pho81 minimum domain25 (a fragment consisting of amino acids 644–723 of Pho81 that is sufficient for the Pi starvation–dependent Pho80-Pho85 regulation in vivo)10, which demonstrates that the inactivation of Pho80-Pho85-Pho81 by 4/6-IP7 is direct.
This result establishes Pho80-Pho85-Pho81 as a cyclin-CDK-CKI system regulated directly by 4/6-IP7. Because there are no known examples of CDK regulation by a natural small-molecule ligand, we sought to further characterize the molecular basis of this inactivation mechanism. This inactivation of cyclin-CDK-CKI by 4/6-IP7 is also noteworthy because it is a rare example in which the molecular targets of IP7 are known and can therefore be studied mechanistically.
In this paper, we demonstrate that Pho80-Pho85-Pho81 is reversibly regulated by 4/6-IP7. This reversible regulation of the cyclin-CDK-CKI is distinct from any of the previously known IP7 functions—competition with PIPs (ref. 21) and covalent modification of proteins22,23. Based on further biochemical analysis, we propose a molecular mechanism for Pho80-Pho85 cyclin-CDK inactivation by the concerted action of 4/6-IP7 and the CKI Pho81.
RESULTS
Reversible regulation of Pho80-Pho85-Pho81 by IP7
Because there is precedent that 5-IP7 and 4/6-IP7 can pyrophosphorylate certain yeast and mammalian proteins in vitro23, we first investigated whether Pho80-Pho85-Pho81 is covalently regulated by 4/6-IP7 (Fig. 1a). We incubated recombinant Pho80-Pho85 bound to recombinant Pho81 minimum domain25 (Pho81-MD) with 4/6-[β-32P]IP7 and analyzed the resulting samples by SDS-PAGE (Fig. 1b). We found no evidence supporting protein phosphorylation. We next determined whether prolonged incubation alters the half-maximal inhibitory concentration (IC50) of 4/6-IP7–mediated Pho80-Pho85-Pho81-MD inactivation (Fig. 1c). In typical irreversible protein inhibition, prolonged incubation with inhibitor decreases the apparent IC50 (ref. 26). However, we observed little change in the IC50 value with different incubation times (IC50 calculated to be 23 ± 6, 14 ± 8, 31 ± 14 and 22 ± 9 μM for 10-, 30-, 60- and 120-min incubation, respectively), which implies that the inhibition is at equilibrium and reversible. Finally, in typical irreversible inhibition, the IC50 decreases with increasing enzyme concentration because the bimolecular reaction rate is faster at higher enzyme concentrations26. Changing the Pho80-Pho85 concentration over two orders of magnitude did not affect the IC50 value (Fig. 1d), which further supports the hypothesis that 4/6-IP7 regulates Pho80-Pho85-Pho81 by a reversible mechanism.
Characterization of IP7 binding to Pho80-Pho85-Pho81
To gain more information on the mechanism of inhibition, we characterized the binding of 4/6-IP7 to the kinase complex. We used an electrophoretic mobility shift assay (EMSA) to test which components of the Pho80-Pho85-Pho81-MD complex are required for binding (Fig. 2a). Although Pho81-MD is required for inactivation of Pho80-Pho85 triggered by 4/6-IP7 (ref. 10), it is unclear whether Pho81-MD is required for binding of 4/6-IP7 to Pho80-Pho85, or only for the inactivation step. In the EMSA experiment, a protein-dependent electrophoretic mobility shift of the 4/6-[β-32P]-IP7 was observed only in the presence of Pho80-Pho85, Pho81-MD and divalent metal cations (Fig. 2a). Among several divalent metal cations tested, Mg2+ and Mn2+ both supported efficient binding. Zn2+ also supported binding but Ca2+ did not; this metal preference is distinct from that for the IP7-mediated protein pyrophosphorylation reaction22,23 (Fig. 2a). All subsequent binding experiments were carried out in the presence of MgCl2. When higher concentrations of Pho80-Pho85-Pho81-MD (μ10 μM) were used, protein–4/6-IP7 complexes did not enter the gel. Subsequent binding analysis was therefore carried out with 8 μM Pho80-Pho85-Pho81. In these conditions, 22 ± 2% (average ± s.d., n = 50) of the 4/6-IP7 was bound, as evidenced by a mobility shift. We also varied the amount of 4/6-IP7 at a constant Pho80-Pho85-Pho81-MD concentration (Supplementary Fig. 1 online). In this experiment, the apparent dissociation constant (Kdapp) was 20.3 μM (Hill coefficient 0.94). These observations suggest that 4/6-IP7 interacts with Pho80-Pho85-Pho81 in a 1:1 complex in a manner dependent on the presence of selected divalent cations and all three proteins.
Figure 2.

Characterization of IP7 binding to Pho80-Pho85-Pho81-MD. (a) Native gel EMSA. Pho80-Pho85 (8 μM), Pho81-MD (8 μM) and divalent metal cations (6 mM each; E, EDTA; Mg, MgCl2; Ca, CaCl2; Mn, MnCl2; Zn, ZnSO4) were mixed and incubated with 0.2 μM 4/6-[β-32P]IP7 (5 μCi mmol−1) for 30 min at 30 °C, followed by electrophoresis at 4 °C. All subsequent binding assays were performed in the presence of 6 mM MgCl2 unless noted otherwise. (b) Isomer selectivity. Pho80-Pho85-Pho81-MD (8 μM each) was incubated with 0.2 μM 5-[β-32P]IP7 or 4/6-[β-32P]IP7 (30 °C, 30 min) and analyzed as in a. (c) Specificity of the binding. Binding of 4/6-[β-32P]IP7 (0.2 μM) to Pho80-Pho85-Pho81-MD (8 μM each) was monitored as in a in the presence of 400 μM competitors. (d) Binding reversibility. After formation of the complex between Pho80-Pho85-Pho81-MD and 4/6-IP7 as in a, reversibility of the binding was tested by the addition of 20 mM EDTA (E), or by heating (B; 90 °C, 1 min). N, not treated. (e) Dissociation kinetics. After formation of the complex, samples were diluted in 400 μM unlabeled 4/6-IP7 at 4 or 30 °C, and the dissociation of the complex was monitored by EMSA as a function of the time following the dilution. (f) Association kinetics. After adding 4/6-[β-32P]IP7 to Pho80-Pho85-Pho81-MD and incubating at 4 or 30 °C, a portion of the sample was diluted into ice-cold (4 °C) unlabeled 4/6-IP7 (400 μM) and analyzed by EMSA. For d and e, data were fit to single exponential equations.
We explored the specificity of binding of different isomers of IP7 (Fig. 2b,c). Previously we reported that 4/6-IP7 regulates Pho80-Pho85 in a Pho81-dependent manner, but 5-IP7 (Fig. 1a) does not significantly affect kinase activity10. We investigated whether this discrimination of the IP7 isomers in the inhibition assay is due to a difference in the specificity of binding or an inability of 5-IP7 to induce conformational changes in Pho80-Pho85-Pho81-MD that are necessary for inhibition. We assayed the binding of 5-[β-32P]-IP7 to Pho80-Pho85-Pho81-MD using EMSA (Fig. 2b) and observed no detectable mobility shift, which indicates that Pho80-Pho85-Pho81-MD binds selectively to 4/6-IP7.
The concentration of IP6 in eukaryotic cells is estimated to be up to several hundred micromolar11, making it far more abundant than 5-IP7 or 4/6-IP7, whose concentration is a few micromolar or less at resting conditions27. Therefore, for 4/6-IP7 binding to Pho80-Pho85-Pho81-MD to be physiologically meaningful, Pho80-Pho85-Pho81-MD should be able to bind to 4/6-IP7 even in the presence of excess IP6. Indeed, when the binding of isotope-labeled 4/6-IP7 (0.2 μM) to Pho80-Pho85-Pho81-MD (8 μM each) was competed with inositol derivatives (400 μM), only the unlabeled 4/6-IP7 showed clear competition (Fig. 2c). This result indicates that the 4/6-IP7 is a specific ligand for Pho80-Pho85-Pho81-MD, and the presence of cellular levels of 5-IP7 or IP6 do not interfere with binding.
Next, we investigated whether the interaction between 4/6-IP7 and Pho80-Pho85-Pho81-MD observed in the EMSA results from reversible binding or from a covalent protein modification (Fig. 2d). To test whether binding requires the folded structure of Pho80, Pho85 and Pho81-MD, the Pho80-Pho85-Pho81-MD-4/6-IP7 quaternary complex was boiled at 90 °C for 1 min (Fig. 2d) and then analyzed by native gel electrophoresis. No detectable electrophoretic shift of 4/6-IP7 was observed after boiling, which suggests that the interaction of 4/6-IP7 with the complex is reversible. Further, when the pre-formed Pho80-Pho85-Pho81-MD-4/6-IP7 complex was treated with EDTA (Fig. 2d), 4/6-IP7 dissociated from the complex, thereby providing further support for the reversible nature of the binding.
Because we observed a reversible interaction of 4/6-IP7 with the kinase complex, we sought to determine how Pho80-Pho85-Pho81 can be purified in an inactive form from Pi-starved cells. Our previous studies4,10 demonstrated that Pho80-Pho85-Pho81 can be immunopurified from Pi-starved cells in a kinase inactive form, which indicates that the inactive form is stable enough to survive the immunopurification steps. However, the enzyme inhibition and 4/6-IP7 binding data (Figs. 1 and 2) indicate that the inhibition is reversible with an IC50 value in the micromolar range, which suggests that the half-life of 4/6-IP7 bound to the cyclin-CDK-CKI complex would not be long enough to remain bound during the immunopurification steps. One possibility is that 4/6-IP7 dissociation from Pho80-Pho85-Pho81-MD is slow enough at low temperature (conditions of the purification) for the complex to survive immunopurification. To test this possibility, the rate of dissociation of 4/6-IP7 was measured by diluting pre-formed 4/6-IP7–protein complex with excess unlabeled 4/6-IP7 equilibrated at 4 or 30 °C and analyzing complex formation by EMSA (Fig. 2e). The disassembly of the 4/6-IP7–protein complex, measured by the loss of the electrophoretic mobility shift, was indeed temperature dependent: at 30 °C, the dilution of the complex with unlabeled 4/6-IP7 caused rapid dissociation; at 4 °C, we observed little disassembly of the pre-formed 4/6-IP7–protein complex. We also measured the association rate by incubating 32P-labeled 4/6-IP7 and proteins at 30 °C followed by quenching the association with excess unlabeled 4/6-IP7 at 4 °C (Fig. 2f). The association rate was also slower at lower temperature. These results indicate that there is a temperature-sensitive step for the association and dissociation of 4/6-IP7, which may explain why we could immunopurify inactive Pho80-Pho85-Pho81 complex from Pi-starved cells.
Effects of IP7 on the kinase-substrate interaction
To obtain a better understanding of the roles that 4/6-IP7 and Pho81 have in Pho80-Pho85 regulation, we examined the effect of 4/6-IP7 on the kinetics of phosphorylation of Pho4 by Pho80-Pho85-Pho81 (Fig. 3). Addition of Pho81-MD up to ~ 1 μM in the absence of 4/6-IP7 did not affect the kinase reaction (Fig. 3a). When kinase assays were carried out in the presence of different amounts of ATP, Pho4 and/or 4/6-IP7, we found that the apparent Km for Pho4 linearly increases as the concentration of 4/6-IP7 increases (Fig. 3a,b; Ki ~ 19 ± 8 μM), which indicates that 4/6-IP7 is an inhibitor that is competitive with Pho4. However, 4/6-IP7 did not alter the Km for ATP (Fig. 3c,d), which indicates that 4/6-IP7 is not competitive with ATP.
Figure 3.
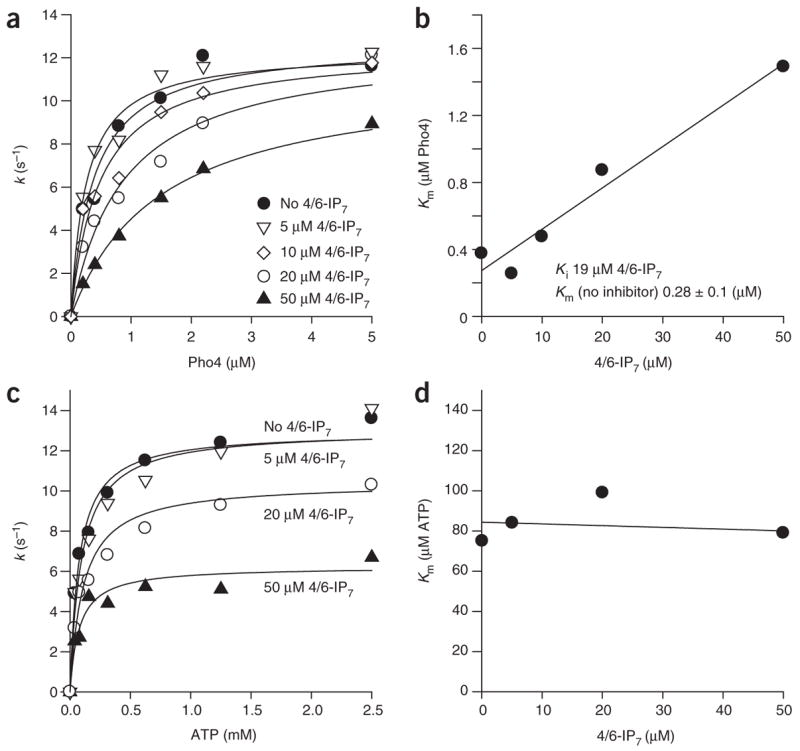
Enzyme kinetic analysis of IP7-mediated Pho80-Pho85-Pho81-MD inactivation. Effect of 4/6-IP7 on Pho80-Pho85-Pho81-MD–catalyzed Pho4 phosphorylation. Pho4 phosphorylation was measured at different Pho4 concentrations (a,b) or ATP concentrations (c,d) in the absence (a) or presence (c) of 4/6-IP7, and the rate constants were plotted against substrate concentrations. Plots of Km, obtained by fitting data shown in a and b to a Michaelis-Menten equation (see Methods), for Pho4 (b) and ATP (d) against the concentration of 4/6-IP7. All reactions were carried out with 0.5 nM Pho80-Pho85 and 450 nM Pho81-MD as described in the Methods.
Although many models can account for these observations, the simplest explanation is that 4/6-IP7 binds to the peptide substrate binding region in the active site and acts as a classical competitive inhibitor. However, we disfavor this possibility because 4/6-IP7 does not bind to or inhibit Pho80-Pho85 in the absence of Pho81-MD (ref. 10). Alternatively, binding of 4/6-IP7 may induce a conformational change in Pho80-Pho85-Pho81 that subsequently prevents the enzyme from binding to Pho4.
The interaction of Pho4 with Pho80-Pho85-Pho81 is complex; there is a distal Pho4 interaction site on Pho80 away from the enzyme active site28,29. The presence of the distal interaction is supported by the observations that Pho80-Pho85 poorly phos-phorylates peptides derived from Pho4 and that Pho80-Pho85 phosphorylates Pho4 semiprocessively, transferring ~ 2 phosphates in each productive kinase-substrate interaction28. Based on yeast two-hybrid analysis30 and glutathione S-transferase pull-down experiments29, Pho4 sequences involved in the distal binding with Pho80-Pho85 were mapped to two regions distant from the five phosphorylation sites29. Pho81-MD and 4/6-IP7 could interfere with either the active site or the distal site interaction between Pho4 and Pho80-Pho85, which would yield similar enzyme kinetic data.
To determine whether the 4/6-IP7– and Pho81-MD–mediated inhibition involves blocking the active site or interfering with the distal Pho4 interaction, we tested the effect of 4/6-IP7 on the phosphorylation of Pho4 mutants whose distal binding sites are mutated29 (Supplementary Fig. 2 online). In comparison with wild-type Pho4, these mutants have five- to ten-fold higher Kms but similar kcats (ref. 29). We observed that phosphorylation of these Pho4 distal site mutants by Pho80-Pho85-Pho81-MD is inhibited by 4/6-IP7 with an IC50 comparable to that observed for wild-type Pho4 (all between 20 and 50 μM; Supplementary Fig. 2a), which suggests that the interaction between Pho4 and its distal binding site is not disrupted in the 4/6-IP7–mediated inactivation of Pho80-Pho85-Pho81-MD. We also tested the effect of 4/6-IP7 on the phosphorylation of peptides corresponding to Pho4 phosphorylation site 6 (ref. 28) (Supplementary Fig. 2b). These peptides cannot interact with the distal binding site on the kinase and are phosphorylated less efficiently. Phosphorylation of these peptides was also inhibited by the presence of 4/6-IP7, with a comparable IC50 (25 μM), which again suggests that the interaction between Pho4 and the distal binding site on Pho80-Pho85-Pho81 is not involved in enzyme inactivation. Based on these results, we conclude that the 4/6-IP7– and Pho81-MD–dependent inhibition of Pho80-Pho85 is likely to be mediated by a change in the interaction of the substrate with the kinase active site.
Secondary interaction between Pho81-MD and Pho80-Pho85
Next, using a fluorescence binding assay10 in which we labeled Pho81-MD with lissamine, we tested whether the presence of 4/6-IP7 alters the interaction between Pho80-Pho85 and Pho81-MD (Fig. 4a). In the presence of 4/6-IP7, the binding of Pho81-MD to Pho80-Pho85 is enhanced by ~ five-fold (Kd ~ 120 and 25 nM in the absence and the presence of 100 μM IP7, respectively). In contrast, 5-IP7 had no significant effect on this interaction (Kd = 182 ± 13, 295 ± 50 and 250 ± 29 nM in the presence of 0, 50 and 100 μM 4/6-IP7, respectively; Supplementary Fig. 3 online). These observations suggest that 4/6-IP7 may interact with both Pho81 and Pho80-Pho85 or may induce structural changes that stabilize interactions between Pho81 and Pho80-Pho85.
Figure 4.

Additional interaction between Pho81-MD and Pho80-Pho85. (a) Binding of Lissamine-labeled Pho81-MD (5 nM) to Pho80-Pho85 in the presence of the indicated concentrations of 4/6-IP7. Protein-binding-dependent lissamine fluorescence quenching was monitored (λex 532 λem 580 nm; data represent mean values ± s.d.; n = 3). (b) Inhibition of Pho80-Pho85 (0.5 nM) by excess Pho81-MD at different concentrations of 4/6-IP7. (c) Additional Pho80-Pho85-Pho81-MD-4/6-IP7 complex formation in the presence of excess Pho81-MD. EMSA experiments were carried out with 8 μM Pho80-Pho85, 0.2 μM 4/6-[β-32P]IP7 and different concentrations of Pho81-MD.
When Pho80-Pho85 kinase activity was measured in different concentrations of 4/6-IP7 and Pho81-MD, we observed that a high concentration of Pho81-MD (μ10 μM) inactivates Pho80-Pho85 in a 4/6-IP7–independent manner (Fig. 4b); this observation is consistent with previous results showing that overexpression of Pho81 inhibits Pho80-Pho85 even in high phosphate conditions (conditions in which 4/6-IP7 levels are low)4,25. Because Pho81-MD binds to Pho80-Pho85 with an apparent Kd of 120 nM (ref. 10) (Fig. 4a), the inactivation of Pho80-Pho85 by micromolar Pho81-MD may indicate that additional molecules of Pho81-MD can bind to Pho80-Pho85 at a high concentration of Pho81-MD, thereby leading to the formation of a higher-order complex. In support of this view, the addition of excess Pho81-MD in the EMSA binding (Fig. 4c) or gel filtration (Supplementary Fig. 4 online) assays resulted in the formation of additional complexes, and also changed the structure of Pho85 (Supplementary Fig. 5 online).
Constitutive binding and IP7-regulated segments of Pho81
What is the relationship between the additional Pho81-MD binding at high concentration and kinase inactivation in the stoichiometric Pho80-Pho85-Pho81 complex? One possibility is that the additional Pho81-MD binding results from additional contacts between Pho80-Pho85 and Pho81-MD that are not observed in the equimolar Pho80-Pho85-Pho81-MD complex in the absence of 4/6-IP7. It is possible that, upon the addition of 4/6-IP7, an additional site on Pho80-Pho85 may be occupied by Pho81-MD, thereby causing kinase inhibition by preventing Pho4 from accessing the active site.
To test this hypothesis, we prepared three nonoverlapping shorter segments derived from Pho81-MD (Fig. 5a) and measured their binding to Pho80-Pho85 in the presence and absence of 4/6-IP7. To obtain quantitative information, we synthesized peptides with an N-terminal Cys-Gly-Gly linker, labeled them site specifically with Alexa Fluor 488 and monitored their interaction with Pho80-Pho85 using fluorescence polarization (Fig. 5b,c). In the absence of 4/6-IP7, only the middle segment (S3; corresponds to amino acids 665–701 of Pho81) demonstrated any evidence of binding (apparent Kd ~ 400 ± 160 nM; Fig. 5b). However, in the presence of 4/6-IP7, the S1 segment corresponding to the C-terminal 22 amino acids of Pho81-MD (amino acids 702–723 of Pho81) showed evidence of binding (apparent Kd ~ 550 ± 170, 30 ± 22 and 25 ± 11 nM for 5, 40 and 200 μM 4/6-IP7, respectively; Fig. 5c). This result suggests that the middle segment (S3) of Pho81-MD binds to Pho80-Pho85 in a manner independent of 4/6-IP7, whereas the S1 segment binds to Pho80-Pho85 only when 4/6-IP7 is present.
Figure 5.
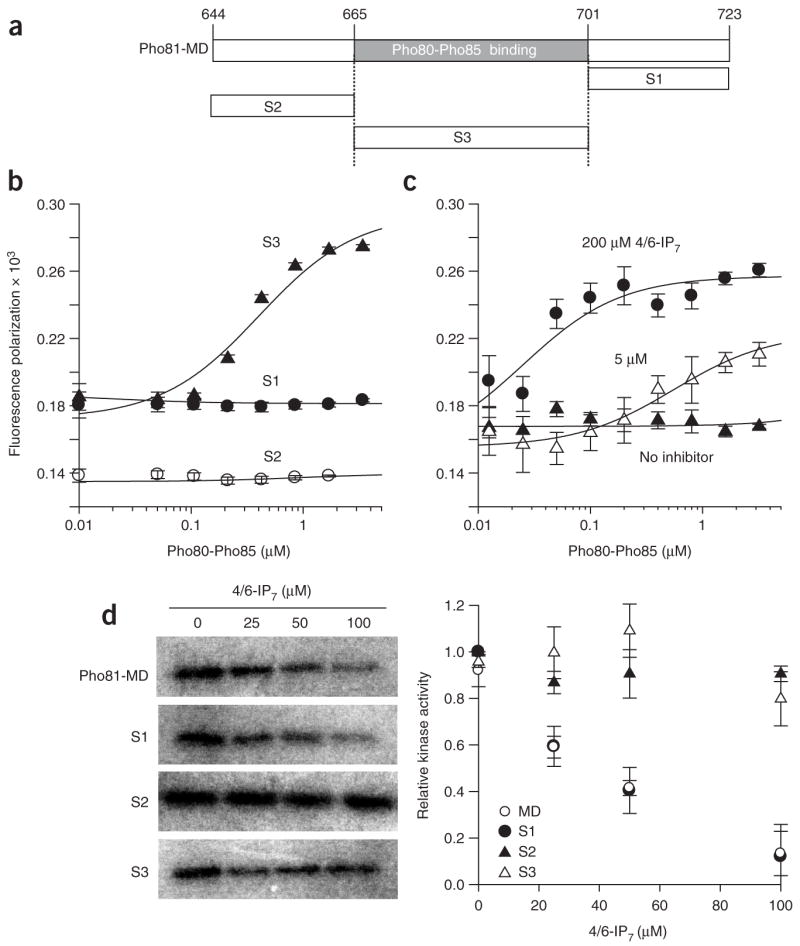
Dissection of Pho81-MD into binding and inhibitory segments. (a) Design of peptide fragments of Pho81-MD. (b,c) Fluorescence polarization binding of Alexa Fluor 488–labeled Pho81-MD (10 nM) segments to Pho80-Pho85 (data represent mean values ± s.d.; n = 3). (b) Binding of S1 (●), S2 (○) and S3 (▲) to Pho80-Pho85 in the absence of 4/6-IP7. (c) Binding of S1 to Pho80-Pho85 in the presence of 0 (▲), 5 (△) and 200 (●) μM 4/6-IP7. (d) Effect of Pho81-MD, S1, S2 and S3 on Pho80-Pho85 inhibition by 4/6-IP7. Experiments were carried out with 1 nM Pho80-Pho85, 400 nM Pho81-MD or shorter segments, 1 μM Pho4, 500 μM ATP, 0.1 μCi μl−1 [γ-32P]ATP for 5 min (data represent mean values ± s.d.; n = 3). Symbols note Pho80-Pho85 activity in the presence of IP7 and Pho81-MD (○), S1 (●), S2 (▲) and S3 (△).
We next assessed the functional relevance of the interaction of the smaller Pho81-MD segments with Pho80-Pho85. We preincubated Pho80-Pho85 with Pho81-MD or the smaller segments and measured 4/6-IP7–dependent inhibition of kinase activity (Fig. 5d). We found that the S3 segment, which is sufficient for binding in the absence of 4/6-IP7, does not show any evidence of Pho80-Pho85 inhibition. The S2 segment, which did not bind to Pho80-Pho85, also did not have any effect on enzyme activity. However, the S1 segment, which binds to Pho80-Pho85 only in the presence of 4/6-IP7 (Fig. 5c), inhibited Pho80-Pho85 in the presence of 4/6-IP7. This result indicates that Pho81-MD can be further divided into two segments: one that binds tightly to Pho80-Pho85 regardless of the concentration of 4/6-IP7, and one that acts as a 4/6-IP7–dependent inhibitor of Pho80-Pho85.
Model of Pho80-Pho85 inhibition by IP7 and Pho81
Based on these biochemical data, we propose a model for the mechanism of inhibition of Pho80-Pho85 (Fig. 6). In the absence of 4/6-IP7 (or in Pi-rich cells), Pho80-Pho85 is constitutively bound to Pho81, but this protein-protein interaction alone is not sufficient to inhibit Pho80-Pho85 kinase activity (Fig. 6a). This constitutive interaction with Pho80-Pho85 is mediated at least in part by the Pho81 S3 segment. Upon starvation of cells for Pi and binding of 4/6-IP7, additional contacts mediated by the Pho81 S1 segment occur, thereby preventing Pho4 from accessing the Pho80-Pho85 active site (Fig. 6a).
Figure 6.

Model of Pho80-Pho85 regulation by 4/6-IP7 and Pho81. (a) The S3 region of Pho81 binds constitutively to Pho80-Pho85 (left); addition of 4/6-IP7 triggers a structural change that results in the S1 segment of Pho81 occluding interaction of Pho4 with the kinase active site (right), without affecting ATP binding (white molecule). (b) The effect of binding additional molecules of Pho81-MD. When a high concentration Pho81-MD is present, it can bind and block substrate access to the active site. Our data suggest (Fig. 4c) that 4/6-IP7 can still bind to this higher order complex.
This model is supported by several observations. First, 4/6-IP7 binds to the Pho80-Pho85-Pho81-MD ternary complex but not to its individual components (Fig. 2). Second, the 4/6-IP7– and Pho81-mediated inhibition competes with Pho4 but not with ATP (Fig. 3). Third, binding of 4/6-IP7 stabilizes the interaction between Pho80-Pho85 and Pho81-MD (Fig. 4a). Finally, Pho81-MD can be divided into two segments: S3 is constitutively bound to Pho80-Pho85 but does not inhibit the kinase, whereas S1 binds and inhibits Pho80-Pho85 only in the presence of 4/6-IP7 (Fig. 5). It is possible that the S1 segment in full-length Pho81 or in Pho81-MD is in an auto-inhibited state (for example, bound to other parts of Pho81-MD) without 4/6-IP7.
Based on this model (Fig. 6), we expect that an effect on kinase activity similar to that seen on addition of 4/6-IP7 can be achieved by the addition of excess Pho81. In the presence of excess Pho81, there may be sufficient amounts of S1 segment to inhibit Pho80-Pho85. In support of this view, addition of excess Pho81-MD inhibits Pho80-Pho85 independent of 4/6-IP7 with a higher IC50 (~ 20 μM, Fig. 4b). It was also shown previously that addition of excess Pho81 to immunopurified Pho80-Pho85 can inhibit kinase activity in vitro4, and that overexpression of Pho81 can partially inhibit the kinase in vivo4,31,32. Our model may help to explain the inhibition of Pho80-Pho85 by overexpressed Pho81 (Fig. 6b). At high concentration, excess Pho81 will weakly bind to Pho80-Pho85-Pho81 and inhibit kinase activity. Consistent with our EMSA assay results (Fig. 4c), 4/6-IP7 can also bind to this higher order complex (Fig. 6b).
DISCUSSION
Since the elucidation of its chemical structure, it has been suggested that IP7 may act as a phosphate donor33. Indeed, both 5-IP7 and 4/6-IP7 were recently shown to pyrophosphorylate mammalian and yeast proteins in vitro22,23. However, because of a lack of suitable assay systems, it has been difficult to determine the physiological relevance of this phosphor transfer reaction. Our previous work demonstrated that 4/6-IP7 inhibits Pho80-Pho85-Pho81 in response to phosphate limitation, thus leading to induction of starvation-response genes10. Because Pho80-Pho85-Pho81 can be immunopurified in a stably regulated form from yeast cells grown in different phosphate concentrations4, we speculated that Pho80-Pho85-Pho81 may be covalently modified by 4/6-IP7. In this report, we provide evidence that 4/6-IP7 reversibly regulates Pho80-Pho85-Pho81. Therefore, we conclude that Pho80-Pho85-Pho81 is unlikely to be a target of an IP7 phosphor transfer reaction; instead, 4/6-IP7 acts as an allosteric regulator of this kinase complex.
Previous structural studies of the CDK inhibitor p27Kip1 bound to cyclin A–CDK2 revealed that the mechanism of inhibition of active cyclin A–CDK2 involves insertion of a 310 helix of p27Kip1 into the ATP-binding pocket of CDK2 (refs. 9,34). In this mode of inhibition, the Km for ATP is expected to increase, possibly without a change in Km for the protein substrate. In contrast, our results indicate that for Pho80-Pho85-Pho81, the Km for the protein substrate (but not the Km for ATP) is affected by 4/6-IP7. These results demonstrate that there are mechanistic differences between the inhibition of Pho80-Pho85 by 4/6-IP7 and Pho81 and cyclin-CDK2 inhibition by Cip/Kip-family CKI proteins.
The allosteric regulation of a cyclin-CDK-CKI by a small-molecule metabolite is a strategy suitable for cells to respond to fluctuating, transient conditions such as nutrient starvation. By using constitutively bound CKI, whose action is regulated by a metabolite, Pho80-Pho85 cyclin-CDK activity can be rapidly modulated in different nutrient or environmental conditions. When the stimulus is reversed in a short period of time, cyclin-CDK activity can be restored. We have shown that 4/6-IP7 dissociates from Pho80-Pho85-Pho81 within minutes at 30 °C in vitro. When the stimulus is persistent, cells may activate an additional inhibition mechanism and make the inactive form of Pho80-Pho85-Pho81 more kinetically inert. We speculate that the interaction of Pho80-Pho85-Pho81 with an additional molecule of Pho81 may be a mechanism for cells to make the inactive form of Pho80-Pho85-Pho81 persistent. Consistent with this model, expression of Pho81 is increased in a Pho4-dependent manner upon Pi starvation31,32.
In addition to its role in the PHO system, Pho85 also regulates other biological processes35. Depending on the identity of its cyclin partners, Pho85 can influence the cell cycle, morphogenesis, carbon source utilization and glycogen metabolism35. In many of these processes Pho85 is believed to be regulated by nutrient or environmental stimuli35. Other Pho85-cyclin complexes may be regulated by metabolite changes in a manner similar to the mechanism described in this paper.
METHODS
Materials
Recombinant proteins used in this study were prepared as described10,28,29. We used both chemically synthesized Pho81-MD (amino acids 644–723 of Pho81; NH2-LLQRPLNLPSAPLNEINSQSSTQRLNTIDLTPNDDK FDLDIQDSI PDFALPPPIIPLRKYGHNFLEKKIFIKLKLRPGLE-CONH2; Biosynthesis, Inc.) and recombinant His6-Pho81-MD (ref. 25), which had similar biochemical properties. Peptide fragments of Pho81-MD were synthesized by Genscript (S1 (amino acids 702–723 of Pho81): NH2-CGGKYGHNFLEKKIFIKLKLRPGLE-CO2H; S2 (amino acids 644–664 of Pho81): NH2-CGGLLQRPLNLPSAPLNEINSQSS-CO2H; S3 (amino acids 665–701 of Pho81): NH2-CGGTQRLNTIDLTPNDDKFDLDIQDSIPDFALPPPIIPLR-CO2H; residues added for fluorophore labeling are underlined). Enzymatic preparations of 5-IP7 and 4/6-IP7 were prepared and characterized as previously reported10,14. See Supplementary Methods online for additional description of materials and methods.
Kinase assays
Pho80-Pho85 kinase assays were performed essentially as previously described10. For typical experiments, Pho80-Pho85 (final concentration 0.1–10 nM after mixing with the kinase assay solution) and Pho81-MD (typically 450 nM final concentration) were mixed with 4/6-IP7 for the noted time period and then diluted in kinase assay solution (40 mM HEPES, pH 7.4, 10 mM MgCl2, 100 mM NaCl, 0.01% NP-40, 1 mM PMSF, 2 mM benzamidine, 10 mM NaF, 10 mM β-glycerophosphate, 0.1 mg ml−1 bovine serum albumin) containing ATP (typically 1 mM with 0.1–0.2 μCi μl−1 [γ-32P]ATP and Pho4 (typically 1–2 μM). After incubation at 30 °C (1–10 min depending on the Pho80-Pho85 concentration), the reaction was quenched by the addition of SDS gel loading buffer and samples were analyzed by SDS-PAGE. Dried gels were exposed to storage phosphor screens and analyzed using a Typhoon imaging system (Molecular Dynamics) equipped with ImageQuant software (GE Healthcare). Enzyme kinetic analysis was done by fitting the data using SigmaPlot software (Systat Software, Inc.) to the following Michaelis-Menten equation:
where [Pho80-Pho85]o, Vo, kcat, Km and [Pho4] indicate initial Pho80-Pho85 concentration, phosphorylation rate, apparent catalytic constant, apparent Michaelis-Menten constant and Pho4 concentration, respectively. Michaelis-Menten constants obtained from these fittings were plotted against the concentration of 4/6-IP7 (Fig. 3b,d) and fitted into the following linear equation36:
where Km,app, Ki and [I] indicate apparent Km in the presence of 4/6-IP7, the dissociation constant for the protein–4/6-IP7 interaction and the concentration of 4/6-IP7. For the analysis of peptide phosphorylation, Pho4 site 6 peptide (400 μM; NH2-SAEGVVVASESPVIAPHGSTHARSY-CO2H; phosphorylation site underlined) was used instead of Pho4 protein. The reaction was carried out for 5 min with Pho80-Pho85 (100 nM) pre-equilibrated with Pho81-MD (450 nM) and 4/6-IP7. After quenching and Tris-Tricine SDS-PAGE, phosphorylation of peptides was analyzed as above.
IP7 binding assays
In typical binding experiments, Pho80-Pho85-Pho81-MD (8 μM each) was mixed with 4/6-[β-32P]IP7 (5 Ci mmole–1; 0.2 μM) in buffer A (20 mM HEPES, pH 7.4, 6 mM MgCl2, 5 mM NaF), and the mixture was incubated for 30 min at 30 °C. The solution was then placed on ice and mixed with glycerol (final concentration 10 % v/v) and a trace amount of bromophenol blue. Samples were loaded onto 4–20% gradient gels (Bio-Rad) in Tris-glycine, pH 8.3 running buffer. After 1 h of electrophoresis (120 V; initial current was approximately 30 mA per gel) at 4 °C, gels were dried and analyzed as above. For the test of covalent phosphotransfer, Pho80-Pho85-Pho81-MD-4/6-IP7 complex solution described above was mixed with SDS gel loading buffer and subjected to SDS-PAGE. The gel was stained with Colloidal Coomassie Blue (Invitrogen), photographed and then dried and exposed to a storage phosphor screen.
Fluorescence binding assays
Pho81-MD binding to Pho80-Pho85 was measured as previously described10. Lissamine-labeled Pho81-MD (ref. 10) (5 nM) was placed in a glass-bottom 96-well plate containing buffer A (100 μl per well), Pho80-Pho85 (0–1 μM) and/or 4/6-IP7. After 1 h incubation at room temperature (22 ± 2 °C), the Lissamine fluorescence was monitored (λex 532 nm; λem 580 nm). Binding of smaller Pho81-MD segments was performed using Alexa Fluor–labeled S1-S3 segments. S1–S3 segments (~ 0.5 mg) in 0.5 ml 20 mM HEPES, pH 7.4, 1 mM tris(2-carboxyethyl)phosphine were mixed with Alexa Fluor 488 C5 maleimide (0.5 mg in 100 μl dimethyl-sulfoxide). After 1 h incubation at room temperature, labeled peptides were purified through a HiTrap Desalt column (5 ml), yielding quantitative labeling. Labeled peptides were analyzed using MALDI-TOF MS (2,5-dihydroxybenzoic acid matrix; 50% acetonitrile and 0.1% trifluoroacetic acid); m/z: MW calcd. for S1, 2889.5; found, 2889.9; MW calcd. for S2, 2549.9; found, 2551.1; MW calcd. for S3, 4474.1; found, 4474.5; MW calcd. for Alexa 488 S1, 3610.2; found, 3611.0; MW calcd. for Alexa 488 S2, 3270.6; found, 3272.8; MW calcd. for Alexa 488 S3, 5195.7; found, 5195.9. For the polarization experiments, samples (100 μl per well) containing 10 nM Alexa Fluor 488–labeled S1–S3 peptides in buffer A supplemented with 125 mM NaCl, Pho80-Pho85 and/or 4/6-IP7 (250 μM) were incubated at room temperature for 30 min, and fluorescence polarization measurements were performed (1 s averaging with fluorescein excitation and emission filters) with a Wallac 1420 Victor3V fluorescence microplate reader.
Supplementary Material
Note: Supplementary information and chemical compound information is available on the Nature Chemical Biology website.
Acknowledgments
We thank J. York (Duke University Medical School) for plasmids encoding Vip1 and hIP6K, I. Carter-O’Connell for protein preparation, O’Shea lab members for helpful discussions and comments on the manuscript, and D. Kahne (Harvard University) for access to equipment. This work was supported by the Helen Hay Whitney Foundation (Y.S.L.), Welch Foundation grant Q0581 (F.A.Q), US National Institutes of Health grant R01 GM051377, the David and Lucile Packard Foundation and the Howard Hughes Medical Institute (E.K.O.).
Footnotes
AUTHOR CONTRIBUTIONS
Y.-S.L. and E.K.O. designed experiments, Y.-S.L. carried out experiments, Y.-S.L. and E.K.O. interpreted data and wrote the manuscript. K.H. and F.A.Q. designed Pho81 peptides and carried out preliminary experiments to study Pho80-Pho85 binding.
Published online at http://www.nature.com/naturechemicalbiology, Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Lindsley JE, Rutter J. Nutrient sensing and metabolic decisions. Comp Biochem Physiol B Biochem Mol Biol. 2004;139:543–559. doi: 10.1016/j.cbpc.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Wilson WA, Roach PJ. Nutrient-regulated protein kinases in budding yeast. Cell. 2002;111:155–158. doi: 10.1016/s0092-8674(02)01043-7. [DOI] [PubMed] [Google Scholar]
- 3.Lenburg ME, O’Shea EK. Signaling phosphate starvation. Trends Biochem Sci. 1996;21:383–387. [PubMed] [Google Scholar]
- 4.Schneider KR, Smith RL, O’Shea EK. Phosphate-regulated inactivation of the kinase PHO80–PHO85 by the CDK inhibitor PHO81. Science. 1994;266:122–126. doi: 10.1126/science.7939631. [DOI] [PubMed] [Google Scholar]
- 5.Kaffman A, Herskowitz I, Tjian R, O’Shea EK. Phosphorylation of the transcription factor PHO4 by a cyclin-CDK complex, PHO80–PHO85. Science. 1994;263:1153–1156. doi: 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- 6.O’Neill EM, Kaffman A, Jolly ER, O’Shea EK. Regulation of PHO4 nuclear localization by the PHO80–PHO85 cyclin-CDK complex. Science. 1996;271:209–212. doi: 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- 7.Komeili A, O’Shea EK. Nuclear transport and transcription. Curr Opin Cell Biol. 2000;12:355–360. doi: 10.1016/s0955-0674(00)00100-9. [DOI] [PubMed] [Google Scholar]
- 8.Springer M, Wykoff DD, Miller N, O’Shea EK. Partially phosphorylated Pho4 activates transcription of a subset of phosphate-responsive genes. PLoS Biol. 2003;1:E28. doi: 10.1371/journal.pbio.0000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287:821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 10.Lee YS, Mulugu S, York JD, O’Shea EK. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. 2007;316:109–112. doi: 10.1126/science.1139080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- 12.Bhandari R, Chakraborty A, Snyder SH. Inositol pyrophosphate pyrotechnics. Cell Metab. 2007;5:321–323. doi: 10.1016/j.cmet.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Falck J, et al. Synthesis and structure of cellular mediators: inositol polyphosphate diphosphates. J Am Chem Soc. 1995;117:12172–12175. [Google Scholar]
- 14.Mulugu S, et al. A conserved family of enzymes that phosphorylate inositol hexa-kisphosphate. Science. 2007;316:106–109. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- 15.Luo HR, et al. Inositol pyrophosphates are required for DNA hyperrecombination in protein kinase c1 mutant yeast. Biochemistry. 2002;41:2509–2515. doi: 10.1021/bi0118153. [DOI] [PubMed] [Google Scholar]
- 16.Dubois E, et al. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J Biol Chem. 2002;277:23755–23763. doi: 10.1074/jbc.M202206200. [DOI] [PubMed] [Google Scholar]
- 17.Ye W, Ali N, Bembenek ME, Shears SB, Lafer EM. Inhibition of clathrin assembly by high affinity binding of specific inositol polyphosphates to the synapse-specific clathrin assembly protein AP-3. J Biol Chem. 1995;270:1564–1568. [PubMed] [Google Scholar]
- 18.Fleischer B, et al. Golgi coatomer binds, and forms K(+)-selective channels gated by, inositol polyphosphates. J Biol Chem. 1994;269:17826–17832. [PubMed] [Google Scholar]
- 19.Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH. Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase-related protein kinases. Proc Natl Acad Sci USA. 2005;102:1911–1914. doi: 10.1073/pnas.0409322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.York SJ, Armbruster BN, Greenwell P, Petes TD, York JD. Inositol diphosphate signaling regulates telomere length. J Biol Chem. 2005;280:4264–4269. doi: 10.1074/jbc.M412070200. [DOI] [PubMed] [Google Scholar]
- 21.Luo HR, et al. Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell. 2003;114:559–572. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 22.Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Phosphorylation of proteins by inositol pyrophosphates. Science. 2004;306:2101–2105. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- 23.Bhandari R, et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci USA. 2007;104:15305–15310. doi: 10.1073/pnas.0707338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol. 1999;9:1323–1326. doi: 10.1016/s0960-9822(00)80055-x. [DOI] [PubMed] [Google Scholar]
- 25.Huang S, Jeffery DA, Anthony MD, O’Shea EK. Functional analysis of the cyclin-dependent kinase inhibitor Pho81 identifies a novel inhibitory domain. Mol Cell Biol. 2001;21:6695–6705. doi: 10.1128/MCB.21.19.6695-6705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marangoni AG. Enzyme Kinetics: a Modern Approach. John Wiley & Sons, Hoboken; New Jersey, USA: 2002. pp. 70–78. [Google Scholar]
- 27.Albert C, et al. Biological variability in the structures of diphosphoinositol polyphosphates in Dictyostelium discoideum and mammalian cells. Biochem J. 1997;327:553–560. doi: 10.1042/bj3270553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeffery DA, Springer M, King DS, O’Shea EK. Multi-site phosphorylation of Pho4 by the cyclin-CDK Pho80-Pho85 is semi-processive with site preference. J Mol Biol. 2001;306:997–1010. doi: 10.1006/jmbi.2000.4417. [DOI] [PubMed] [Google Scholar]
- 29.Byrne M, Miller N, Springer M, O’Shea EK. A distal, high-affinity binding site on the cyclin-CDK substrate Pho4 is important for its phosphorylation and regulation. J Mol Biol. 2004;335:57–70. doi: 10.1016/j.jmb.2003.10.035. [DOI] [PubMed] [Google Scholar]
- 30.Jayaraman PS, Hirst K, Goding CR. The activation domain of a basic helix-loop-helix protein is masked by repressor interaction with domains distinct from that required for transcription regulation. EMBO J. 1994;13:2192–2199. doi: 10.1002/j.1460-2075.1994.tb06496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogawa N, et al. Promoter analysis of the PHO81 gene encoding a 134 kDa protein bearing ankyrin repeats in the phosphatase regulon of Saccharomyces cerevisiae. Mol Gen Genet. 1993;238:444–454. doi: 10.1007/BF00292004. [DOI] [PubMed] [Google Scholar]
- 32.Ogawa N, et al. Functional domains of Pho81p, an inhibitor of Pho85p protein kinase, in the transduction pathway of Pi signals in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:997–1004. doi: 10.1128/mcb.15.2.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voglmaier SM, et al. Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc Natl Acad Sci USA. 1996;93:4305–4310. doi: 10.1073/pnas.93.9.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 35.Carroll AS, O’Shea EK. Pho85 and signaling environmental conditions. Trends Biochem Sci. 2002;27:87–93. doi: 10.1016/s0968-0004(01)02040-0. [DOI] [PubMed] [Google Scholar]
- 36.Fersht A. Enzyme Structure and Mechanism. 2. Freeman; New York: 1984. pp. 107–109. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information and chemical compound information is available on the Nature Chemical Biology website.