

Abstract
Introduction
Atrial fibrillation (AF) is a heritable disorder with male predilection, suggesting a sex chromosome defect in certain patients. Loss-of-function truncation mutations in EMD, encoding the nuclear membrane protein emerin, cause X-linked Emery-Dreifuss muscular dystrophy (EDMD) characterized by localized contractures and skeletal myopathy in adolescence, sinus node dysfunction (SND) in early adulthood, and atrial fibrillation as a variably associated trait. This study sought to identify the genetic basis for male-restricted, nonsyndromic sinus node dysfunction and AF in a multigenerational family.
Methods and Results
Genealogical and medical records, and DNA samples, were obtained. Progressive SND and AF occurred in four males related through maternal lineages, consistent with X-linked inheritance. Skeletal myopathy was absent, even at advanced ages. Targeted X chromosome genotyping mapped the disease locus to Xq28, implicating EMD as a positional candidate gene. DNA sequencing revealed hemizygosity for an in-frame 3-bp deletion in EMD (Lys37del) in affected males, disrupting a residue within the LEM binding domain critical for nuclear assembly but leaving the remainder of the protein intact. Buccal epithelial cell staining with emerin antibody demonstrated near-total functional loss of emerin. Female relatives underwent prospective electrocardiographic and genetic testing. Those heterozygous for Lys37del had ~50–70% emerin-positive nuclei and variable degrees of paroxysmal supraventricular arrhythmia.
Conclusions
Mutation of EMD can underlie X-linked familial AF. Lys37del is associated with epithelial cell emerin deficiency, as in EDMD, yet it causes electrical atriomyopathy in the absence of skeletal muscle disease. Targeted genetic testing of EMD should be considered in patients with SND-associated AF and/or family history suggesting X-linked inheritance.
Keywords: atrial fibrillation, sinus node dysfunction, X-linked, emerin, LEM domain
Introduction
Recognition of heritable risk for both lone and acquired AF1-4 has coincided with discovery of disease-causing mutations5-7 and risk-conferring functional polymorphisms.8-10 Moreover, genes for cardiomyopathic11,12 and myopathic13 syndromes in which AF occurs as a variably expressed trait have been identified. Collectively, these studies highlight genetic heterogeneity in AF susceptibility and a pathogenic role of ion channel, gap junction, and nuclear membrane defects in atrial arrhythmogenesis. Emerging insights into the molecular and cellular basis of AF, however, have not explained its striking male predilection. In each of three large, population-based, follow-up studies of lone AF,14-16 a three- to four-fold greater number of males than females were reported. Even in patients with acquired (non-lone) AF, males within each of several risk factor subgroups consistently had greater risk of AF.4 These observations raise the possibility that mutations or functional polymorphisms within sex chromosome-linked gene(s) may partially account for this gender difference. The majority of reported pedigrees with familial AF has an autosomal dominant mode of inheritance.1-3 X-linked inheritance of AF may be underrecognized, however, due to mild or absent disease in female relatives resulting in generation skipping. Here we describe a cardioselective mutation in EMD, encoding the nuclear membrane protein emerin, in a family with nonsyndromic X-linked AF.
Methods
Study Subjects
Family members provided written, informed consent under a research protocol approved by the Mayo Clinic Institutional Review Board. Extensive genealogical records provided by a family member were utilized for pedigree construction. Medical records and study-specific questionnaires were reviewed, with particular focus on cardiac and neurologic evaluations and symptoms. Prospective electrocardiographic screening, if not done for clinical indications, was performed in females at risk as mutation carriers. Phenotypic classification of familial AF required documentation on an electrocardiographic tracing, normal echocardiogram, and absence of clinical risk factors for arrhythmia. Unrelated spouses and individuals with a normal electrocardiogram who lacked symptoms of palpitations, racing heart rate, dizziness, or syncope were phenotypically classified as normal. Family members for whom clinical data were incomplete or unavailable were classified as uncertain. Genomic DNA was isolated from peripheral white blood cells using the Puregene® DNA Purification Kit (Gentra Systems, Minneapolis, MN, USA).
Locus Mapping and DNA Sequencing
A set of 18 polymorphic short tandem repeat (STR) DNA markers spanning the X chromosome were utilized for initial locus mapping (ABI PRISM Linkage Mapping Set-MD10, Applied Biosystems, Foster City, CA, USA). Markers were amplified from genomic DNA samples by the polymerase chain reaction (PCR) on DNA Engine Tetrad Peltier Thermal Cyclers (MJ Research, Watertown, MA, USA) using the fluorescently labeled primer pairs, reagents, and optimized experimental conditions provided by the manufacturer. PCR-amplified fragments were resolved on an ABI PRISM 3100 Genetic Analyzer and scored using GeneScan Analysis and Genotyper Software. Fine locus mapping utilized additional closely spaced STR markers localized on genetic and physical maps accessible at the National Center for Biotechnology Information website (www.ncbi.nlm.nih.gov). Genotyping was accomplished by PCR amplification of genomic DNA radio-labeled with alpha-(32P)-dCTP, resolution of alleles by polyacrylamide gel electrophoresis, and visualization by autoradiography, as previously described.17 Scored genotypes were assembled as haplotypes to define the critical region of complete linkage among affected individuals. For sequencing, primer pairs for PCR amplification of the six translated exons of EMD were designed using OLIGO v6.51 Primer Analysis Software (National Biosciences, Plymouth, MN, USA):
EMD1F: 5′-TGCTCGGCCGGTTTTGGTAG-3′;
EMD2R: 5′-CCTTTCTCCAGTGCCGCTCT-3′;
EMD3F: 5′-GAGAAAGGGGAGGGAAGTCT-3′;
EMD4R: 5′-GCCCAAGAGCCACCATTTGT-3′;
EMD5F: 5′-GTCCCCTCGCCCTGACTCTC-3′;
EMD6R: 5′-CCCCCACCCCCACTGCTAAG-3′.
Amplified products were treated with the PCR Product Pre-sequencing Kit (USB Corporation, Cleveland, OH, USA) and sequenced by the dye-terminator method in a core facility, using an ABI PRISM 3730 XL DNA Analyzer (Applied Biosystems). DNA sequences were viewed and analyzed using the Sequencher computer program (Gene Codes Corporation, Ann Arbor, MI, USA).
Immunocytochemistry
Epithelial cells were scraped from the buccal mucosa with a cheek swab brush (Cyto-Pak, Medical Packaging Corporation, Camarillo, CA, USA), collected in plastic tubes containing sterile buffer, and transported at room temperature. Cells were resuspended and fixed on glass microscope slides, as previously described.18 Slides were incubated with an anti-emerin monoclonal antibody, generated in mice by a synthetic peptide corresponding to a 222 amino acid region near the amino-terminus of emerin (Vector Laboratories, Burlingame, CA, USA). Secondary antibody staining was carried out with Alexa Fluor 488 goat anti-mouse IgG, labeled with a greenfluorescent dye (Invitrogen, Carlsbad, CA, USA). A second set of emerin antibodies was also used: rabbit anti-human polyclonal antibody against amino acids 157–173 (Orbigen, San Diego, CA, USA) and Alexa Fluor 350 goat anti-rabbit IgG, labeled with a blue-fluorescent dye (Invitrogen). Chromatin counterstaining was performed with propidium iodide red-fluorescent nucleic acid stain (Invitrogen). Cells were visualized on a laser scanning confocal microscope and presence/absence of emerin staining was determined in ≥200 nuclei.
Results
Case Summary
The proband (V.2: Fig. 1 and Table 1) described frequent palpitations in his youth and first underwent comprehensive cardiology evaluation at 33 years of age for complaint of dizzy spells. He had worked as a farmer and construction worker with no disability. Initial assessment by a neurologist, including a detailed neurological examination, revealed no neuromuscular disease. An ECG, however, showed an irregular, chaotic atrial rhythm with first-degree atrioventricular block. Ambulatory ECG monitoring revealed frequent premature atrial complexes with heart rates of 35–100 beats per minute and episodes of sinus arrest with junctional escape rhythm. Echocardiography revealed normal left atrial and ventricular size and systolic function. He was diagnosed with sick sinus syndrome and subsequently developed atrial flutter/fibrillation with a very slow ventricular response. At 40 years of age, an endocardial VVIR pacing system was implanted. Over 17 years of follow-up, his arrhythmia evolved from paroxysmal to chronic AF, and he continues to be ventricularly paced. He developed several comorbidities, including hypertension, hyperlipidemia, type II diabetes, and coronary artery disease, but at 57 years of age he had no clinically evident skeletal myopathy. Family history, however, indicated other male relatives had AF and pacemakers.
Figure 1.
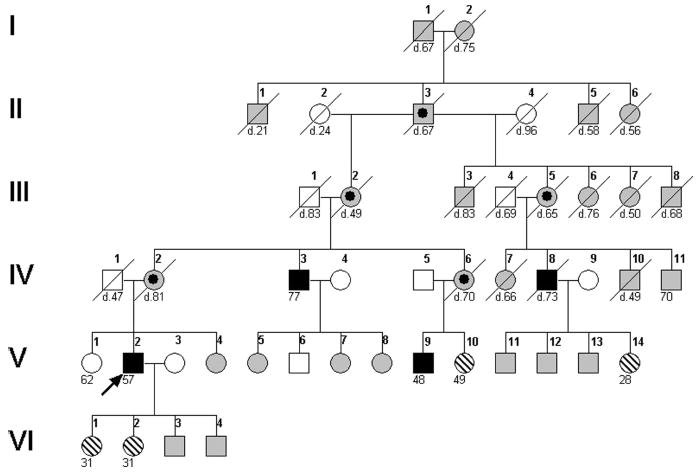
Pedigree structure. Genealogical records facilitated construction of this six-generation pedigree. The proband, V.2 (indicated by arrow), a maternal uncle, IV.3, a maternal cousin, V.9, and a distant male relative, IV.8, each had early-onset sinus bradycardia degenerating into SND and AF (filled symbols). X-linked inheritance was implicated by male-restricted disease transmitted by interrelated carrier females through a common founder male, II.3 (bulls-eye symbols). Six other family members underwent clinical, genetic, and immunocytochemical evaluation: four females with symptoms of paroxysmal arrhythmia (V.10, V.14, VI.1, VI.2), one asymptomatic female (V.1), and one asymptomatic male (V.6). Age at last evaluation or death (diagonal line) is indicated. Other pedigree symbols: hatched, rhythm abnormality except AF; unfilled, normal phenotype; gray, unknown medical history.
TABLE 1.
Clinical, Cellular, and Molecular Genetic Characteristics of Family Members
| Ped No. |
Sxs onset (yrs) |
Cardiac Sxs* |
Myopathy Sxs† |
Initial eval (yrs) |
AF onset (yrs) |
Pace maker (yrs) |
ECG∥ | Cardiac phenotype |
Emerin positive nuclei |
Emerin Lys37del genotype |
|---|---|---|---|---|---|---|---|---|---|---|
| IV.3 | teens (brady) | 2 | none | 73 | 70 | DDDR (73) | A-V pacing | SND, PAF | 4% | +/ |
| IV.8 | teens (faint) | 1,3 | none‡ | 44 | 44 | VVIR (44) | SB@53, NVT | SND, PAF | – | +/ |
| V.1 | – | none | none | 62 | – | – | NSR | Normal | 96% | −/− |
| V.2 | teens (palp) | 1,2 | none§ | 33 | 40 | VVIR (40) | SB@35 | SND, CAF | 1% | +/ |
| V.6 | – | none | none | 49 | – | – | NSR | Normal | 98% | −/ |
| V.9 | teens (brady) | 1,2 | none | 42 | 41 | DDDR (42) | AF, NVT | SND, CAF | 0% | +/ |
| V.10 | 30’s | 1,2 | none | 49 | – | – | SB@46 | SB | 71% | +/− |
| V.14 | 28 | 1,3 | none | 28 | – | – | NSR, PACs | Palp | 62% | +/− |
| VI.1 | 31 | 1,2 | none | 31 | – | – | NSVT | NSVT | 63% | +/− |
| VI.2 | 31 | 1 | none | 31 | – | – | NSR | Palp | 53% | +/− |
1, palpitations; 2, irregular pulse; 3, syncope.
Stiffness, weakness, or difficulty walking.
Normal lower extremity electromyography and exam by neurologist at age 66 years for chronic back pain; worked as farmer, shingled roofs, and climbed silos into his sixties.
Normal exam by neurologist at age of 33 and 46 years for dizziness; worked as farmer and construction worker.
Including ambulatory electrocardiographic and event monitor recordings.
+ / = hemizygous male; −/ = normal male; +/− = heterozygous female; −/− = normal female.
A-V = atrioventricular; brady = bradycardia; CAF = chronic AF; eval = evaluation; NSR = normal sinus rhythm; NSVT = non-sustained supraventricular tachycardia; NVT = nonsustained ventricular tachycardia; PACs = premature atrial complexes; PAF = paroxysmal AF; palp = palpitations; Ped = pedigree; SB = sinus bradycardia; Sxs = symptoms.
Pedigree Analysis
Genealogical records traced the proband’s ancestry to his great-great maternal grandparents (I.1 and I.2: Fig. 1), born in the Pommern Province of Prussia (modern day Germany, Russia, and Poland) in the early 1800s following the defeat of Napoleon. The obligate founder (II.3) was found dead on his family farm at age 67 of a presumed heart attack suffered while chasing a run-away horse team. While clinical data are unavailable for deceased family members, three other male relatives had documented arrhythmia similar to the proband’s (Table 1). Each described having had a slow pulse and/or syncope in their teenage years. SND and AF were diagnosed in the fourth to eighth decades of life, necessitating permanent pacemaker implantation. Aside from variable degrees of atrial enlargement, none had structural heart disease. The contractures and progressive muscle weakness that characterize EDMD in adolescence were absent, even in the sixth to eighth decades of life. One individual (IV.8) developed obstructive sleep apnea and pulmonary hypertension, and died following an out of hospital cardiac arrest. Autopsy was not performed. X-linked inheritance was implicated by male-restricted disease transmitted by interrelated carrier females through a common founder male (Fig. 1). While no female family member was diagnosed with SND and/or AF, four had variable symptoms of palpitations, irregular pulse, and/or syncope (Fig. 1 and Table 1). Electrocardiography, performed for clinical or research study indications, revealed sinus bradycardia, premature atrial complexes, and nonsustained supraventricular tachycardia.
Genetic Investigations
Genotyping in the four affected males and an unaffected female sibling (V.1) was performed with 18 polymorphic markers spanning the X chromosome. Genotypes for two markers on the distal q-arm suggested genetic linkage, prompting fine mapping with additional closely spaced polymorphic markers in this region. A haplotype comprised markers DXS8069, DXS8061, and DXS1073 at the q-terminus was inherited by all four males but not by the female (Fig. 2A). Individual V.9 had a genetic recombination event, defining DXS1193 as the centromeric flanking marker. EMD, encoding the nuclear membrane protein emerin and located within the 6.7 mega-base critical region on Xq28, was selected as a positional candidate gene. DNA sequence analysis revealed an in-frame 3-bp deletion in exon 2 of the single copy EMD gene in each affected male (Fig. 2B), resulting in deletion of the amino acid lysine at position 37 (Lys37del) of the 254 residue protein. The mutation disrupted the highly conserved LEM domain, critical for interaction with intranuclear DNA-binding proteins.19 Each of the four female family members, who exhibited variable degrees of paroxysmal atrial arrhythmia, was heterozygous for the Lys37del mutation (Fig. 1 and Table 1).
Figure 2.
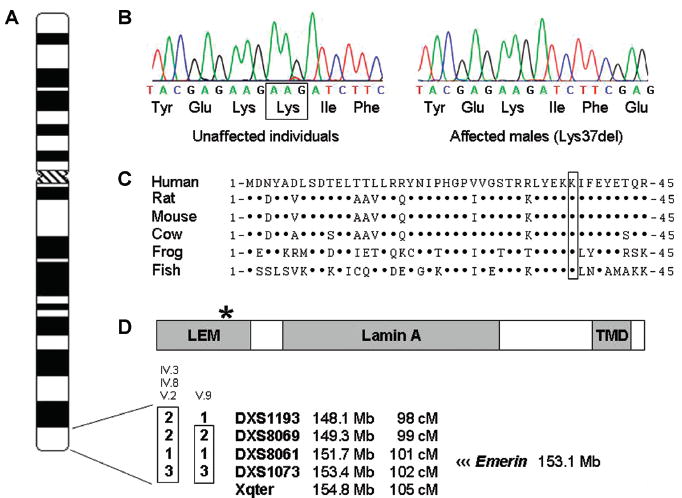
Genetic analyses. Genotyping with polymorphic DNA markers mapped a locus for SND and AF to chromosome Xq28. A: A haplotype identical in all four affected family members was identified, defining a 6.7 megabase critical region between marker DXS1193 and the q-arm telomere. B: Sequence analyses of EMD, a positional candidate gene, demonstrated a 3-bp deletion (109–111delAAG) in exon 2, resulting in deletion of the codon for lysine at residue 37 of the 254 amino acid protein. C: The mutation removes a highly conserved amino acid in emerin. Dots designate aligned amino acids identical to the human protein sequence. D: Lys37del disrupts a highly conserved region of the LEM domain at the N-terminus (residues 1–50), distinct from the lamin A and transmembrane (TMD) domains.19 The LEM domain is a critical binding site for intranuclear proteins that function as an interface between the nuclear membrane and DNA.
Immunocytochemistry
Lack of emerin staining of buccal epithelial cell nuclei has been described as a noninvasive alternative to skeletal muscle or skin biopsy to diagnose EDMD.18 Accordingly, we stained buccal cells with an antiemerin monoclonal antibody to determine whether the identified EMD mutation affected nuclear localization of emerin (Fig. 3). In contrast to a normal control (Fig. 3A), the three males hemizygous for Lys37del from whom cells were procured exhibited absent or nearly absent nuclear staining (Fig. 3B). Specifically, only 0–4% of nuclei demonstrated positive staining, albeit less intense than staining observed in controls. Among the four female carriers of Lys37del (one mutant and one normal copy of EMD), positive staining was seen in 53–71% of nuclei consistent with relatively random X chromosome inactivation (Fig. 3C). To exclude the possibility of disruption of primary antibody binding by the Lys37del mutation, a different primary emerin antibody against amino acids remote from the deleted residue was also tested. Staining patterns were the same as those observed with the initial antibody (data not shown).
Figure 3.
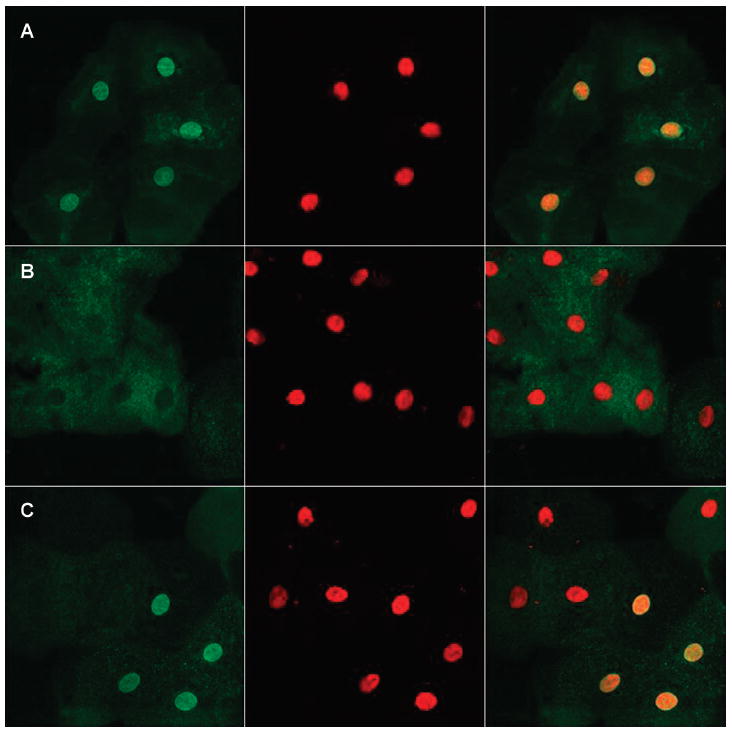
Immunocytochemistry. The EMD-Lys37del mutation impaired nuclear localization of the mutant protein. Left panels, emerin staining (green); middle panels, chromatin staining (red); right panels, combined emerin and chromatin staining. A: In a normal female control (V.1), nuclear membranes of exfoliated oral epithelial cells showed robust staining with an anti-emerin antibody. B: Buccal cell nuclei from an affected male (IV.3) exhibited lack of emerin staining. C: A female carrier (VI.1) heterozygous for the Lys37del mutation demonstrated staining of ~60% of nuclear membranes.
Discussion
Genotype-Phenotype Correlation
Emerin is a ubiquitously expressed protein, yet mutation of EMD is associated with a specific, localized pattern of contractures and muscle wasting and electrical heart disease largely confined to the atria.20 Moreover, intrafamilial variability in expression of cardiac and skeletal muscle pathologies can occur in X-linked EDMD.21-24 In some cases, arrhythmias are prominent and myopathy is mild. A majority of EDMD-associated defects are nonsense or frameshift mutations that introduce premature stop codons, resulting in truncated, nonexpressed emerin protein.20 Consequently, most male patients with EDMD have complete absence of emerin, and specific phenotypic traits cannot be attributed to mutation-specific effects. These observations implicate stochastic factors and/or disease-modifying effects of other genes in EDMD. Rare EMD missense mutations and in-frame deletions have been associated with normal emerin expression and localization, yet these patients have typical EDMD phenotypes.19 Notwithstanding, such mutations point to a distinct disease mechanism based on altered protein-protein interactions at the nuclear membrane, and raise the possibility that certain domain-specific mutations could have cardioselective effects. Indeed, the Lys37del mutation we identified disrupts the highly conserved LEM domain of emerin while preserving the open reading frame for the remainder of the protein, including the domains required for interaction with lamin A/C and cytoskeletal proteins. The LEM domain is a distinct binding site for intranuclear DNA-binding proteins, like BAF (barrier-to-autointegration factor).19 Targeted mutagenesis of the LEM domain results in reduced emerin-BAF binding in a biochemical assay19 and failed localization to the nuclear membrane in cultured HeLa cells.25 Furthermore, structural analysis of the BAF-emerin complex at the atomic level has localized the positively charged lysine residue at position 37 (deleted in our family) to the interface between proteins, mediating electrostatic interaction with negatively charged residues on BAF.26
A specific genotype-phenotype correlation between emerin LEM domain disruption and AF is implicated by isolated cardiac disease in the four males hemizygous for Lys37del. While sharing a common male ancestor and inheriting the same 6.7 Mb segment of chromosome Xq28 harboring EMD, the genetic backgrounds of these individuals are otherwise divergent and a codominant autosomal modifier gene(s) is unlikely. For example, the proband (V.2) and individual IV.8 are fifth-degree relatives, predicting that they inherited only 1/32 of the same genes. Moreover, two females heterozygous for Lys37del (V.14 and VI.1), who exhibited early manifestations of cardiac electrical instability in the absence of clinical myopathy, are seventh-degree relatives (1/128 of genes in common).
Remarkably, the same Lys37del mutation was reported recently by Ben Yaou et al. in an Algerian family with digenic inheritance of both EMD and LMNA mutations and variable clinical features.27 AF in the absence of skeletal muscle disease was found in one male hemizygous for the EMD mutation alone, three males hemizygous for the EMD mutation and heterozygous for the LMNA mutation, and two females heterozygous for both mutations. The cardioselective effects—AF and pacemaker dependence—of the Lys37del mutation exhibited within two ethnically distinct families provides compelling evidence for this genotype-phenotype relationship.
Irrespective of LMNA mutation status, Ben Yaou et al. found highly diminished but not absent emerin protein in lymphoblastoid cells and skin fibroblasts from hemizygous and heterozygous Lys37del-carriers.27 Correspondingly, immunofluorescence studies in fibroblasts demonstrated that nuclear emerin staining was reduced but not completely abolished.27 These observations, together with our findings of total or near-total lack of nuclear staining of buccal epithelial cells in hemizygous males, raise the possibility of different degrees of expression, nuclear localization, and functional effects of emerin-Lys37del depending on cell-type and tissue-specific interacting proteins.28 This, in turn, could account for a disease phenotype involving atrial myocardium but not skeletal muscle. Skeletal and cardiac tissues were unavailable in the present study and in the study by Ben Yaou et al., however, to enable emerin studies in skeletal and cardiac myocytes.
Clinical Implications
AF is a heritable, genetically heterogeneous disorder; no single AF gene yet identified is a common basis for disease. Associated phenotypic traits like prolonged QT interval,5 dilated cardiomyopathy,11,12 and adrenergic-sensitivity29 have been linked to certain gene defects in lone and syndromic AF. In principle, these reports demonstrate that clinical subclassification has the potential to guide mutational analysis of candidate genes. In this study, eliciting a detailed family history and pedigree analysis revealed X-linked inheritance and enabled us to target the X chromosome for locus mapping and select EMD as a positional candidate gene, despite lack of pathognomonic neuromuscular features of EDMD. The frequency and spectrum of EMD mutations in lone AF are unknown, but our findings provide a rationale for EMD mutation testing in cases of X-linked inheritance and/or pacemaker-dependent SND. Once an EMD mutation is identified, genetic testing should be offered to at-risk male and female relatives alike, recognizing risk for SND and AF in both genders.
Conclusion
Mutation of EMD can cause nonsyndromic, X-linked SND and AF. While the mechanism for atrial electrical instability caused by heritable defects in emerin remains unknown, our findings suggest a cardiospecific role for the LEM domain and DNA-binding protein interaction. Diagnosis of EMD-associated AF has important implications for family counseling and clinical follow-up.
Acknowledgments
We gratefully acknowledge the family who participated in this study. We also thank Lois Rowe for help with the immunocytochemistry experiments and Katharine Brauch for critical review of the manuscript.
This work was supported by a grant from the National Institutes of Health, National Heart Lung and Blood Institute (R01 HL075495), and by the Mayo Foundation.
References
- 1.Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A, Bachinski LL, Roberts R. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997;336:905–911. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- 2.Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, Hammill SC, Packer DL, Olson TM. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. 2003;41:2185–2192. doi: 10.1016/s0735-1097(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 3.Ellinor PT, Yoerger DM, Ruskin JN, MacRae CA. Familial aggregation in lone atrial fibrillation. Hum Genet. 2005;118:179–184. doi: 10.1007/s00439-005-0034-8. [DOI] [PubMed] [Google Scholar]
- 4.Fox CS, Parise H, D’Agostino RB, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 5.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 6.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 7.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–2688. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich JR, Zicha S, Coutu P, Hebert TE, Nattel S. Atrial fibrillation-associated minK38G/S polymorphism modulates delayed rectifier current and membrane localization. Cardiovasc Res. 2005;67:520–528. doi: 10.1016/j.cardiores.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Chen LY, Ballew JD, Herron KJ, Rodeheffer RJ, Olson TM. A common polymorphism in SCN5A is associated with lone atrial fibrillation. Clin Pharmacol Ther. 2007;81:35–41. doi: 10.1038/sj.clpt.6100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juang JM, Chern YR, Tsai CT, Chiang FT, Lin JL, Hwang JJ, Hsu KL, Tseng CD, Tseng YZ, Lai LP. The association of human connexin 40 genetic polymorphisms with atrial fibrillation. Int J Cardiol. 2007;116:107–112. doi: 10.1016/j.ijcard.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 11.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Spudich S, De Girolami U, Seidman JG, Seidman C. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 12.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 14.Brand FN, Abbott RD, Kannel WB, Wolf PA. Characteristics and prognosis of lone atrial fibrillation. 30-year follow-up in the Framingham Study. JAMA. 1985;254:3449–3453. [PubMed] [Google Scholar]
- 15.Kopecky SL, Gersh BJ, McGoon MD, Whisnant JP, Holmes DR, Jr, Ilstrup DM, Frye RL. The natural history of lone atrial fibrillation. A population-based study over three decades. N Engl J Med. 1987;317:669–674. doi: 10.1056/NEJM198709103171104. [DOI] [PubMed] [Google Scholar]
- 16.Scardi S, Mazzone C, Pandullo C, Goldstein D, Poletti A, Humar F. Lone atrial fibrillation: Prognostic differences between paroxysmal and chronic forms after 10 years of follow-up. Am Heart J. 1999;137:686–691. doi: 10.1016/s0002-8703(99)70224-3. [DOI] [PubMed] [Google Scholar]
- 17.Olson TM, Keating MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest. 1996;97:528–532. doi: 10.1172/JCI118445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabatelli P, Squarzoni S, Petrini S, Capanni C, Ognibene A, Cartegni L, Cobianchi F, Merlini L, Toniolo D, Maraldi NM. Oral exfoliative cytology for the non-invasive diagnosis in X-linked Emery-Dreifuss muscular dystrophy patients and carriers. Neuromuscul Disord. 1998;8:67–71. doi: 10.1016/s0960-8966(97)00147-8. [DOI] [PubMed] [Google Scholar]
- 19.Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci. 2001;114:4567–4573. doi: 10.1242/jcs.114.24.4567. [DOI] [PubMed] [Google Scholar]
- 20.Zacharias AS, Wagener ME, Warren ST, Hopkins LC. Emery-Dreifuss muscular dystrophy. Semin Neurol. 1999;19:67–79. doi: 10.1055/s-2008-1040827. [DOI] [PubMed] [Google Scholar]
- 21.Waters DD, Nutter DO, Hopkins LC, Dorney ER. Cardiac features of an unusual X-linked humeroperoneal neuromuscular disease. N Engl J Med. 1975;293:1017–1022. doi: 10.1056/NEJM197511132932004. [DOI] [PubMed] [Google Scholar]
- 22.Bialer MG, McDaniel NL, Kelly TE. Progression of cardiac disease in Emery-Dreifuss muscular dystrophy. Clin Cardiol. 1991;14:411–416. doi: 10.1002/clc.4960140509. [DOI] [PubMed] [Google Scholar]
- 23.Boriani G, Gallina M, Merlini L, Bonne G, Toniolo D, Amati S, Biffi M, Martignani C, Frabetti L, Bonvicini M, Rapezzi C, Branzi A. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: A long-term longitudinal study. Stroke. 2003;34:901–908. doi: 10.1161/01.STR.0000064322.47667.49. [DOI] [PubMed] [Google Scholar]
- 24.Sakata K, Shimizu M, Ino H, Yamaguchi M, Terai H, Fujino N, Hayashi K, Kaneda T, Inoue M, Oda Y, Fujita T, Kaku B, Kanaya H, Mabuchi H. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation. 2005;111:3352–3358. doi: 10.1161/CIRCULATIONAHA.104.527184. [DOI] [PubMed] [Google Scholar]
- 25.Haraguchi T, Koujin T, Segura-Totten M, Lee KK, Matsuoka Y, Yoneda Y, Wilson KL, Hiraoka Y. BAF is required for emerin assembly into the reforming nuclear envelope. J Cell Sci. 2001;114:4575–4585. doi: 10.1242/jcs.114.24.4575. [DOI] [PubMed] [Google Scholar]
- 26.Cai M, Huang Y, Suh JY, Louis JM, Ghirlando R, Craigie R, Clore GM. Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J Biol Chem. 2007;282:14525–14535. doi: 10.1074/jbc.M700576200. [DOI] [PubMed] [Google Scholar]
- 27.Ben Yaou R, Toutain A, Arimura T, Demay L, Massart C, Peccate C, Muchir A, Llense S, Deburgrave N, Leturcq F, Litim KE, Rahmoun-Chiali N, Richard P, Babuty D, Récan-Budiartha D, Bonne G. Multitissular involvement in a family with LMNA and EMD mutations: Role of digenic mechanism? Neurology. 2007;68:1883–1894. doi: 10.1212/01.wnl.0000263138.57257.6a. [DOI] [PubMed] [Google Scholar]
- 28.Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol. 2005;6:21–31. doi: 10.1038/nrm1550. [DOI] [PubMed] [Google Scholar]
- 29.Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, Seino S, Asirvatham SJ, Jahangir A, Terzic A. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;4:110–116. doi: 10.1038/ncpcardio0792. [DOI] [PMC free article] [PubMed] [Google Scholar]