

Abstract
We present a general-purpose model for biomolecular simulations at the molecular level that incorporates stochasticity, spatial dependence, and volume exclusion, using diffusing and reacting particles with physical dimensions. To validate the model, we first established the formal relationship between the microscopic model parameters (timestep, move length, and reaction probabilities) and the macroscopic coefficients for diffusion and reaction rate. We then compared simulation results with Smoluchowski theory for diffusion-limited irreversible reactions and the best available approximation for diffusion-influenced reversible reactions. To simulate the volumetric effects of a crowded intracellular environment, we created a virtual cytoplasm composed of a heterogeneous population of particles diffusing at rates appropriate to their size. The particle-size distribution was estimated from the relative abundance, mass, and stoichiometries of protein complexes using an experimentally derived proteome catalog from Escherichia coli K12. Simulated diffusion constants exhibited anomalous behavior as a function of time and crowding. Although significant, the volumetric impact of crowding on diffusion cannot fully account for retarded protein mobility in vivo, suggesting that other biophysical factors are at play. The simulated effect of crowding on barnase-barstar dimerization, an experimentally characterized example of a bimolecular association reaction, reveals a biphasic time course, indicating that crowding exerts different effects over different timescales. These observations illustrate that quantitative realism in biosimulation will depend to some extent on mesoscale phenomena that are not currently well understood.
INTRODUCTION
Biology at the molecular scale is shifting from the collation of genes, proteins, and metabolites to the study of the interactions of molecular players within complex networks and systems. At this system-level view of organisms, computational simulation is required for both quantitative and qualitative prediction of system behavior. Current network approaches tend to represent cells as homogenous deterministic systems, ignoring spatial processes and randomness at multiple scales. The cytoplasmic environment, however, is crowded and spatially heterogeneous in molecular population types and numbers (1), a milieu that is remote from the conditions of typical in vitro experiments.
The relevance of spatial partition to cellular physiology is immediately evident from examples such as compartmentalization, cell division, morphogenesis, intracellular trafficking, and signal transduction (2–6). The effects of crowding on cellular physiology are less obvious (7–11) and include, among other phenomena, increased association rates (12,13), metabolic channeling (14), phase separation (15), and slower rates of diffusion (16–19), with documented consequences for cell volume regulation (20), lens formation (21,22), reaction kinetics (23–26) and signal transduction (27,28). Thus, there is growing awareness of the importance of space when constructing realistic cellular models (11,29–32).
There are currently a variety of dynamic spatial modeling approaches (30). They include strategies based on partial differential equations (PDEs) (5,33–35), cellular automata (24,36,37), on-lattice reaction diffusion (5,27,38–41), and off-lattice diffusing particles (24,42–44). These approaches vary in their range of application and in their limitations. PDEs provide the most straightforward extension of ordinary differential equations into spatial degrees of freedom, but have difficulty including stochasticity or accounting for macromolecular crowding. Lattice approaches provide a computationally simple treatment of space. However, the artificial nature of the lattice limits spatial resolution, introduces lattice anisotropy, and may (if lattice occupancy is unlimited) discount volume exclusion. As an alternative, particle-based approaches explicitly track every molecule off-lattice. Although these models are closer to physical reality, they are computationally expensive, resulting in compromises within existing models based on research focus. In MCell, for example, particles react only with surfaces (42), whereas Smoldyn ignores excluded volume by treating particles as points (43). At an even finer scale are coarse-grain molecular dynamics approaches (45,46), which have been used in membrane simulation (47–49), diblock copolymer self-assembly (50,51), and protein folding and dynamics (52,53), among other areas. Although these submolecular models are even more expensive than molecular scale models, they allow the prediction of molecular properties and dynamics at the cost of limited timescale.
There are many important systems to which an improved model could be applied. We consider two here, an investigation of the nature of hindrance to in vivo protein diffusion in Escherichia coli, and an assessment of the impact of crowding on a prototypical transport-limited process, the diffusion-limited dimerization of the barnase-barstar pair.
It has been known for some time that in vivo diffusion rates of soluble proteins differ dramatically from those observed in vitro, but the detailed mechanisms involved are still a matter of debate (19). Possible explanations include altered viscosity, nonspecific interactions, hindrance due to crowding, and caging effects from cytoplasmic polymers. Although some of these explanations can be partially illuminated by appropriate experiments, the ability to directly simulate the processes involved has the obvious potential to determine the sufficiency of any proposed alternatives.
In vivo, the function of the bacterial ribonuclease barnase is to cleave extracellular RNA, as a defense against infectious viral RNA. This functional requirement of high catalytic efficiency for organism defense is complicated by toxicity to the cell's own RNA, thus requiring a highly effective mechanism for minimizing the activity of barnase in the host cell. This is the role of barstar, which forms a rapid, tight, nearly irreversible association with barnase that deactivates it. This process has evolved to be as fast as possible, and has therefore become a prototypical example of a diffusion-limited protein-protein interaction, and thus forms an excellent system for studying the impact of crowding on in vivo reactions.
Here, we describe and validate a high-performance, parallelized particle-based simulation where particles capable of diffusion and reaction take on physical dimensions. Using this model in combination with a proteomic-scale evaluation of protein abundance, we approximated the populational and diffusive characteristics of the E. coli cytoplasm to study the volumetric impact of macromolecular crowding on biomolecular diffusion and diffusion-limited reactions. The observed dependence of diffusion and reaction “constants” on crowding highlights the value this type of approach toward the development of realistic biological network simulations.
METHODS
Particle-based hard-sphere reaction-diffusion model
We model reactions in the cytoplasm at the one particle/molecule level, with the discrete computational model based on a microscopic continuous reaction-diffusion system. The center of mass of each particle obeys the diffusion equation
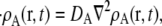 |
(1) |
where r is the location of the molecule, t is the time, ρA is the local particle number density, and DA is the diffusion constant. Reactions occur via the Collins-Kimball boundary condition between reacting pairs of particles (54):
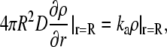 |
(2) |
where ka is the microscopic reaction rate, R is the contact distance, and D is the relative diffusion constant of the pair. Our model is an off-lattice discretized version of this system. A conceptual flow chart of the simulation appears in Fig. 1. All source code and input files can be downloaded from http://projectcybercell.ca/Downloads/.
FIGURE 1.

Flow chart for model algorithm.
Movement and collisions
The simulation proceeds in discrete timesteps of fixed size 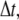 and particles move off-lattice via discrete moves of a fixed length,
and particles move off-lattice via discrete moves of a fixed length, 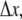 in a random direction uniformly distributed over the surface of a sphere. The move probability, PA, for a species A is connected to its dilute aqueous diffusion constant, DA, through the formula
in a random direction uniformly distributed over the surface of a sphere. The move probability, PA, for a species A is connected to its dilute aqueous diffusion constant, DA, through the formula 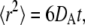 using
using 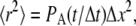 giving
giving
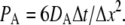 |
(3) |
After movement, collisions for moved particles are determined by comparing center-to-center distances, based on new particle positions, with the sum of particle radii. If, upon collision, no reaction occurs (see below), the move is rejected, and the particle is returned to its original position, enforcing hard-sphere exclusion.
Reactions
If, however, the colliding species can undergo a bimolecular reaction, the collision triggers that reaction with probability γ. This probability may be connected to the intrinsic reaction rate, which in a continuum reaction-diffusion model is specified via the partially absorbing boundary condition (Eq. 2) that connects the diffusive flux of particles across the contact distance to the local concentration. We have computed a discrete form of this equation as follows. Since the projection of the spherical move distribution onto any axis is uniform, the probability per move of a particle B initially located at z = z′ crossing a surface at z = 0 is pB/2 (1 − z′/Δx) for z′ < Δx, where pB is the probability of movement. The total flux of B particles crossing the contact radius, R = RA + RB, near a fixed A particle is computed by integrating over both the surface of the sphere and initial separations. If the B concentration is constant on the scale of Δx (valid for small γ), the result is 4πR2pB/4 Δx ρB. Of these particle collision events, a fraction γ react, and the resulting reaction events per timestep equals the total reacting flux from Eq. 2 times, Δt. We then solve for ka in terms of γ as
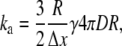 |
(4) |
where the move probability has been replaced with the diffusion constant via Eq. 3. A similar formula is used by the MCell model (42), where, however, moves are normally distributed. If both particles can move simultaneously, the calculation gives the same expression as Eq. 4, where D is replaced by a combined diffusion probability, Dc:
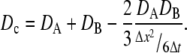 |
(5) |
Dissociation events for complexes are handled with a fixed probability/timestep/complex,
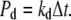 |
(6) |
Products are positioned at their contact distance, in a random orientation, where the location of the center of mass of the dissociating complex is conserved.
Environment
Simulations occur in a rectangular box. Boundary conditions at the walls can be 1), periodic, 2), reflecting, 3), absorbing, or 4), absorbing with nonzero fixed species concentrations on the other side, acting as a particle source. The latter case corresponds to Dirichlet boundary conditions for a PDE.
Model validation
Diffusion
We validate diffusion by measuring mean-square distance traveled versus time for a heterogeneous dilute collection of particles with a size distribution corresponding to the virtual cytoplasm (Table 1, and discussed below). Diffusion constants are computed from a reference to the in vitro diffusion of green fluorescent protein (GFP), D = 87 μm2/s (55), via the scalings 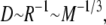 valid if it is assumed that all particle classes share equal specific volumes and the hydrodynamic radius is proportional to the contact radius. Move probabilities are calculated from the diffusion constants via Eq. 3. Simulations were conducted in a (1000 nm)3 box with reflecting boundary conditions where the move length Δx = 0.5 nm, and the timestep Δt = 0.35 ns for a total simulation time of 1 μs. The system was initialized with random nonoverlapping particles to a 1% occupied volume fraction. Convergence was checked against additional shorter simulations with twofold- and fourfold-smaller time and space steps.
valid if it is assumed that all particle classes share equal specific volumes and the hydrodynamic radius is proportional to the contact radius. Move probabilities are calculated from the diffusion constants via Eq. 3. Simulations were conducted in a (1000 nm)3 box with reflecting boundary conditions where the move length Δx = 0.5 nm, and the timestep Δt = 0.35 ns for a total simulation time of 1 μs. The system was initialized with random nonoverlapping particles to a 1% occupied volume fraction. Convergence was checked against additional shorter simulations with twofold- and fourfold-smaller time and space steps.
TABLE 1.
Particle distribution and properties for virtual cytoplasm
| Molecular mass range (kDa) | Weight fraction (%) | Average molecular mass (kDa) | Radius (nm) | D (μm2/s) |
|---|---|---|---|---|
| 0–20 | 0.7 | 11.4 | 1.7 | 156.8 |
| 20–40 | 3.5 | 29.1 | 2.3 | 83.7 |
| 40–60 | 3.9 | 46.0 | 2.7 | 61.7 |
| 60–80 | 4.1 | 67.2 | 3.0 | 47.9 |
| 80–100 | 4.8 | 91.4 | 3.4 | 39.0 |
| 100–120 | 1.9 | 107.1 | 3.5 | 35.1 |
| 120–140 | 2.5 | 132.8 | 3.8 | 30.4 |
| 140–160 | 3.5 | 156.1 | 4.0 | 27.3 |
| 160–180 | 0.6 | 161.7 | 4.1 | 26.7 |
| 180–200 | 1.3 | 186.5 | 4.3 | 24.3 |
| 200+ | 37.7 | 346.0 | 5.2 | 16.1 |
| Ribosomes | 35.4 | 2700 | 10.4 | 4.1 |
Association reactions
Assuming the movement length and timestep are both unity (always possible via a change of units), the remaining variables for an irreversible 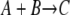 reaction are the radii, RA and RB, of the A and B particles, the probability of reaction γ, and the initial concentration of each species. The corresponding macroscopic rate coefficients are given by Eq. 4.
reaction are the radii, RA and RB, of the A and B particles, the probability of reaction γ, and the initial concentration of each species. The corresponding macroscopic rate coefficients are given by Eq. 4.
To validate the reaction model over a wide range of parameters, we conducted simulations over a range of combinations of microscopic constants including: reaction probability (0.001, 0.01, 0.1, and 1), move probability (0.1 and 1), and reactant radii (10 and 100), for a total of 40 independent cases. Initial particle numbers are NA = NB = 5000 for each case, where the simulation volume is adjusted so that the total occupied volume is 1%, corresponding to uncrowded conditions. The results are averaged over 10 runs of 100,000 iterations each. Survival probability is assessed as the fraction of species A remaining at a given time. The time-dependent rate coefficient is calculated from the concentration time curves as
 |
(7) |
where tfinal is selected to ensure that a minimum of 100 events have occurred when summed over all runs. This threshold provides a means of controlling the tradeoff between noise and time resolution in a manner that is independent of rate coefficient.
We used the standard Smoluchowski theory of diffusion-limited reactions to compute predicted survival curves and time-dependent reaction coefficients as follows (56,57):
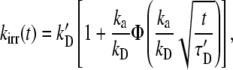 |
(8) |
where ka is the association reaction rate constant; kD = 4πDR is the maximally diffusion-limited reaction rate in terms of the contact distance, R, and the combined diffusion constant, D; k′D = kakD/(ka + kD) is the asymptotic reaction rate, τ′D= (k′D/kDR)2/D, and the function Φ(x) = exp(x2)erfc(x). In terms of kirr(t), the survival probability for an A particle is given as
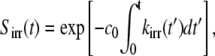 |
(9) |
where c0 is the concentration of B particles.
Reversible reactions
For evaluation of a prototypical diffusion-influenced reversible reaction 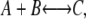 with forward association rate ka and reverse dissociation rate kd, we consider problems with the following parameters: ka = 125, RA = RC =1, RB = 0, DA = DC = 0, cB = 1, DB = 1, and kd = 5 or 500. These parameters are selected to correspond to the limitations of current theoretical approaches to reversible reactions, in particular the target approximation, dilute As, and noninteracting Bs. The association reaction is primarily diffusion-limited (ka/4πDR ≈ 10), so a small
with forward association rate ka and reverse dissociation rate kd, we consider problems with the following parameters: ka = 125, RA = RC =1, RB = 0, DA = DC = 0, cB = 1, DB = 1, and kd = 5 or 500. These parameters are selected to correspond to the limitations of current theoretical approaches to reversible reactions, in particular the target approximation, dilute As, and noninteracting Bs. The association reaction is primarily diffusion-limited (ka/4πDR ≈ 10), so a small 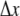 is required for the validity of Eq. 4. We use
is required for the validity of Eq. 4. We use 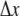 = 1.5 × 10−3, resulting in γ = 0.01; and set
= 1.5 × 10−3, resulting in γ = 0.01; and set 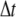 = 3.80 × 10−7, giving PB = 1. The dissociation probability Pd = 1.9 × 10−4 and 1.9 × 10−6 for kd = 5 and 500, respectively. The system simulated consists of a single A particle and 125 B particles in a cubic box of volume 53, with reflecting boundary conditions, and the average survival probability is computed over 1000 runs with independent seeds.
= 3.80 × 10−7, giving PB = 1. The dissociation probability Pd = 1.9 × 10−4 and 1.9 × 10−6 for kd = 5 and 500, respectively. The system simulated consists of a single A particle and 125 B particles in a cubic box of volume 53, with reflecting boundary conditions, and the average survival probability is computed over 1000 runs with independent seeds.
Simulation results are compared with theoretical curves, computed assuming either mass action or applying the best available approximate theory, the MPK1 multiparticle kernel (57). The survival kernel is computed numerically in Laplace space followed by a numerical inverse Laplace transform.
Numerical experiments
Transport in a crowded environment
We examine the impact of crowding on diffusion by conducting diffusive transport experiments incorporating particles distributed according to the virtual cytoplasm at an occupied volume fraction of 0.01, 0.10, 0.20, 0.30, 0.34, 0.40, and 0.50. Conditions are the same as for the diffusion validation except for the occupied volume. The total number of particles ranges up to 1.7 × 106 particles for the 0.50 case. Time-dependent self-diffusion coefficients were computed through measurements of mean-squared displacement over elapsed time. Total simulation time was generally 100 μs, with 245 μs used for the 0.34 case.
Crowding in barnase-barstar dimerization
A (400 nm)3 box was initialized with equal numbers of randomly positioned barnase and barstar particles at 300 μM, corresponding to ∼11,000 particles and an occupied volume fraction of 4%. Radii and diffusion coefficients for barnase and barstar were both set as equal, at 3.0 nm and 150 μm2 s−1 (58). The virtual cytoplasm particles were added to this system at an additional occupied volume of 0%, 30%, and 50%. The timestep, Δt, was 0.1 ns, the move length was 0.3 nm, and reactions between barnase and barstar were irreversible with an association probability of 1. Simulations were run in periodic boundary conditions for 10 μs.
The virtual cytoplasm
In terms of its dry weight, the cytosolic content of E. coli consists mainly of ribosomes (62% RNA, 38% protein, constituting 40–50% of cell weight) and free protein (20–30%). To construct a volumetrically accurate size distribution of the E. coli cytoplasm, we used the catalog of proteins and their abundances originally reported in our proteomic analysis of E. coli K–12 (59). From this analysis, we initially selected 159 of the major polypeptides representing >90% of the cytosolic protein mole-percentage. From this pool, we removed from the analysis 1), ribosomal polypeptides (whose quantification was deemed unreliable); 2), polypeptides that transiently associate with other cell structures (i.e., the inner membrane) or with other proteins in noncomplex arrangements (i.e., elongation factors); and 3), proteins representing cross-compartment contamination. The stoichiometric relationships of the remaining 118 polypeptides were found by bioinformatic analysis to include 82 species involved in homocomplexes, 19 species involved in 13 heterocomplexes, and 17 monomers (60,61).
The relative molar abundance for monomers and homo complexes were calculated as follows:
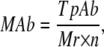 |
(10) |
where TpAb is the total protein abundance (49); Mr is the experimentally determined molecular mass (59), and n is the number of protein subunits within the complex. For heterocomplexes the abundance of the complex was taken as the relative abundance of the most abundant identified member.
A cytoplasm mass distribution was created by grouping monomers and complexes into size classes separated by 20 kDa to an upper class of 200 kDa and greater. The resulting distribution (Table 1, and illustrated at three packing densities in Fig. 2) is smooth and has (mass-weighted) peaks of ∼80 kDa and 200 kDa. The largest single complex by mass is the 374 kDa RNA polymerase complex, at 11.4% by molar abundance.
FIGURE 2.

Virtual cytoplasm, with 1% (a), 10% (b), and 50% (c) occupied volume fraction.
Since ribosomes constitute 40–50% of the total cell mass of exponentially growing E. coli (62), a separate ribosomal class was introduced that assumed a copy number of 18,000 ribosomes per cell (60) contributing to a total density of 0.34 g/ml (10).
The effective molecular radius of each class was determined by assuming spherical particles of weight corresponding to the class average and a specific volume of 1.0 cm3/g (10). In the simulation, each particle class was assigned diffusive behavior appropriate to its molecular mass.
RESULTS
Model validation
In this model, spherical molecules of different sizes diffuse through space and are capable of reversible reactions upon collision. The realism of the model in terms of its diffusive and reaction properties was therefore evaluated according to known empirical behavior and existing theory, validated against the underlying microscopic description of Eqs. 1 and 2.
For realistic diffusion, we expect that the movement of molecules within the simulation will conform to the relationship 〈R2〉 = 6Dt, where 〈R2〉 is the mean-squared distance traveled by the molecule at time t, and D is the diffusion constant. Fig. 3 shows the mean-squared path length versus time under free diffusion for a population of hard spheres with different sizes. The results agree well with the expected formula over the duration shown.
FIGURE 3.

Diffusion validation. 〈R2〉 versus t for diffusion of particles distributed according to the virtual cytoplasm (Table 1) at 1% occupied volume. Solid lines, simulation; dashed lines, 6Dt, where D is the input diffusion constant of the species from Table 1.
As a prelude to evaluating the model's performance with respect to reversible reactions, we confirmed that irreversible association reactions exhibited expected behavior for the simple bimolecular case A + B → C over a wide range of parameters (Methods, Eq. 8). Since the predicted reactant survival curves given by Eq. 8 vary for every case, only several selected cases are illustrated (Fig. 4, a and b). In general, the agreement is very good for all conditions examined. The worst discrepancies apply to cases with the smallest collision distances, all of which are included among the curves plotted. The agreement indicates that when applying Eq. 4, these simulations are able to reproduce reactions in all ranges, including maximally to minimally diffusion-limited reactions. An even more precise test can be made by rearrangement of Eq. 8 so that time-dependent rate coefficients fall on a single universal curve Φ(t) (after the asymptotic value has been subtracted and k and t have been rescaled). This resulting collapse of data is shown in Fig. 4 c. Since the subtraction emphasizes the noise, the smallest collision distance cases were removed for clarity.
FIGURE 4.

Reaction model conforms with theory for irreversible reactions, A + B → C . (a–c) Survival probability of reactant A as measured in simulations (solid lines) and predicted from Eqs. 3–9 (dashed lines) versus iterations for the selected cases (a) γ = 0.01, (b) γ = 0.1, and (c) γ = 1.0, with other parameters as indicated. (d) Rescaled time-dependent reaction rate k(t) versus rescaled t, for all cases. The shaded line is the universal predicted curve from Eq. 9.
Since the simulations cover parameter space in terms of microscopic move and reaction probabilities, they also illustrate convergence properties of the model. As Δx becomes larger, approaching the collision distance, the collision surface can no longer be treated as planar, as required in Eq. 4, resulting in a discrepancy visible between Fig. 4, a and b, for some of the cases with smallest radii. In the opposite limit, as Δx becomes much smaller than the collision distance, the discrete model will approach a continuous reaction-diffusion system.
When applying the above observations to reversible reactions, we note that, among other problems, reversibility is complicated by the possibility that proximally dissociated molecules will tend to recombine (geminate recombination). To validate the simulation of reversible reactions, we therefore limited consideration to theoretically tractable cases with both high and low dissociation constants. We compared our simulation results for the cases of the simple reversible reaction A + B ↔ C with well-stirred mass-action chemical kinetics and with continuum reaction-diffusion using the accepted MPK1 approximation (Fig. 5). It is worthy of note that our simulations agree well with MPK1 up to noise limits, thereby providing validation of diffusion-influenced reversible reactions. However, mass action was found to deviate substantially from both simulation and MPK1 theory, indicating the failure of conventional ODE approaches to this system (see Discussion).
FIGURE 5.

Reaction model conforms with theory for reversible reactions. Survival probability S(t) for a single target A particle in the bimolecular reversible reaction A + B ↔ C, plotted as the normalized deviation from the predicted equilibrium value S∞ = kd/(ka + kd), with DA = DC = 0, DB = cB = 1, ka = 125, and box size 53. Solid lines are simulation results averaged over 1000 repetitions, dashed lines are theoretical predictions calculated according to MPK1 multiparticle kernel theory, and dotted lines are theoretical predictions calculated according to mass action. (a) kd = 500. (b) kd = 5.
Simulating the effects of crowding on diffusion and reaction
We next applied the validated model to study the impact of macromolecular crowding on diffusion and reaction using a size distribution of mobile but inert particles that approximates the volumetric composition of the E. coli cytoplasm. The move length was selected to be a fraction of the smallest particle size, and the timestep was maximized given the constraint that the maximum diffusion coefficient Δx2/6Δt must be greater than the largest modeled diffusion constant. The time-dependent behavior of self-diffusion in this simulated cytoplasm is shown in Fig. 6 a at an occupied volume of 34%. Convergence and stepsize dependence was tested by comparison of test cases with shorter simulations at higher resolution, with Δx reduced two- and fourfold, and Δt reduced to match (data not shown). The higher-resolution runs displayed quantitatively similar behavior, within a few percent, indicating partial, albeit not total, convergence. The higher-resolution runs lie slightly above the data shown, unsurprisingly indicating that step size discreteness results in a slightly higher effective collision radius for the particles.
FIGURE 6.

Anomalous self-diffusion in the cytoplasm. (a) Self-diffusion coefficient versus time at 34% cytoplasm occupied volume. Lines represent different particle sizes, from smallest (top) to largest (bottom). (b) Measured diffusion coefficients at t = 10−4 s normalized by dilute values versus cytoplasm density.
The simulated diffusion coefficients in Fig. 6 are time-dependent. There is a crossover in diffusion rate between 10−7 s and 10−6 s, corresponding roughly to the diffusion time for cytoplasm particles across a ribosome, the largest species in the model. At the longest times available, the rate of change of diffusion for most species has slowed but not stopped, leaving it unclear whether the remaining time dependence is a crossover between two regimes of normal (if slowed) diffusion, or if the asymptotic long-time behavior is that of anomalous subdiffusion. For the smallest species, a distinct kink starting above 10−5 s is a finite size effect, due to the limited box size of the simulation. Since the box volume was selected to roughly match E. coli cytoplasm volume at 1 fl, this is showing the beginning of equilibration across the entire cell.
The hindrance to diffusion at a fixed time as a function of crowding is displayed in Fig. 6 b. At low levels of crowding, the diffusion approaches the aqueous level, as expected, reflecting the validity of the movement model in this regime. At higher levels of crowding, however, mobility becomes time-dependent, with longer times corresponding to effectively lower diffusion rates, thereby displaying a type of anomalous diffusion.
We next examined the effect of crowding on the barnase system, a well-characterized, diffusion-limited biochemical association reaction. In this reaction, barnase is known to associate rapidly and tightly with its inhibitor, barstar. The survival probability curves for free barnase in a system with equal levels of barnase and barstar at three levels of crowding are shown in Fig. 7. Crowding had biphasic impact on barnase survival. At early times, the reaction was accelerated, and barnase survival in the crowded systems was below that of the dilute system. At later times, the crowded survival curves were above the dilute curve, showing the impact of hindered diffusion upon the ability of a barnase to find a matching barstar. As demonstrated by this simulation, the impact of crowding can therefore be to either increase or decrease reaction rate, even in the same reaction, and in a time-dependent manner, and thus introduces a level of complexity that is difficult to account for without explicit simulation.
FIGURE 7.

Barnase-barstar dimerization in a crowded environment. Barnase survival probability versus time at 0%, 30%, and 50% added inert cytoplasmic occupied volume fraction.
DISCUSSION
The model
There are a variety of spatially sensitive biochemical processes whose dynamic behavior cannot be easily captured at the atomic scale of molecular dynamics nor by the macroscopic description of mass-action kinetics afforded by continuous approaches. The biochemical impact of macromolecular crowding is one of several issues that illustrate the paucity of understanding of this biomolecular “no man's land”. Unlike other particle-based simulation approaches (42,43), we have accounted for these important volumetric effects in this model by assigning molecules their physical radius.
Before comparing our model with the underlying microscopic model, it is worth discussing its limits of applicability. Equations 1 and 2 of our reaction-diffusion model imply that the particles engage in free diffusion, without intermolecular forces, until they approach closely enough to define a collision. At sufficiently short length scales, this viewpoint is obviously invalid: many macromolecules have charges that exert electrostatic forces on each other, and the free diffusion equation applied here includes no electrostatics. Although in a vacuum electrostatic forces are long-range power laws, in physiological media these forces are muted by counterions from dissolved salts. The question then becomes what length scales are relevant to what type of model. Debye-Hückel theory gives the potential around a charge q as
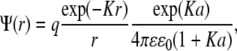 |
(11) |
where ɛ is the dielectric constant of the media, ɛ0 is the permittivity of the vacuum, a is the closest approach distance of the ion and the counterions, and 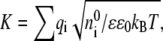 the reciprocal of the Debye length, is defined in terms of the number density of the counterions,
the reciprocal of the Debye length, is defined in terms of the number density of the counterions, 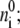 their charges, qi; Boltzmann's constant, kB; and the temperature, T (63). Bare ions are exponentially screened with a length scale 1/K, which sets a limit on the distance over which electrostatic forces can act. At a typical physiological salt concentration of 150 mM, the Debye length is 0.8 nm. Setting four Debye lengths as a maximum, we conclude that local electrostatics will exert effects only inside∼3 nm. On longer spatial scales, free diffusion is the appropriate description. This is not to say that electrostatics has no effect on the reactions. On the contrary, on short scales, electrostatic effects can steer proteins into appropriate alignment (64), and are ultimately responsible for the eventual binding. This guiding and alignment are accounted for in the experimentally measured binding constants and therefore in reaction rate constants derived from them.
their charges, qi; Boltzmann's constant, kB; and the temperature, T (63). Bare ions are exponentially screened with a length scale 1/K, which sets a limit on the distance over which electrostatic forces can act. At a typical physiological salt concentration of 150 mM, the Debye length is 0.8 nm. Setting four Debye lengths as a maximum, we conclude that local electrostatics will exert effects only inside∼3 nm. On longer spatial scales, free diffusion is the appropriate description. This is not to say that electrostatics has no effect on the reactions. On the contrary, on short scales, electrostatic effects can steer proteins into appropriate alignment (64), and are ultimately responsible for the eventual binding. This guiding and alignment are accounted for in the experimentally measured binding constants and therefore in reaction rate constants derived from them.
It is evident from Fig. 4 that the model can accurately represent bimolecular reactions over a wide range of reaction rates, in systems ranging from strongly diffusion-limited to completely reaction-limited. We note, however, that the convergence to continuum results is limited, with a quantitative discrepancy at the largest move lengths with the highest reaction probability. As Δx decreases, of course, the results approach the continuous reaction-diffusion model. The demand for numerical precision must therefore be balanced with simulation speed.
Reversible reactions represent a particular challenge for hard-sphere reaction models and are worth some discussion. When a bimolecular reaction A + B ↔ C is reversible, the associated complex C can dissociate into a pair, which may either recombine with each other (geminate recombination) or escape and combine with other particles. This process, called diffusion-influenced dissociation or diffusion-influenced reversible reaction, has attracted a considerable degree of theoretical attention (56,65–67). The most precisely measured experimental systems are in fluorescence quenching and excited-state proton transfer reactions (57,68), though biological implications beyond the obvious impact on reversible enzyme catalysis have also been explored (69,70). In principle, full analysis of the problem requires the solution of a complex coupled N-body reaction-diffusion system. The best available theories are able to make predictions of the resulting reaction kinetics only for systems satisfying three conditions: 1), dilute A particles, 2), noninteracting B particles, and 3), either the A or the B particles held stationary (called the target and trap approximations, respectively). In the standard Smoluchowski approach to irreversible reactions, a change of variables is used to fix all A particles at the origin, resulting in the B particles moving with a combined diffusion constant, D = DA + DB. This, however, induces correlation into the relative motions of all of the B particles, which, for reversible reactions, affects the analysis of the dynamics after the dissociation of the complex. The problem with both species diffusing in the irreversible case has received some attention (71,72), but we consider only the target problem described above.
At steady state, a reversible diffusion-influenced reaction will approach the equilibrium value given by
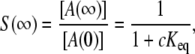 |
(12) |
where c = [B] ≫ [A] and Keq = ka/kd, where ka and kd are the intrinsic rate constants for association and dissociation. Note that this is the same equilibrium value that would be reached in a well-stirred system, the diffusion-influenced aspect of the dynamics being the dynamics of the relaxation to that equilibrium value. At long times, nearing equilibrium, the relaxation is known to ultimately obey a power law ∼t−3/2 (66). When our simulation results are compared with well-stirred mass-action chemical kinetics (Fig. 5), it is clear that mass action fails dramatically. It approaches the correct asymptotic value not as a power law, but exponentially, deviating by orders of magnitude from the simulated system. The best available approximate theory, on the other hand, tracks the simulation precisely within noise limits (Fig. 5 and (66)). The discrepancy between mass action and the actual diffusion-influenced system demonstrates the importance of incorporating diffusion effects, either through theory or simulation.
The effects of crowding on diffusive transport
The potential effects of a crowded intracellular environment on cellular processes are multifactorial and far from understood. They may include impediments to diffusion such as elevated viscosity, nonspecific intermolecular interactions, and volumetric effects associated with space occupancy. Realistically, the model described here is, for the moment, limited to volumetric effects; however, as discussed below, even these are not obvious.
Muramatsu and Minton introduced a version of scaled particle theory (SPT) as an approach for examining the volumetric effects of crowding on diffusion that was later modified and extended by Han and Herzfeld (73–75). The decrease of the diffusion coefficient as a function of crowding in Fig. 6 b is roughly exponential, in accordance with the predictions of SPT. It is worthy of note that large complexes such as ribosomes become almost stationary at higher levels of crowding. However, attempts to fit SPT to the simulation data presented do not quantitatively match the dependence on crowding (data not shown). At first sight, this is surprising, considering that SPT models involve molecular movement that is the same as in the model described here (fixed moves that may succeed or fail). However, SPT differs from this model by assuming that all attempts at movement are independent. In fact, failure of a particle to move reveals information about the location of other nearby particles with the consequence that successive movement attempts become correlated. Indeed, without such correlations, diffusion would be constant in time, in contradiction to the results shown in Fig. 6 a, as well as numerous experiments (18,76,77). In conclusion, there are risks to applying SPT to systems exhibiting anomalous diffusion that do not apply to this model.
The ability to simulate diffusion in a crowded, cytosol-like environment provides some insight into protein mobility in vivo. It has been reported that the diffusion of GFP expressed in E. coli ranges from 6.1 to 7.7 μm2 s−1, ∼10-fold slower than its measured in vitro value of 87 μm2 s−1 (16,78). The volumetric contribution to retardation as a result of crowding can be estimated using our model. Although estimates differ as to the level of excluded volume in E. coli cytoplasm, the value 34% lies central to the range (10). A visual rendering of the simulated cytosolic environment is shown in Fig. 2. At this level, the diffusion of a particle with the approximate mass of GFP (29 kDa vs. 27 kDa for GFP) is reduced only by a factor of 2, from 84 μm2 s−1 to 39 μm2 s−1 at t = 10−4 s. There are five possible explanations for this discrepancy: 1), underestimation of in vivo excluded volume; 2), a high intracellular viscosity; 3), nonspecific binding to other cytoplasmic constituents; 4), GFP dimerization; and, finally, 5), caging or confinement. Despite the difficulty of estimating excluded volume in vivo, from Fig. 6 b it is clear that the volume packing would need to be >50% to achieve 10-fold reduction in diffusion, a value that is not consistent with the range of experimental estimates available. In vivo viscosity has been measured via multiple methods, with the general consensus being that cytoplasmic viscosity is near aqueous, with a value around 1 cP, and certainly <2 cP (1,19). Although nonspecific binding has been shown to have minimal impact on the hindrance of the small globular protein BCECF in vivo (19), GFP has a hydrophobic patch that could potentially interact nonspecifically with other cellular substituents. This hydrophobic patch also allows GFP to dimerize, with Km ∼ 100 μM (78). If the dimerization state differs in vivo from the in vitro reference, diffusion attributed to the monomeric state will be reduced. Finally, caging effects that are known to occur in gels may be present. We note that biopolymers capable of gel formation, such as DNA and mRNA, constitute <4% of dry E. coli cell mass and therefore contribute little to volumetric effects. Nonetheless, it is known that gel states can be created at extremely low occupied volume. Although this simulation alone does not provide sufficient evidence to distinguish among these potential effects and their combinations, it does make clear that the volumetric component of crowding alone is insufficient to account for magnitude of this effect. A similar conclusion was reached by Konopka et al. based on comparison of the SPT crowding model with experimental hindrance measurements at varying levels of hyperosmolality (78). Furthermore, these poorly defined facets of cytoplasmic architecture illustrate the range of biophysical considerations that could ultimately be incorporated into models such as this.
The impact of crowding on a simple diffusion-limited reaction
As an illustration of the impact of crowding on a realistic biochemical reaction, we chose to study the barnase-barstar system. The extracellular bacterial ribonuclease barnase is highly cytotoxic, and must be inhibited within the cell by dimerization with barstar. Due to the high toxicity level of unbound barnase, the binding between barstar and barnase is both fast and tight, thereby minimizing the period during which barnase could act on the cell's own RNA. The barnase-barstar system has thus become an exemplar of a diffusion-limited protein dimerization reaction and has attracted substantial attention due to the impact of local electrostatic steering on the final stages of contact and binding (79). Although local electrostatic forces contradict the pure diffusion with hard-sphere repulsion model applied here, the effect is relatively short range, as a typical Debye length for electrostatic screening in the cytoplasm is 1 nm, and the net effect of a local interaction potential on a diffusion-influenced reaction could in any case be accounted for through a rescaling of the contact radius (56). More important, the hindrance due to crowding in the cell will affect the speed with which barnase can be inactivated, and acts at a longer range than any local electrostatic effects.
In all cases, barnase will eventually bind to barstar (irreversibly on the timescale of the simulation). Therefore, crowding will only affect the rate at which association occurs. At early times, crowding increases the rate of association (Fig. 7). This is an excluded-volume effect, in which the activity (effective local concentration) of the reactants has been increased by the crowders. At later times, however, the association rate actually decreases as the diffusion-limited nature of the reaction takes hold and it becomes more difficult for partners to find one another. Thus crowding results in antagonistic effects whose net impact depends on the degree of diffusion limitation and the timescale examined.
These results may be surprising from some perspectives. The impact of macromolecular crowding is often consigned to be merely an excluded-volume effect, with the increase of activity of the molecular players increasing equilibrium constants and speeding reactions. Although this effect is valid, it is not dominant in all reactions. For systems where transport is important (diffusion-limited enzyme catalysis, spatial effects in signal transduction) the hindrance due to alterations in cytoplasmic geometry can cause a substantial decrease in reaction rates as reduced diffusion becomes relevant. This effect is in accordance with predictions (73), but is now demonstrated in simulation by these results.
Even in this specific example, not only the rate but also the kinetics is altered. Diffusion-limited systems cannot be described by a rate-coefficient alone, as the spatial correlations induced by diffusion limitation result in nonmass-action kinetics. This effect is altered by the changes induced by crowding agents, and the system no longer can be described by the approximate theory applied to the dilute system in Fig. 5.
The in vivo implications of these results are interesting. Although the situation modeled is not directly representative of in vivo levels, either in terms of absolute concentrations or details of initial conditions, it does provide insight into how macromolecular crowding affects transport-driven processes. Biologically, the relevant criterion is how many RNA strands are cut by a newly translated barnase before it meets and is inactivated by a barstar. The impact of crowding on explored volumes shown in Fig. 6 and on reaction rates on the case simulated in Fig. 7 demonstrates that the effective hindrance due to crowding is more pronounced at longer distances and longer timescales. It should therefore be no surprise that barnase and barstar production are regulated separately, which allows the organism to adjust barstar levels to ensure that a barstar is sufficiently close to a barnase to prevent excessive cellular damage.
In a broader sense, these simulations show the difficulties faced by simple theories of the spatial effects of protein-protein interactions. Many reactions approach the diffusion limit, but that limit is affected by the crowded intracellular environment. The complex time dependence of diffusion-limited reactions does have well-studied theoretical approximations that are effective in certain limiting cases (target approximation, dilute systems, uncrowded systems), but these cases are far from typical conditions for the cell. Simulations are therefore a necessary tool for studying the impact of cytoplasmic environment on transport-limited biological reaction processes.
CONCLUSION
It is becoming clearer that spatially distributed particle-based models have a role to play in elucidating cellular behavior at scales that are for the moment beyond the computational capability of molecular dynamics, and lie outside of the conceptual context of continuum-based models (24,42–44,80,81). They do so by sacrificing detail at the atomic level in favor of both endurance and population complexity. This model, for example, is able to simulate complex populations of several million molecules (the biomolecular complement of a simple cell) for up to 10−4 s of real-time duration, a system 10 orders of magnitude or more beyond atomic-scale molecular dynamics.
A feature that is unique to this model, compared with others of its type, is the assignment of physical size to its particle components. This feature has allowed us to explore the volumetric consequences on molecular diffusion and reaction of a simulated approximation of the E. coli cytosol. Under these conditions, diffusion becomes limiting, with significant consequences for biochemical reactions. Moreover, the impact of crowding is anomalous, whereby diffusion and reaction coefficients lose their time-constant nature, an effect that increases disproportionately with increased molecular mass and decreased copy number. One obvious application of this simulation approach, therefore, is in the calibration of existing in vitro experimental measurements to more accurately reflect the crowded conditions of the cytoplasm.
The inability to fully account for the reduced in vivo diffusion rate of GFP by simulation illustrates that excluded volume is only one of several contributors to a potentially complicated mix of biophysical factors. This point emphasizes the value of simulation in defining the magnitude and complexity of natural phenomena in terms of what is known and what remains to be known.
The mathematical simplicity of the particle-based approach means that its application to large and complex physiological systems is limited by computational capacity alone. Continued adherence to Moore's Law would translate into incremental advances in model sophistication and capability. Conversely, major advancements in hardware performance would have an immediate and dramatic impact on model evolution, combining greater molecular detail at the lower scale with the capability of handling larger and longer problems at the higher scale. If the computational challenges of particle-based modeling can be overcome, then the approach provides an accessible and flexible strategy for the construction of spatially dynamic networks that can be moved seamlessly from the stochastic behavior of small biomolecular populations to the continuous behavior of large biomolecular populations. Time will tell.
Acknowledgments
The support of the Canada Foundation for Innovation and IBM Canada is gratefully acknowledged, as is helpful discussion with Peter Tieleman.
Douglas Ridgway and Gordon Broderick contributed equally to this work.
Editor: Gregory A. Voth.
References
- 1.Luby-Phelps, K. 2000. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 192:189–221. [DOI] [PubMed] [Google Scholar]
- 2.Bhalla, U. S. 2004. Signaling in small subcellular volumes. I. Stochastic and diffusion effects on individual pathways. Biophys. J. 87:733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maly, I. V., H. S. Wiley, and D. A. Lauffenburger. 2004. Self-organization of polarized cell signaling via autocrine circuits: computational model analysis. Biophys. J. 86:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wylie, D. C., Y. Hori, A. R. Dinner, and A. K. Chakraborty. 2006. A hybrid deterministic-stochastic algorithm for modeling cell signaling dynamics in spatially inhomogeneous environments and under the influence of external fields. J Phys Chem B. 110:12749–12765. [DOI] [PubMed] [Google Scholar]
- 5.Mayawala, K., D. G. Vlachos, and J. S. Edwards. 2006. Spatial modeling of dimerization reaction dynamics in the plasma membrane: Monte Carlo vs. continuum differential equations. Biophys. Chem. 121:194–208. [DOI] [PubMed] [Google Scholar]
- 6.Kholodenko, B. N. 2006. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis, R. J. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597–604. [DOI] [PubMed] [Google Scholar]
- 8.Minton, A. P. 2006. How can biochemical reactions within cells differ from those in test tubes? J. Cell Sci. 119:2863–2869. [DOI] [PubMed] [Google Scholar]
- 9.Minton, A. P. 2001. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276:10577–10580. [DOI] [PubMed] [Google Scholar]
- 10.Zimmerman, S. B., and S. O. Trach. 1991. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 222:599–620. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi, K., S. N. Arjunan, and M. Tomita. 2005. Space in systems biology of signaling pathways: towards intracellular molecular crowding in silico. FEBS Lett. 579:1783–1788. [DOI] [PubMed] [Google Scholar]
- 12.Hall, D. 2006. Protein self-association in the cell: a mechanism for fine tuning the level of macromolecular crowding? Eur. Biophys. J. 35:276–280. [DOI] [PubMed] [Google Scholar]
- 13.Kozer, N., and G. Schreiber. 2004. Effect of crowding on protein-protein association rates: fundamental differences between low and high mass crowding agents. J. Mol. Biol. 336:763–774. [DOI] [PubMed] [Google Scholar]
- 14.Ovadi, J., and P. A. Srere. 2000. Macromolecular compartmentation and channeling. Int. Rev. Cytol. 192:255–279. [DOI] [PubMed] [Google Scholar]
- 15.Walter, H., and D. E. Brooks. 1995. Phase separation in cytoplasm, due to macromolecular crowding, is the basis for microcompartmentation. FEBS Lett. 361:135–139. [DOI] [PubMed] [Google Scholar]
- 16.Elowitz, M. B., M. G. Surette, P. E. Wolf, J. B. Stock, and S. Leibler. 1999. Protein mobility in the cytoplasm of Escherichia coli. J. Bacteriol. 181:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dauty, E., and A. S. Verkman. 2004. Molecular crowding reduces to a similar extent the diffusion of small solutes and macromolecules: measurement by fluorescence correlation spectroscopy. J. Mol. Recognit. 17:441–447. [DOI] [PubMed] [Google Scholar]
- 18.Banks, D. S., and C. Fradin. 2005. Anomalous diffusion of proteins due to molecular crowding. Biophys. J. 89:2960–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verkman, A. S. 2002. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 27:27–33. [DOI] [PubMed] [Google Scholar]
- 20.Wehner, F., H. Olsen, H. Tinel, E. Kinne-Saffran, and R. Kinne. 2003. Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev. Physiol. Biochem. Pharmacol. 148:1–80. [DOI] [PubMed] [Google Scholar]
- 21.Clark, J. I., and J. M. Clark. 2000. Lens cytoplasmic phase separation. Int. Rev. Cytol. 192:171–187. [DOI] [PubMed] [Google Scholar]
- 22.Bloemendal, H., W. de Jong, R. Jaenicke, N. H. Lubsen, C. Slingsby, and A. Tardieu. 2004. Ageing and vision: structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol. 86:407–485. [DOI] [PubMed] [Google Scholar]
- 23.Schnell, S., and T. E. Turner. 2004. Reaction kinetics in intracellular environments with macromolecular crowding: simulations and rate laws. Prog. Biophys. Mol. Biol. 85:235–260. [DOI] [PubMed] [Google Scholar]
- 24.Grima, R., and S. Schnell. 2006. A systematic investigation of the rate laws valid in intracellular environments. Biophys. Chem. 124:1–10. [DOI] [PubMed] [Google Scholar]
- 25.Dzubiella, J., and J. A. McCammon. 2005. Substrate concentration dependence of the diffusion-controlled steady-state rate constant. Chem. Phys. 122:1–7. [DOI] [PubMed] [Google Scholar]
- 26.Tucci, K., and R. Kapral. 2004. Mesoscopic model for diffusion-influenced reaction dynamics. J. Chem. Phys. 120:8262–8270. [DOI] [PubMed] [Google Scholar]
- 27.Eide, J. L., and A. K. Chakraborty. 2006. Effects of quenched and annealed macromolecular crowding elements on a simple model for signaling in T lymphocytes. J Phys Chem B. 110:2318–2324. [DOI] [PubMed] [Google Scholar]
- 28.Lipkow, K., S. S. Andrews, and D. Bray. 2005. Simulated diffusion of phosphorylated CheY through the cytoplasm of Escherichia coli. J. Bacteriol. 187:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrews, S. S., and A. P. Arkin. 2006. Simulating cell biology. Curr. Biol. 16:R523–R527. [DOI] [PubMed] [Google Scholar]
- 30.Ridgway, D., G. Broderick, and M. J. Ellison. 2006. Accommodating space, time and randomness in network simulation. Curr. Opin. Biotechnol. 17:493–498. [DOI] [PubMed] [Google Scholar]
- 31.Lemerle, C., B. Di Ventura, and L. Serrano. 2005. Space as the final frontier in stochastic simulations of biological systems. FEBS Lett. 579:1789–1794. [DOI] [PubMed] [Google Scholar]
- 32.Bork, P., and L. Serrano. 2005. Towards cellular systems in 4D. Cell. 121:507–509. [DOI] [PubMed] [Google Scholar]
- 33.Slepchenko, B. M., J. C. Schaff, J. H. Carson, and L. M. Loew. 2002. Computational cell biology: spatiotemporal simulation of cellular events. Annu. Rev. Biophys. Biomol. Struct. 31:423–441. [DOI] [PubMed] [Google Scholar]
- 34.Slepchenko, B. M., J. C. Schaff, I. Macara, and L. M. Loew. 2003. Quantitative cell biology with the virtual cell. Trends Cell Biol. 13:570–576. [DOI] [PubMed] [Google Scholar]
- 35.Francke, C., P. W. Postma, H. V. Westerhoff, J. G. Blom, and M. A. Peletier. 2003. Why the phosphotransferase system of Escherichia coli escapes diffusion limitation. Biophys. J. 85:612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wishart, D. S., R. Yang, D. Arndt, P. Tang, and J. Cruz. 2005. Dynamic cellular automata: an alternative approach to cellular simulation. In Silico Biol. 5:139–161. [PubMed] [Google Scholar]
- 37.Weimar, J. R. 2002. Cellular automata approaches to enzymatic reaction networks. In Cellular Automata. S. Bandini, B. Chopard, and M. Tomassini, editors. Springer, Berlin. 294–303.
- 38.Hattne, J., D. Fange, and J. Elf. 2005. Stochastic reaction-diffusion simulation with MesoRD. Bioinformatics. 21:2923–2924. [DOI] [PubMed] [Google Scholar]
- 39.Ander, M., P. Beltrao, B. Di Ventura, J. Ferkinghoff-Borg, M. Foglierini, A. Kaplan, C. Lemerle, I. Thomas-Oliveira, and L. Serrano. 2004. SmartCell, a framework to simulate cellular processes that combines stochastic approximation with diffusion and localisation: analysis of simple networks. Syst. Biol. 1:129–138. [DOI] [PubMed] [Google Scholar]
- 40.Vidal Rodriguez, J., J. A. Kaandorp, M. Dobrzynski, and J. G. Blom. 2006. Spatial stochastic modelling of the phosphoenolpyruvate-dependent phosphotransferase (PTS) pathway in Escherichia coli. Bioinformatics. 22:1895–1901. [DOI] [PubMed] [Google Scholar]
- 41.Lee, K. H., A. R. Dinner, C. Tu, G. Campi, S. Raychaudhuri, R. Varma, T. N. Sims, W. R. Burack, H. Wu, J. Wang, O. Kanagawa, M. Markiewicz, P. M. Allen, M. L. Dustin, A. K. Chakraborty, and A. S. Shaw. 2003. The immunological synapse balances T cell receptor signaling and degradation. Science. 302:1218–1222. [DOI] [PubMed] [Google Scholar]
- 42.Stiles, J. R., and T. M. Bartol. 2001. Monte Carlo methods for simulating realistic synaptic microphysiology using MCell. In Computational Neuroscience. E. De Schutter, editor. CRC Press, Boca Raton, FL. 87–127.
- 43.Andrews, S. S., and D. Bray. 2004. Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys. Biol. 1:137–151. [DOI] [PubMed] [Google Scholar]
- 44.van Zon, J. S., and P. R. Ten Wolde. 2005. Green's-function reaction dynamics: A particle-based approach for simulating biochemical networks in time and space. J. Chem. Phys. 123:1–16. [DOI] [PubMed] [Google Scholar]
- 45.Marrink, S. J., H. J. Risselada, S. Yefimov, D. P. Tieleman, and A. H. deVries. 2007. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111:7812–7824. [DOI] [PubMed] [Google Scholar]
- 46.Nielsen, S. O., C. F. Lopez, G. Srinivas, and M. L. Klein. 2004. Coarse grain models and the computer simulation of soft materials. J. Phys. Condens. Matter. 16:R481–R512. [Google Scholar]
- 47.Lopez, C. F., P. B. Moore, J. C. Shelley, M. Y. Shelley, and M. L. Klein. 2002. Computer simulation studies of biomembranes using a coarse grain model. Comput. Phys. Commun. 147:1–6. [Google Scholar]
- 48.Baoukina, S., L. Monticelli, M. Amrein, and D. P. Tieleman. 2007. The molecular mechanism of monolayer-bilayer transformations of lung surfactant from molecular dynamics simulations. Biophys. J. 93:3775–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venturoli, M., M. Maddalena Sperotto, M. Kranenburg, and B. Smit. 2006. Mesoscopic models of biological membranes. Phys. Rep. 437:1–54. [Google Scholar]
- 50.Srinivas, G., D. E. Discher, and M. L. Klein. 2004. Self-assembly and properties of diblock copolymers by coarse-grain molecular dynamics. Nat. Mater. 3:638–644. [DOI] [PubMed] [Google Scholar]
- 51.Balazs, A. C. 2007. Modeling self-assembly and phase behavior in complex mixtures. Annu. Rev. Phys. Chem. 58:211–233. [DOI] [PubMed] [Google Scholar]
- 52.Liwo, A., M. Khalili, and H. A. Scheraga. 2005. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc. Natl. Acad. Sci. USA. 102:2362–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tozzini, V. 2005. Coarse-grained models for proteins. Curr. Opin. Struct. Biol. 15:144–150. [DOI] [PubMed] [Google Scholar]
- 54.Collins, F. S., and G. E. Kimball. 1949. Diffusion-controlled reaction rates. J. Colloid Sci. 4:425–437. [Google Scholar]
- 55.Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444–456. [DOI] [PubMed] [Google Scholar]
- 56.Agmon, N., and A. Szabo. 1990. Theory of reversible diffusion-influenced reactions. J. Chem. Phys. 92:5270–5284. [Google Scholar]
- 57.Popov, A. V., and N. Agmon. 2001. Three-dimensional simulations of reversible bimolecular reactions: the simple target problem. J. Chem. Phys. 115:8921–8932. [Google Scholar]
- 58.Spaar, A., C. Dammer, R. R. Gabdoulline, R. C. Wade, and V. Helms. 2006. Diffusional encounter of barnase and barstar. Biophys. J. 90:1913–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lopez-Campistrous, A., P. Semchuk, L. Burke, T. Palmer-Stone, S. J. Brokx, G. Broderick, D. Bottorff, S. Bolch, J. H. Weiner, and M. J. Ellison. 2005. Localization, annotation, and comparison of the Escherichia coli K-12 proteome under two states of growth. Mol. Cell Proteomics. 4:1205–1209. [DOI] [PubMed] [Google Scholar]
- 60.Sundararaj, S., A. Guo, B. Habibi-Nazhad, M. Rouani, P. Stothard, M. Ellison, and D. S. Wishart. 2004. The CyberCell Database (CCDB): a comprehensive, self-updating, relational database to coordinate and facilitate in silico modeling of Escherichia coli. Nucleic Acids Res. 32:293–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Apweiler, R., A. Bairoch, C. H. Wu, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H. Huang, R. Lopez, M. Magrane, M. J. Martin, D. A. Natale, C. O'Donovan, N. Redaschi, and L.-S. L. Yeh. 2004. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32:D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yun, H. S., J. Hong, and H. C. Lim. 1991. Analysis of functioning ribosome content in Escherichia coli at different growth rates. Biotechnol. Tech. 5:471–474. [Google Scholar]
- 63.Daun, M. 1999. Molecular Biophysics: Structures in Motion. Oxford University Press, Oxford, UK.
- 64.Gabdoulline, R. R., and R. C. Wade. 2001. Protein-protein association: investigation of factors influencing association rates by Brownian dynamics simulations. J. Mol. Biol. 306:1139–1155. [DOI] [PubMed] [Google Scholar]
- 65.Berg, O. G. 1978. On diffusion-controlled dissociation. Chem. Phys. 31:47–57. [Google Scholar]
- 66.Popov, A. V., and A. I. Burshtein. 2003. Theories of reversible dissociation: A comparative study. J. Phys. Chem. A. 107:9688–9694. [Google Scholar]
- 67.Burshtein, A. I. 2000. Unified theory of photochemical charge separation. Adv. Chem. Phys. 114:419–587. [Google Scholar]
- 68.Agmon, N. 2005. Elementary steps in excited-state proton transfer. J. Phys. Chem. A. 109:13–35. [DOI] [PubMed] [Google Scholar]
- 69.Agmon, N., and A. L. Edelstein. 1997. Collective binding properties of receptor arrays. Biophys. J. 72:1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shvartsman, S. Y., H. S. Wiley, W. M. Deen, and D. A. Lauffenburger. 2001. Spatial range of autocrine signaling: modeling and computational analysis. Biophys. J. 81:1854–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blumen, A., G. Zumofen, and J. Klafter. 1984. Target annihilation by random walkers. Phys. Rev. B. 30:5379–5382. [Google Scholar]
- 72.Szabo, A., R. Zwanzig, and N. Agmon. 1988. Diffusion-controlled reactions with mobile traps. Phys. Rev. Lett. 61:2496–2499. [DOI] [PubMed] [Google Scholar]
- 73.Zimmerman, S. B., and A. P. Minton. 1993. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 22:27–65. [DOI] [PubMed] [Google Scholar]
- 74.Muramatsu, N., and A. P. Minton. 1988. Tracer diffusion of globular proteins in concentrated protein solutions. Proc. Natl. Acad. Sci. USA. 85:2984–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han, J., and J. Herzfeld. 1993. Macromolecular diffusion in crowded solutions. Biophys. J. 65:1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weiss, M., M. Elsner, F. Kartberg, and T. Nilsson. 2004. Anomalous subdiffusion is a measure for cytoplasmic crowding in living cells. Biophys. J. 87:3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Golding, I., and E. C. Cox. 2006. Physical nature of bacterial cytoplasm. Phys. Rev. Lett. 96:098102–098104. [DOI] [PubMed] [Google Scholar]
- 78.Konopka, M. C., I. A. Shkel, S. Cayley, M. T. Record, and J. C. Weisshaar. 2006. Crowding and confinement effects on protein diffusion in vivo. J. Bacteriol. 188:6115–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gabdoulline, R. R., and R. C. Wade. 2002. Biomolecular diffusional association. Curr. Opin. Struct. Biol. 12:204–213. [DOI] [PubMed] [Google Scholar]
- 80.Materi, W., and D. S. Wishart. 2007. Computational systems biology in drug discovery and development: methods and applications. Drug Discov. Today. 12:295–303. [DOI] [PubMed] [Google Scholar]
- 81.Broderick, G., M. Ru'aini, E. Chan, and M. J. Ellison. 2004. A life-like virtual cell membrane using discrete automata. In Silico Biol. 5:163–178. [PubMed] [Google Scholar]