

Abstract
The distribution and movement of elemental ions in biologic tissues is critical for many cellular processes. In contrast to chemical techniques for imaging the intracellular distribution of ions, however, techniques for imaging the distribution of ions across tissues are not well developed. We used time-of-flight secondary ion mass spectrometry (TOF-SIMS) to obtain nonlabeled high-resolution analytic images of ion distribution in ischemic retinal tissues. Marked changes in Ca2+ distribution, compared with other fundamental ions, such as Na+, K+, and Mg2+, were detected during the progression of ischemia. Furthermore, the Ca2+ redistribution pattern correlated closely with TUNEL-positive (positive for terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick end-labeling) cell death in ischemic retinas. After treatment with a calcium chelator, Ca2+ ion redistribution was delayed, resulting in a decrease in TUNEL-positive cells. These results indicate that ischemia-induced Ca2+ redistribution within retinal tissues is associated with the order of apoptotic cell death, which possibly explains the different susceptibility of various types of retinal cells to ischemia. Thus, the TOF-SIMS technique provides a tool for the study of intercellular communication by Ca2+ ion movement.
INTRODUCTION
Several biologic disorders result from or induce abnormal levels of essential ions (1,2), and monitoring these ions can assist in the diagnosis and prevention of disorders. There are several available methodologies to measure both free and bound cations in cells, including fluorescent indicators, radiographic crystallography, and radiographic microanalysis. However, there are currently several technical problems in the direct detection of ion movement in tissues. In this investigation, we used nonlabeling time-of-flight secondary ion mass spectrometry (TOF-SIMS) to obtain high-resolution analytic images of ion distribution in ischemic mice retinal tissues.
TOF-SIMS, a highly sensitive surface analysis technique originally developed for material science, is presently applied to the imaging of biomolecules (3–6). TOF-SIMS has several advantages over conventional imaging techniques. No treatment is necessary to acquire ion images, which allows the integrity of biologic samples to be preserved. Thus, nonlabeled tissue samples are detected without fixatives, markers, or stains. Additionally, with TOF-SIMS technology, all molecular ions as well as elemental ions can be detected simultaneously with high sensitivity and excellent spatial resolution. Until recently, in vivo or ex vivo ion imaging of biologic samples mostly using chemical imaging techniques has been reported, but this approach has significant limitations in the context of complex multicellular organs because of the inadequate stability of fluorescent dye and because only a single ion can be detected at a time. Especially with fluorescent probes, it is critical to be aware of the specificity and relative selectivity of a probe for the targeted ion (7). For example, although Fura-2 is best known as Ca2+ indicator, its use is actually complicated by competitive binding of other cations, such as the Zn2+ ion (8,9). Moreover, properties such as the selectivity and sensitivity of a metallofluorescent probe are influenced by factors in the experimental environment. Therefore, more selective probes are needed to define the targeted ion signaling. Because TOF-SIMS detects ions using the intrinsic property of molecular mass, the information obtained from TOF-SIMS could be excluded from consideration because of specificity. We used TOF-SIMS imaging on ischemia-induced mice retina tissues to directly evaluate changes in ion distribution in ischemia conditions.
The retina is composed of three layers of nerve cell bodies—ganglion cell layer (GCL), inner nuclear layer (INL), and outer nuclear layer (ONL)—and two layers of synapses—inner plexiform layer (IPL) and outer plexiform layer (OPL)—so that six different types of cells contact one another; thus, it serves as a good model for intercellular communication between various neuronal cells. Retinal ischemia is a common cause of visual impairment and blindness (10,11). Ischemia affects all cell types in the retina, and leads to loss of cells within retinal tissue. Mammalian retinal ischemia results in irreversible morphologic and functional changes mediated by a number of pathophysiologic processes, including glutamate toxicity and free-radical formation, which ultimately end in damage to retinal neurons and cell death by apoptosis (10,12–15). Neuronal cells in the inner nuclear layer of the retina display significantly enhanced susceptibility to retinal ischemia, compared with outer layer neurons (13,16). However, little is known about the specific molecular pathways involved in the degree of susceptibility of different neurons to ischemia and apoptotic cascade between different neuronal cells in intact retinal tissue.
MATERIALS AND METHODS
Ischemic model
All protocols were approved by the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research. We induced permanent focal cerebral ischemia by occlusion of the common carotid artery with 6-0 Vicryl sutures (Johnson & Johnson, New Brunswick, NJ) in male c57BL mice, following modified ligation procedures (17,18). While the mice were under halothane anesthesia, a small longitudinal incision was made along the midline of neck and the left common carotid artery was exposed. The artery was permanently double-ligated and transected in the middle between two sites. The incision was sutured using 6-0 Vicryl sutures, and the animals were left to recover for 2 h. For calcium chelation, 20 mM of BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-n,n,n′,n′-tetraacetic acid-acetoxymethyl ester] was injected intraocularly before the induction of ischemia.
Tissue preparation
Eyeballs were cut into semi-thin sections and covered with Tissue-Tek O.C.T. Compound (Sakura Finetek Europe, Zoeterwoude, the Netherlands). Serial 8-μm sections were dissected with a cryo-ultramicrotome (HM560, Zeiss, Oberkochen, Germany) and air-dried.
Analysis by time-of-flight secondary ion mass spectrometry
TOF-SIMS was performed using a TOF-SIMS V instrument (ION-TOF GmbH, Münster, Germany) with 25-keV 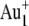 primary ions (average current of 0.94 pA, pulse width of 16.8 ns, repetition rate of 10 kHz) or 25-keV
primary ions (average current of 0.94 pA, pulse width of 16.8 ns, repetition rate of 10 kHz) or 25-keV 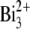 primary ions (average current of 0.2 pA, pulse width of 25.0 ns, repetition rate of 10 kHz). TOF-SIMS images for the retina samples were obtained by randomly rastering the 25-keV
primary ions (average current of 0.2 pA, pulse width of 25.0 ns, repetition rate of 10 kHz). TOF-SIMS images for the retina samples were obtained by randomly rastering the 25-keV 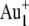 beam with a primary ion influence of 5 × 1012 ions·cm−2 or the
beam with a primary ion influence of 5 × 1012 ions·cm−2 or the 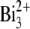 beam with a primary ion influence of 1.5 × 1012 ions·cm−2, in either case over a surface area of ∼300 × 300 μm2. To remove possible surface contamination layers, we presputtered each retina surface for 3 min (
beam with a primary ion influence of 1.5 × 1012 ions·cm−2, in either case over a surface area of ∼300 × 300 μm2. To remove possible surface contamination layers, we presputtered each retina surface for 3 min (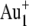 DC current of 18 nA) or 15 s (
DC current of 18 nA) or 15 s (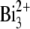 DC current of 10 nA) before image acquisition. The analysis area was charge-compensated by low-energy electron flooding. Mass resolution was usually higher than 6000 at m/z 27 in the positive mode. Positive ion spectra were internally calibrated using
DC current of 10 nA) before image acquisition. The analysis area was charge-compensated by low-energy electron flooding. Mass resolution was usually higher than 6000 at m/z 27 in the positive mode. Positive ion spectra were internally calibrated using 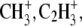 and
and 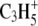 peaks. The average area profiles of Na+, K+, Mg+, and Ca+ images were obtained for the total retina area using IonImage (ION-TOF GmbH) software.
peaks. The average area profiles of Na+, K+, Mg+, and Ca+ images were obtained for the total retina area using IonImage (ION-TOF GmbH) software.
Immunohistochemistry
Terminal deoxynucleotidyl transferase-mediated nick end-labeling (TUNEL) assays using 2′-deoxyuridine 5′-triphosphate were performed using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore, Billerica, MA) and In Situ Cell Death Detection Kit (Roche, Basel, Switzerland). All immunostaining experiments were performed using standard procedures.
Ca2+ imaging
For simultaneously recording the changes in intracellular Ca2+, Neuro2A was loaded with 5 μM fluo-4/AM (Molecular Probes, Invitrogen, Carlsbad, CA) for 40 min at 37°C. Cells were pretreated with 10 μM BAPTA/AM for Ca2+ chelation, then with 10 mM NaN3 in a glucose-free buffer for chemical ischemia. Images were acquired using confocal microscopy (Leica, Wetzlar, Germany).
Western blot analysis
Total protein (30 μg) was extracted from cells immunoblotted with specific antibodies to active caspase-3 (cell signaling), poly(ADP)ribose polymerase (PARP) cell signaling, and α-tubulin (Sigma, St. Louis, MO).
RESULTS
Ion mapping of ischemic retinas by time-of-flight secondary ion mass spectrometry
Normal neuronal membrane processes and ionic homeostasis are markedly affected by the lack of cellular energy that results from ischemic insult. Thus, we assessed ischemia-induced changes in the most abundant cations in eukaryotic cells—specifically, Na+, K+, Mg2+, and Ca2+. These cations are fundamental for multiple cellular processes in every phase of life, including cell growth and differentiation, development, cell-to-cell interactions, morphology, motility, and apoptosis leading to cell death (1,2,19).
Fig. 1 a shows the schematic diagram of an experimental setup and cross-sectional view of the retina, which is composed of three layers of nerve cell bodies (GCL, INL, ONL) and two layers of synapses (IPL, OPL). Normal and ischemic mice retinas were dissected, cryostat-sectioned, and analyzed by TOF-SIMS. Among the various ions, we initially detected Na+, K+, Mg2+, and Ca2+ ions in normal and ischemic mouse retinal samples and profiled their intensities. Intensities are presented in Fig. 1 b by a color scale where bright colors correspond to high, and dark colors correspond to low, intensities. The average area profiles of Na+, K+, Mg+, and Ca2+ images were obtained for the total retina area. In TOF-SIMS studies of normal and ischemic retinas, significant amounts of Na+ and K+ were detected in all layers of retina, so we could not detect any significant differences in the distribution of these ions between normal and ischemic retinas. Singly ionized Mg2+ (labeled as Mg+ in Fig. 1 b) was detected mainly in the nuclear layers, including GCL, INL, and ONL, and no changes in its distribution were evident between normal and ischemic retinas. However, distribution of singly ionized Ca2+ (labeled as “Ca+” in Fig. 1 b) was markedly altered in ischemic retinas. Area profiles obtained through molecular-specific images to compare the relative distribution of each ion across the retinal cell layers show distinct changes in Ca2+ during retinal ischemia (Fig. 1 b).
FIGURE 1.

Experimental setup and ion mapping of mouse ischemia by TOF-SIMS. (a) Schematic drawing of the experimental setup. Cryostat-sectioned mice retinas were air-dried, and then analyzed by TOF-SIMS. (b) TOF-SIMS images of mouse retina sections. (Squares indicate area of retina detected.) Na+ (m/z 22.99), K+ (m/z 38.96), Mg+ (m/z 23.98), and Ca+ (m/z 39.96) ion mapping and their area profiles. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
Ca2+ redistribution leads to retinal cell death
As shown in Fig. 2 a, the Ca2+ ion was mainly located in nuclear layers, including GCL, INL, and ONL in control mice, but was detected specifically in GCL and plexiform layers, including IPL and OPL, which contain axons and dendrites, at 12 h after ischemia induction. Ca2+ ion began to be diffusely dispersed in retinal INL at 24 h after ischemia induction. In 48-h and 72-h post-ischemic retinas, Ca2+ was diffusely distributed without discrimination of layers and its expression decreased with a relatively linear slope from GCL. Calcium ions, which are cellular messengers, mediate many cellular functions. Reports of investigations by other researchers have suggested that various apoptotic stimuli affect the intracellular Ca2+ concentration and that the Ca2+ ion is a major mediator of neuronal cell death (20,21). Accordingly, we assessed neuronal cell death during retinal ischemia using the TUNEL assay, which allowed the detection of controlled DNA cleavage in apoptotic cells. TUNEL-positive cells were not detected in control retinas at any of the timepoints analyzed, whereas a time-dependent increase in TUNEL-positive cells within different retinal cell layers was observed in ischemic retinas (Fig. 2 a). Specifically, the distribution of TUNEL-positive cells progressed from GCL to ONL, which was detected in GCL at the 12-h point, INL at the 24-h point, and in ONL at the 48-h point after ischemia.
FIGURE 2.

Correlation between Ca2+ redistribution and retinal cell death. (a) Changes in Ca2+ ion distribution during retinal ischemia were measured by TOF-SIMS, and their intensities were profiled. Ischemic retinas displayed TUNEL evidence of neuronal death, compared with the sham-operated control retina, in a time-dependent manner. (Squares indicate area of retina detected; dotted lines indicate borders of each layer of retina.) (b) Schematic representation of the relationship between Ca2+ ion movement and TUNEL-positive cell death in retinal ischemia. Ca2+ ion movement induced by ischemia within retinal layers leads to cell death.
Fig. 2 b shows the scheme of the relationship between Ca2+ distribution and retina neuronal cell death. Ca2+ was initially detected in nuclear layers (GCL, INL, and ONL) in normal retina, plexiform layers (IPL, OPL) and GCL at 12 h of ischemia progressed to INL at 24 h of ischemia, and finally diffused into ONL at 48 h and 72 h of ischemia. Ca2+ redistribution with ischemic progression shows that before induction of active apoptosis, dynamic movement of Ca2+ in the tissue is followed by definite neuronal cell death.
Ca2+ signals trigger apoptosis and lead to the activation of calpains and caspases (22,23). Calpains and caspases are cysteine proteases that are important in cell death, and they display crosstalk in the regulation of apoptosis. To confirm the effects of Ca2+ movement on neuronal cell death in ischemic retinas, we determined the expression of Ca2+-activated proapoptotic proteases, caspase-3, and μ-calpain (Fig. 3). Immunoreactivity of active caspase-3 was altered in correlation with the redistribution of Ca2+ detected in ischemic retinas, whereby apoptosis progressed from GCL to INL and ONL. Active caspase-3 was initially detected in the plexiform layers at 12 h, and in the nuclear layers (GCL, INL, and ONL) at 12 h, 24 h, and 48 h, respectively. Furthermore, μ-calpain distribution changed over the course of ischemia, and corresponded to the state of pathology of each retinal layer. Slight immunostaining of μ-calpain was detected in the plexiform layers of normal retina and more intensively in the nuclear layers during ischemia. Therefore, the patterns of caspase-3 and μ-calpain activation correlate positively with results of the TUNEL assay. The possible translocation of caspase-3 and μ-calpain to the nucleus implicates these proteases in apoptotic machinery at the nuclear level (24,25). Translocation of caspase-3 and μ-calpain to nuclear layers of the retina is associated with Ca2+ distribution during ischemia, which may play a role in apoptosis.
FIGURE 3.

Upregulation of active caspase-3 and μ-calpain after retinal ischemia. (a) TUNEL staining and immunoreactivities of (b) active caspase-3 and (c) μ-calpain in mice ischemic retinas. Sections of mice retina were immunostained with antibodies specific for active caspase-3 (green) and μ-calpain (red). Nuclei were stained with propidium iodide (red, b) or Hoechst (blue, c), respectively.
Calcium chelator reduces apoptotic cell death
To assess the biologic meaning of changes in Ca2+ distribution detected by TOF-SIMS, we observed Ca2+ mobilization in vitro (Fig. 4, a–c). Neuro2A, a mouse neuroblastoma cell line, was rendered chemically ischemic by use of mitochondrial inhibitor NaN3, and by the omission of glucose. Fluo-4-labeled Ca2+ fluorescence began to increase after the onset of ischemia, which was blocked by pretreatment with calcium chelator BAPTA-AM (Fig. 4 a). The Neuro2A nucleus displayed a distinctly lower Ca2+ concentration than the perinuclear region and other areas of the cytoplasm. In contrast, after exposure to chemical ischemia conditions, increase in Ca2+ fluorescence was detected in both cytoplasmic and intranuclear regions of the same cell, which was blocked by pretreatment with BAPTA-AM. The changes in intracellular Ca2+ distribution are accompanied by cell death, as shown in Fig. 4 b, further supporting the hypothesis that these alterations occur much earlier than apoptotic cell death. However, Ca2+ chelation with BAPTA-AM for 5 h reduced the number of TUNEL-positive apoptotic cells by ∼56%, compared with untreated control retinas. Furthermore, ischemia-induced caspase-3 activation and PARP cleavage were reduced after pretreatment with BAPTA-AM (Fig. 4 c). This in vitro Ca2+ distribution pattern is similar to Ca2+ movement detected by TOF-SIMS in that Ca2+ ion distribution is altered before the induction of apoptosis.
FIGURE 4.

Calcium chelation with BAPTA-AM suppresses retinal cell death under in vitro and in vivo ischemic conditions. (a) Chemical ischemia induced the intracellular Ca2+ content in fluo-4-loaded Neuro2A cells, which was blocked by pretreatment with the calcium chelator BAPTA-AM (10 μM). (b) The rate of ischemia-mediated TUNEL-positive cell death was reduced by ∼56%, after pretreatment with BAPTA-AM (10 μM), compared with untreated control retinas. *p < 0.05. (c) Ischemia-induced caspase-3 activation and PARP cleavage was reduced upon pretreatment with BAPTA-AM (10 μM). (d) Ischemic retinas treated with 20 mM BAPTA-AM displayed different patterns of Ca+ ion movement from ischemic retinas. Furthermore, Ca2+ chelation with BAPTA-AM reduced the number of TUNEL-positive dead cells, particularly in INL and ONL. (Squares indicate area of retina detected; dotted lines indicate borders of each layer of retina.) (e) Schematic representation showing that in the presence of the calcium chelator, Ca2+ ion redistribution was delayed, and diffusion of Ca2+ into entire layers of retina was not detected under ischemic conditions, which was associated with decreased TUNEL-positive cell death.
With intravitreous injection of BAPTA-AM before the induction of ischemia, Na+, Mg+, and K+ distribution was similar to that in control retinas (data not shown), whereas Ca+ distribution was distinct (Fig. 4 d). Compared with ischemic retinas in the absence of BAPTA-AM, delayed Ca2+ ion movement into plexiform layers was detected in BAPTA-AM-treated retinas, after ischemia induction (24 h of ischemia). Moreover, diffusion of Ca2+ into entire layers was not detected, even at 72 h of ischemia. Thus, Ca2+ chelation with BAPTA-AM led to a reduction in the number of TUNEL-positive cells in INL and ONL. Specifically, Ca2+ chelation with BAPTA-AM blocked Ca2+ diffusion, thus contributing to the prevention of neuronal cell death (Fig. 4 e). These results support the hypothesis that Ca2+ redistribution occurs during retinal ischemia, which may participate in retinal cell death.
DISCUSSION
We observed Ca2+ ion movement, which progresses from GCL to ONL during apoptotic retinal cell death, using TOF-SIMS. This is a label-free technique, which overcomes the necessity for chemical labeling, insufficient chemical specificity, and low spatial resolution.
In the animal model of carotid ligation, all retinal layers are simultaneously damaged by occlusion of retinal vessels. However, the differences of susceptibility to ischemia between retinal cells of each layer would be a cause of successive progression of cell death in ischemia. The susceptibility of retinal cells to ischemia is dependent on each cell's repertoire of receptors and/or ion channels. The degree of depolarization of the neuronal cells during ischemia will depend on the quantity and type of the combination of excitatory versus inhibitory input. Other factors also play a part in susceptibility during ischemia, including the ability to buffer intracellular Ca2+, extracellular pH, and the cells' ability to quench free radicals. It is commonly believed, because of numerous in vitro experiments, that Ca2+ is a major mediator of cell death in ischemia. However, it is not known to what extent results obtained in vitro can be extrapolated to the in vivo conditions.
TOF-SIMS allowed us to examine the order of cell death under ischemic conditions in retinal tissues, which is closely related to changes in Ca2+ ion distribution. As previously reported, the inner retina seems to be more sensitive than the outer retina to ischemia, possibly because Ca2+ movement occurs from GCL to ONL. Although why ONL photoreceptors appear to be less sensitive to ischemia remains unknown, one possibility is that photoreceptors might buffer Ca2+ changes more efficiently than other layers do. In the same way, Ca2+ movement into ONL was detected later than in other layers, including GCL and INL, in the ischemic retinas, which results in the cell death later in ONL than in other layers.
Currently, this technique is limited only to resolving intracellular versus extracellular ions, identifying the compartmental localization of ions, and determining the compartment from which ions have come when changes are seen. However, changes in Ca2+ distribution within the affected layers were closely related to apoptotic events, which indicate relevant coordination of ionic movement in the tissue with biochemical reactions. Thus, our study demonstrates the suitability of TOF-SIMS for monitoring the movement of specific ions in retinal tissue. Our findings have also elucidated the relationship between Ca2+ ion movement and ischemia-induced cell death.
Acknowledgments
This work was supported by the Biosignal Analysis Technology Innovation Program (grant No. M1064501001-06n4501-00110) of the Ministry of Science and Technology (MOST)/Korea Science and Engineering Foundation (KOSEF) and by the Creative Research Initiatives (Neurovascular Coordination Research Center) of MOST/KOSEF.
Jin Hyoung Kim and Jeong Hun Kim contributed equally to this work.
Editor: Paul H. Axelsen.
References
- 1.Hansen, A. J. 1985. Effect of anoxia on ion distribution in the brain. Physiol. Rev. 65:101–148. [DOI] [PubMed] [Google Scholar]
- 2.Lipton, P. 1999. Ischemic cell death in brain neurons. Physiol. Rev. 79:1431–1568. [DOI] [PubMed] [Google Scholar]
- 3.Strick, R., P. L. Strissel, K. Gavrilov, and R. Levi-Setti. 2001. Cation-chromatin binding as shown by ion microscopy is essential for the structural integrity of chromosomes. J. Cell Biol. 155:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ostrowski, S. G., C. T. Van Bell, N. Winograd, and A. G. Ewing. 2004. Mass spectrometric imaging of highly curved membranes during Tetrahymena mating. Science. 305:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulp, K. S., E. S. Berman, M. G. Knize, D. L. Shattuck, E. J. Nelson, L. Wu, J. L. Montgomery, J. S. Felton, and K. J. Wu. 2006. Chemical and biological differentiation of three human breast cancer cell types using time-of-flight secondary ion mass spectrometry. Anal. Chem. 78:3651–3658. [DOI] [PubMed] [Google Scholar]
- 6.Börner, K., H. Nygren, B. Hagenhoff, P. Malmberg, E. Tallarek, and J. E. Månsson. 2006. Distribution of cholesterol and galactosylceramide in rat cerebellar white matter. Biochim. Biophys. Acta. 1761:335–344. [DOI] [PubMed] [Google Scholar]
- 7.Martin, J. L., C. J. Stork, and Y. V. Li. 2006. Determining zinc with commonly used calcium and zinc fluorescent indicators, a question on calcium signals. Cell Calcium. 40:393–402. [DOI] [PubMed] [Google Scholar]
- 8.Grynkiewicz, G., M. Poenie, and R. Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- 9.Tsien, R., and T. Pozzan. 1989. Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 172:230–262. [DOI] [PubMed] [Google Scholar]
- 10.Osborne, N. N., R. J. Casson, J. P. Wood, G. Chidlow, M. Graham, and J. Melena. 2004. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 23:91–147. [DOI] [PubMed] [Google Scholar]
- 11.Biousse, V., and J. D. Trobe. 2005. Transient monocular visual loss. Am. J. Ophthalmol. 140:717–721. [DOI] [PubMed] [Google Scholar]
- 12.Rosenbaum, D. M., P. S. Rosenbaum, A. Gupta, M. D. Michaelson, D. H. Hall, and J. A. Kessler. 1997. Retinal ischemia leads to apoptosis which is ameliorated by aurintricarboxylic acid. Vision Res. 37:3445–3451. [DOI] [PubMed] [Google Scholar]
- 13.Rosenbaum, P. S., H. Gupta, S. I. Savitz, and D. M. Rosenbaum. 1997. Apoptosis in the retina. Clin. Neurosci. 4:224–232. [PubMed] [Google Scholar]
- 14.Adachi, K., S. Kashii, H. Masai, M. Ueda, C. Morizane, K. Kaneda, T. Kume, A. Akaike, and Y. Honda. 1998. Mechanism of the pathogenesis of glutamate neurotoxicity in retinal ischemia. Graefes Arch. Clin. Exp. Ophthalmol. 236:766–774. [DOI] [PubMed] [Google Scholar]
- 15.Luo, X., G. N. Lambrou, J. A. Sahel, and D. Hicks. 2001. Hypoglycemia induces general neuronal death, whereas hypoxia and glutamate transport blockade lead to selective retinal ganglion cell death in vitro. Invest. Ophthalmol. Vis. Sci. 42:2695–2705. [PubMed] [Google Scholar]
- 16.Szabo, M. E., M. T. Droy-Lefaix, M. Doly, C. Carré, and P. Braquet. 1991. Ischemia and reperfusion-induced histologic changes in the rat retina. Demonstration of a free radical-mediated mechanism. Invest. Ophthalmol. Vis. Sci. 32:1471–1478. [PubMed] [Google Scholar]
- 17.Levine, S. 1960. Anoxic-ischemic encephalopathy in rats. Am. J. Pathol. 36:1–17. [PMC free article] [PubMed] [Google Scholar]
- 18.Rice, J. E., R. C. Vannucci, and J. B. Brierley. 1981. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 9:131–141. [DOI] [PubMed] [Google Scholar]
- 19.Boynton, A. L., W. L. McKeehan, and J. F. Whitfield. 1982. Ions, Cell Proliferation, and Cancer. Academic Press, New York.
- 20.Orrenius, S., B. Zhivotovsky, and P. Nicotera. 2003. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4:552–565. [DOI] [PubMed] [Google Scholar]
- 21.Demaurex, N., and C. Distelhorst. 2003. Apoptosis—the calcium connection. Science. 300:65–67. [DOI] [PubMed] [Google Scholar]
- 22.Neumar, R. W., Y. A. Xu, H. Gada, R. P. Guttmann, and R. Siman. 2003. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J. Biol. Chem. 278:14162–14167. [DOI] [PubMed] [Google Scholar]
- 23.Gómez-Vicente, V., M. Donovan, and T. G. Cotter. 2005. Multiple death pathways in retina-derived 661W cells following growth factor deprivation: crosstalk between caspases and calpains. Cell Death Differ. 12:796–804. [DOI] [PubMed] [Google Scholar]
- 24.Kubbutat, M. H., and K. H. Vousden. 1997. Proteolytic cleavage of human p53 by calpain: a potential regulator of protein stability. Mol. Cell. Biol. 17:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellgren, R. L. 1997. Evidence for participation of a calpain-like cysteine protease in cell cycle progression through late G1 phase. Biochem. Biophys. Res. Commun. 236:555–558. [DOI] [PubMed] [Google Scholar]