

Abstract
It has been found that with mutation of two surface residues (Lys22 → Glu and His104 → Arg) in human purine nucleoside phosphorylase (hPNP), there is an enhancement of catalytic activity in the chemical step. This is true although the mutations are quite remote from the active site, and there are no significant changes in crystallographic structure between the wild-type and mutant active sites. We propose that dynamic coupling from the remote residues to the catalytic site may play a role in catalysis, and it is this alteration in dynamics that causes an increase in the chemical step rate. Computational results indicate that the mutant exhibits stronger coupling between promotion of vibrations and the reaction coordinate than that found in native hPNP. Power spectra comparing native and mutant proteins show a correlation between the vibrations of Immucillin-G (ImmG):O5′⋯ImmG:N4′ and H257:Nδ⋯ImmG:O5′ consistent with a coupling of these motions. These modes are linked to the protein promoting vibrations. Stronger coupling of motions to the reaction coordinate increases the probability of reaching the transition state and thus lowers the activation free energy. This motion has been shown to contribute to catalysis. Coincident with the approach to the transition state, the sum of the distances of ImmG:O4′⋯ImmG:O5′⋯H257:Nδ became smaller, stabilizing the oxacarbenium ion formed at the transition state. Combined results from crystallography, mutational analysis, chemical kinetics, and computational analysis are consistent with dynamic compression playing a significant role in forming the transition state. Stronger coupling of these pairs is observed in the catalytically enhanced mutant enzyme. That motion and catalysis are enhanced by mutations remote from the catalytic site implicates dynamic coupling through the protein architecture as a component of catalysis in hPNP.
INTRODUCTION
The investigation of detailed mechanisms by which enzymes catalyze chemical reactions is one of the most vigorously studied areas of modern biochemistry and biophysics. It is widely accepted from both experimental and computational studies that protein motions can be intimately connected to enzymatic function. Such fluctuations are often necessary for substrate binding (1–6). In addition, there is convincing experimental evidence that the height of the chemical barrier is different in different conformational substates (7).
A more controversial question is whether there are motions in the enzyme on the timescale of barrier passage that directly couple to the reaction coordinate and affect catalysis. Both computational and theoretical evidence suggest the plausibility of such motions. We have called these enzyme motions “promoting vibrations” (8–15). These motions are different from the networks of coupled motions that have been suggested to facilitate chemical reactions. The latter refer to conformational changes on a relatively slow timescale (16,17). The “promoting vibration” described here refers to dynamic motion within a single global protein conformation. The two effects are different and occur on separate timescales.
Previous studies on rate-promoting enzymatic motions in the human purine nucleoside phosphorylase (hPNP) (EC 2.4.2.1) reaction had shown that promoting vibrations in the complex of hPNP·guanosine·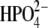 cause the O5′, O4′, and Op oxygens to compress, and this motion contributed to catalysis (13). This article provides evidence that remote mutations in purine nucleoside phosphorylase (PNP) affects active site dynamics, which in turn correlate with the catalytic properties.
cause the O5′, O4′, and Op oxygens to compress, and this motion contributed to catalysis (13). This article provides evidence that remote mutations in purine nucleoside phosphorylase (PNP) affects active site dynamics, which in turn correlate with the catalytic properties.
PNP catalyzes the reversible phosphorolysis of (deoxy)ribonucleosides to generate the corresponding purine base and (deoxy)ribose 1-phosphate (Fig. 1) (18). Although the equilibrium of this reaction favors nucleoside synthesis, the enzyme operates in the phosphorolysis direction in vivo because of metabolic removal of products by purine phosphoribosyl transferase (19,20). hPNP functions to remove deoxyguanosine, and its genetic deficiency causes apoptosis of T-cells as a consequence of the accumulation of deoxyguanosine in the circulation and dGTP in the cells (21–24). Inhibition of hPNP is useful in targeting undesirable cell proliferation in T-cell cancers, autoimmune diseases, and tissue transplant rejection. Hence, the discovery of powerful inhibitors for PNP has been an important goal (25,26). Transition state (TS) theory applied to PNP has resulted in picomolar TS analogs that have entered human clinical trials (8,27,28).
FIGURE 1.

PNP-catalyzed phosphorolysis of guanosine yielding guanine and α-d-ribose 1-phosphate. ImmG is a TS analog mimic of the TS oxacarbenium ion character and is a picomolar inhibitor of human PNP.
Kinetic isotope effect (KIE) studies of the arsenolysis reaction catalyzed by hPNP suggested a reaction proceeding via an SN1-like stepwise mechanism (DN*AN) to form a TS with a substantial oxacarbenium ion character. The TS is completely dissociated with the purine leaving-group departure from the ribosyl group complete before the attack by the arsenate nucleophile (27,28). The rate-limiting step is release of purine, and the ribosyl group is proposed to migrate several times between the purine and phosphate before release of products (8). Compounds that resemble the enzymatic TS capture the enzymatic forces used for catalysis and convert catalytic energy into binding energy, resulting in powerful inhibition (29–32). The features of the TS structure from studies of the chemical mechanism and KIE analysis were used to design chemically stable analogs to act as TS analog inhibitors, resulting in discovery of the Immucillins (Fig. 1) (27,33–36).
hPNP and bovine PNP (bvPNP) show 88% sequence identity and are homotrimers (Fig. 2 a). The catalytic site is located near the subunit-subunit interface (Fig. 2, b and c), where residue F159* is a catalytic site residue donated to the active site from an adjacent subunit. However, the catalytic activity and the crystallographic packing properties of bvPNP compared with hPNP are different. A mutant protein in which the surface residues of hPNP were changed to the identity of those in the bovine enzyme was made to improve the crystallographic properties of hPNP to match the superior diffraction properties of bvPNP. The two surface residues remote from the active site, Lys22 and His104 of hPNP, were mutated to Glu and Arg, respectively (Fig. 2 a). The crystal structure of Lys22Glu, His104Arg hPNP (E:R hPNP) exhibits no significant changes in the catalytic site and only small structural differences from native hPNP. The E:R hPNP is catalytically competent with little change in Km, kcat, or inhibition constants for TS analogs (37). However, as will be shown, the temperature dependence of catalysis by E:R hPNP is altered.
FIGURE 2.

(a) Asymmetric unit of trimer hPNP with the catalytic site of subunit-A located near the trimer interface and covered with the loop including Phe-159C of subunit C. Mutated residues His104Arg and Lys22Glu are located 25 Å and 45 Å away from the catalytic site of the adjacent subunit and 26 Å and 32 Å from the catalytic site of their own subunit. (b) The active site residues near ImmG and the phosphate nucleophile. (c) Structure of ImmG at the catalytic site of hPNP. Active site residues in contact with ImmG and the nucleophile phosphate are shown. The ImmG:O5′, ImmG:N4′, H257:Nδ, and 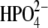 :Op atoms are highlighted in red.
:Op atoms are highlighted in red.
On the basis of these background data, we hypothesized that dynamic coupling from the remote residues to the catalytic site may play a role in catalysis. In this study, we investigated by both computation and experiment how “promoting vibrations” in normal and E:R hPNP might influence the rate of reaction and the dynamic properties. We carried out single-turnover rapid reaction kinetics as a function of temperature to establish the rate of the chemical step. Molecular dynamics (MD) simulations of trimer hPNP and E:R hPNP complexed with a TS analog and also with guanosine and phosphate provide a model of protein dynamics to compare with experimental chemistry. The power spectrum of the distance-distance cross-correlation function between O5′⋯N4′, N4′⋯Op, and H257:Nδ⋯O5′ were compared. These analyses of chemistry, structures, and dynamics were used to gain insight into the mechanism by which distant residue mutations affect both dynamics and catalysis.
EXPERIMENTAL METHODS
Computational method
Model of enzyme
The starting structure for the human enzyme was taken from the crystal structure of hPNP complexed with the TS analog, Immucillin-H (ImmH), and phosphate at 2.5 Å resolution (Protein Data Bank (pdb) code 1RR6). The crystal structures of hPNP provide coordinates for the monomer as the asymmetric unit. Therefore, we computationally generated the hPNP homotrimer based on the rotation and translation along the threefold crystallographic axis using the Crystallographic Object-Oriented Toolkit (COOT) program (38). Validation of the modeled trimer of hPNP comes from comparison with the coordinates of bvPNP in the trimeric form (pdb code 1LVU) (39). The molecular threefold axis of hPNP is coincident with the crystallographic threefold axis in cubic space group P213, enforcing a fully symmetric homotrimer. The global root mean-square deviation (RMSD) of the backbone atoms between the trimer bvPNP (pdb code 1LVU) and the modeled trimer hPNP is 0.7 Å with identical alignment of the TS inhibitor and residues at the active site (Fig. 3 a).
FIGURE 3.

(a) Superposition of trimeric bvPNP complexed with acyclic nucleoside phosphonate inhibitor (2,6-diamino-(S)-9-[2-(phosphonomethoxy)propyl]purine) (pdb code 1LVU, blue) and the modeled trimer of hPNP complexed with ImmH·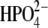 (pdb code 1RR6, orange). (b) Superposition of the crystal structure of monomeric bvPNP (ribbon enzyme blue; stick ImmH·
(pdb code 1RR6, orange). (b) Superposition of the crystal structure of monomeric bvPNP (ribbon enzyme blue; stick ImmH·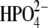 cyan) with hPNP (ribbon enzyme orange; stick ImmH·
cyan) with hPNP (ribbon enzyme orange; stick ImmH·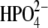 red).
red).
The trimer of the E:R hPNP mutant structure was derived from the hPNP structure. It was modeled by mutation of Lys22Glu and His104Arg using the Biopolymer module in the InsightII program (Accelrys, San Diego, CA). Comparing the in silico mutated protein with the now available crystal structure of the mutant protein (37), the heavy atom RMSD is ∼0.9 Å, but in all important sections, such as the catalytic site and the loop conformation in contact with two mutated residues, the atomic positions are exactly the same. The difference between the two is likely caused by divergences in the minimization process necessary for both structures. The crystal structure of E:R hPNP exhibits no significant changes in the catalytic site and small structural differences from native hPNP to give a RMSD heavy atom deviation (including side-chain residues) of only 0.6 Å. All backbone atoms are perfectly superimposable. Although these two mutated residues are distant from the catalytic site of the adjacent subunit (25 Å and 45 Å for H104R and K22E, respectively), they indirectly affect this subunit through an interface loop contact. Residue H104 is in van der Waals contact with the F159 loop (residues 155–166), which interacts at the catalytic site of the adjacent subunit. Catalysis is sequential at each subunit in PNP, and a one-third molar concentration of TS analog provides complete enzymatic inhibition (36); thus, the only subunit (subunit A) of trimer PNP and E:R hPNP was filled in the catalytic site with molecules of TS inhibitor or substrate and phosphate (Fig. 2 a).
Based on the H-bond pattern to Immucillin-G (ImmG), the catalytic-site residue Glu201 is deprotonated at neutral pH to stabilize the purine base (Fig. 2 c) (40). The protonation states for all other ionizable residues were set to their solution pKa values assuming a pH of 7. Thus, histidine residues were modeled as neutral residues with the proton on the nitrogen atom at the δ-or ɛ-position (Nδ or Nɛ) determined by hydrogen bond patterns deduced from the crystal structure. The catalytic site His257 with protonation at Nɛ was assigned so that Nδ could stabilize the O5′-hydroxyl of the ribose ring (8).
The CHARMM22 all-atom force field (41) was used to represent the protein amino acid residues, guanosine, and phosphate. The TIP3P model (42) was used to represent the crystallographic water molecules. The potential for ImmG was generated using the structure of guanosine, which is well parameterized in the nucleic acid force field of CHARMM22. ImmG is a transition-state analog of PNP, and the substrate guanosine and ImmG are isosteric, differing by only two atoms, N9 → C9 and O4′ → N4′. These atom types and partial charges were assigned from the atom types available in the CHARMM22 topology file. The C9 carbon was assigned as the CG carbon type from the pyrrole ring of tryptophan (CY type 28). Atom N4′ and its attached hydrogen atoms in ImmG were assigned from the protonated 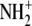 of a proline N-terminal (NP type 59). Imms bound at the catalytic site of hPNP are known to be cationic (43).
of a proline N-terminal (NP type 59). Imms bound at the catalytic site of hPNP are known to be cationic (43).
MD simulations
MD simulations of hPNP and E:R hPNP in complex with 1), ImmG and phosphate and 2), guanosine and phosphate were performed with the CHARMM molecular mechanics program. The initial minimization removed van der Waals contacts using 5000 steps of steepest descent followed by the adopted-basis Newton-Raphson algorithm until convergence in the RMSD gradient of 0.001 kcal/mol·Å and energy changes of <0.001 kcal/mol. We applied a weak restraining harmonic potential of 5 kcal/mol·Å2 to ImmG, guanosine, and phosphate to retain their positions in the crystallographic structures. Nonbonded interactions were calculated within a 12-Å cutoff distance using a smoothing function to gradually reduce to zero this contribution between 12 Å and 14 Å. The SHAKE algorithm, with a tolerance of 10−8, was applied to constrain all bonds involving hydrogen atoms (44). The time step for the integration was 1 fs. During the heating phase, the temperature of the system was increased over the first 10 ps from 0 to 300 K and remained constant through the rest of simulation. The system was equilibrated for 50 ps at 300 K, with the velocities rescaled every 100 steps to the target temperature of 300 K, using a single scaling factor for all atoms. Finally, the simulation was continued for another 200 ps as the production run with coordinates saved every 10 fs. To reiterate: 10 ps heating, 50 ps equilibration, and 200 ps for the production run, for a total of 260 ps. Energetic conservation was monitored by recording energetic properties including the potential energy, kinetic energy, and total energy as well as temperature as a function of time. Plots of both the absolute values and the fluctuations of these energies are stable. The trajectory from the last 100 ps of the production run was used for the promoting vibration analysis.
The root mean-square fluctuation (RMSF) of peptide backbone residues was used to identify the relative displacements of residues in the trimer PNP. This analysis reflects the flexibility and mobility of residues in protein but does not provide the details of correlated motions within protein. Principal component analysis (PCA), also known as essential dynamics (45–47), is a method commonly used for dissecting the large-scale dynamics of proteins and their importance in biological processes such as loop motion or substrate binding. We implemented PCA to identify correlated motions in the subunits of E:R hPNP and hPNP complexed with ImmG and phosphate. A previous essential dynamics study of correlated motions in hPNP has shown that the correlated motions observed in a 200 ps simulation were comparable to those of a 1 ns simulation (48). With that observation in mind, we applied the PCA to 200 ps production MD simulations.
In addition, 900-ps solvated simulations under cubic periodic boundary conditions were also performed for the hPNP·ImmG·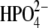 and E:R hPNP·ImmG·
and E:R hPNP·ImmG·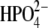 systems to demonstrate that a total of 260 ps from the initial heating step to the production run is an adequate simulation time. The conclusion drawn from both simulations is similar. The details are given in the Supplementary Material (Fig. S1 and Fig. S2 in Data S1).
systems to demonstrate that a total of 260 ps from the initial heating step to the production run is an adequate simulation time. The conclusion drawn from both simulations is similar. The details are given in the Supplementary Material (Fig. S1 and Fig. S2 in Data S1).
Experimental procedure
Site-directed mutagenesis
Site-directed mutagenesis was carried out using a QuikChange Multi Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) to prepare hPNP (E:R hPNP). The method used the native hPNP gene inserted into pCRT7/NT-TOPO (28) as a template and
K22E forward 5′-GCTTCTGTCTCATACTGAGCAGCGACCTCAAGT-3′,
K22E reverse 5′-ACTTGAGGTCGGTGCTCAGTATGAGCAAGAAC-3′,
H104R forward 5′-AGTGAGGGTTTTCCGCCTTCTGGGTGTGG-3′,
H104R reverse 5′-CCACACCCAGAAGGCGGAAAACCCTCACT-3′ oligonucleotides.
The DNA sequence was confirmed and the plasmid was transformed into Escherichia coli strain BL21(DE3)pLysS competent cells (Invitrogen, Carlsbad, CA).
Expression, purification, and assay of E:R hPNP
E:R hPNP was expressed and purified to homogeneity as judged by SDS-PAGE using the same procedure used previously for the purification of the native hPNP (28). Activity assays for the E:R hPNP mutant monitored the conversion of hypoxanthine to uric acid (ɛ293 = 12.9 mM−1 cm−1) (49) in a coupled assay containing 60 milliunits xanthine oxidase, variable concentrations of inosine, and 50 mM KH2PO4, pH 7.4, at 25°C (50). E:R hPNP was catalytically competent with little change in Km or kcat compared with native hPNP.
Stopped-flow fluorescence
The temperature dependence of single-turnover catalysis with guanosine was determined using an Applied Photophysics (Leatherhead, UK) Stopped-Flow Spectrophotometer (model π*-180 spectrometer) equipped with a thermostat-controlled water bath. The fluorescence increase caused by enzyme-bound guanine was followed by excitation at 280 nm and emission above 290 nm (2.5-nm slit widths, 2-mm path length). The rates (k3) represent the conversion of PNP·guanosine·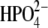 to PNP·guanine·R1-P (Fig. 4). Equal volumes of∼4 μM enzyme in 50 nM KH2PO4, pH 7.4, were rapidly mixed with saturated guanosine in the same buffer. The temperature was varied from 4° to 20°C. Two hundred data points were collected over 0.1 to 0.5 s, and at least three runs were averaged at each temperature.
to PNP·guanine·R1-P (Fig. 4). Equal volumes of∼4 μM enzyme in 50 nM KH2PO4, pH 7.4, were rapidly mixed with saturated guanosine in the same buffer. The temperature was varied from 4° to 20°C. Two hundred data points were collected over 0.1 to 0.5 s, and at least three runs were averaged at each temperature.
FIGURE 4.

Reaction scheme of PNP (E) for conversion of guanosine to guanine and ribose 1-phosphate (R1-P). Only the enzyme-bound guanine is fluorescent under these conditions. Product release (k7) is rate limiting.
RESULTS AND DISCUSSION
Crystal structures comparison
Immucillins are tightly bound TS analogs of PNPs that immobilize the protein domains involved in catalysis as established by H-D exchange (36). This structure is thought to be closely related to the TS geometry (8). TS structures have been reported for bvPNP and hPNP based on KIE studies (27,28). Both enzymes employ SN1-like mechanisms and have TSs with substantial ribooxacarbenium ion character. The bvPNP proceeds through an early SN1-like TS, whereas the hPNP proceeds through a more dissociative TS. ImmG and other TS analogs were developed to mimic these TS features.
The crystal structures of bvPNP and hPNP with bound ImmH and 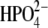 were superimposed (pdb codes 1RR6 and 1B8O(50), respectively). The superposition gave structures with highly conserved features: a global RMSD of 0.7 Å and orientation of the catalytic site residues, which are identical within crystallographic error (structures are 1.5 Å and 2.5 Å resolution, respectively). The structural superimposition is shown in Fig. 3 b. The greatest structural difference is contributed by the loop of residues 57–65 (RMSD of ∼5–7 Å), which is closed in bvPNP but more open in hPNP (RMSD of the backbone atoms is given in Fig. S3 in Data S1). In the closed-loop form, residue His64 H-bonds to the PO4 group and induces a H-bond between the carbonyl backbone atom of Pro62 and the Nɛ of His257. However, the conformation of Immucillin binding is the same, within crystallographic error, in hPNP and bvPNP. The trimeric hPNP and bvPNP exhibit one-third the site inhibitor binding and are proposed to function by sequential site catalysis. Site-site interactions occur because the active sites are located at the subunit interfaces. Most residues at the active sites are contributed from one subunit, but residue F159* is contributed from a loop on the neighboring subunit.
were superimposed (pdb codes 1RR6 and 1B8O(50), respectively). The superposition gave structures with highly conserved features: a global RMSD of 0.7 Å and orientation of the catalytic site residues, which are identical within crystallographic error (structures are 1.5 Å and 2.5 Å resolution, respectively). The structural superimposition is shown in Fig. 3 b. The greatest structural difference is contributed by the loop of residues 57–65 (RMSD of ∼5–7 Å), which is closed in bvPNP but more open in hPNP (RMSD of the backbone atoms is given in Fig. S3 in Data S1). In the closed-loop form, residue His64 H-bonds to the PO4 group and induces a H-bond between the carbonyl backbone atom of Pro62 and the Nɛ of His257. However, the conformation of Immucillin binding is the same, within crystallographic error, in hPNP and bvPNP. The trimeric hPNP and bvPNP exhibit one-third the site inhibitor binding and are proposed to function by sequential site catalysis. Site-site interactions occur because the active sites are located at the subunit interfaces. Most residues at the active sites are contributed from one subunit, but residue F159* is contributed from a loop on the neighboring subunit.
Protein promoting vibrations
Structural stabilities of hPNP and E:R hPNP during the simulation were determined as a function of time using the RMSD of the backbone atomic positions and the radius of gyration (Fig. 5). The analysis suggests that the systems are relatively stable over the final 100 ps of the analysis and do not significantly deviate from their respective crystal structures. The radius of gyration centered on the center of mass of the trimeric PNP remains essentially constant.
FIGURE 5.

RMSD of the backbone atoms (a) and radius of gyration (b) of the trimeric complexes of hPNP·ImmG·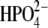 and E:R hPNP·ImmG·
and E:R hPNP·ImmG·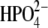 in the last 100 ps of 260 ps molecular dynamic simulations.
in the last 100 ps of 260 ps molecular dynamic simulations.
Fourier transforms (FTs) of distance-distance cross-correlation functions for several previously studied distances (13) were performed to compare promoting vibrations identified in hPNP and E:R hPNP. PNP complexed with ImmG and 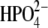 was used as the transition-state model to examine promoting vibrations. The dynamics inherent in promoting vibrations are subpicosecond motions that couple to the reaction coordinate and facilitate formation of the TS. We examined the vibration of the
was used as the transition-state model to examine promoting vibrations. The dynamics inherent in promoting vibrations are subpicosecond motions that couple to the reaction coordinate and facilitate formation of the TS. We examined the vibration of the 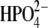 :Op⋯ImmG:N4′ distances (Op⋯N4′) and correlated this with the ImmG:O5′⋯ImmG:N4′ (O5′⋯N4′) distance and the H257:Nδ⋯ImmG:O5′ (H257:Nδ⋯O5′) distances (Fig. 2 c) and then computed their FTs to obtain power spectra. From the active site geometry, it is proposed that H257 participates in the formation of a three-oxygen stack (O5′⋯O4′⋯Op) when the guanosine substrate is bound near the TS geometry.
:Op⋯ImmG:N4′ distances (Op⋯N4′) and correlated this with the ImmG:O5′⋯ImmG:N4′ (O5′⋯N4′) distance and the H257:Nδ⋯ImmG:O5′ (H257:Nδ⋯O5′) distances (Fig. 2 c) and then computed their FTs to obtain power spectra. From the active site geometry, it is proposed that H257 participates in the formation of a three-oxygen stack (O5′⋯O4′⋯Op) when the guanosine substrate is bound near the TS geometry.
The power spectra of the distance autocorrelation function of Op⋯N4′ and the distance-distance cross-correlation functions of Op⋯N4′ with O5′⋯N4′ were calculated for hPNP and E:R hPNP (Fig. 6). The frequency and pattern of the Op⋯N4′ distance in the two protein complexes are significantly different (Fig. 6 a). From comparison of the peak resonance features and the pattern of the power spectrum, the coupling of the distances variation is stronger in E:R hPNP than in native hPNP. In E:R hPNP, a similar pattern of the power spectrum resonance peaks is found for the two distances at∼200 cm−1, 300 cm−1, 420 cm−1, and 520 cm−1 (which translates into 160–60 fs motions). In hPNP, the dominant vibrational frequencies of the Op⋯N4′ distance are found at 100 cm−1 and 300 cm−1 (which translates into 330–100 fs motions). In contrast to E:R hPNP, the resonance vibrations between the two distances (Op⋯N4′ with O5′⋯N4′) are found only at 100 cm−1.
FIGURE 6.

Power spectra of the Op⋯N4′ distance autocorrelation function and the Op⋯N4′ and O5′⋯N4′ distance-distance cross-correlation functions of hPNP·ImmG·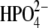 (a) and E:R hPNP·ImmG·
(a) and E:R hPNP·ImmG·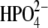 (b).
(b).
The power spectra of the autocorrelation function of the O5′⋯N4′ distance and the distance-distance cross-correlation function of the O5′⋯N4′ distance with the H257:Nδ⋯O5′ distance are shown in Fig. 7. In hPNP, the vibration of the O5′⋯N4′ distance and the H257:Nδ⋯O5′ distance shows weak correlation with the only resonant peak at 490 cm−1. In E:R hPNP, the FT of the correlation functions is similar, showing that O5′⋯N4′ and H257:Nδ⋯O5′ vibrate at the same frequencies: at ∼150 cm−1, ∼330 cm−1, and ∼410 cm−1. Greater spectral similarity between these motions in the E:R hPNP than in hPNP suggests more effective dynamic coupling. Experimental data support the observation of the protein promoting vibration from residue H257. The data include several crystal structures of PNP complexed with substrate or transition-state inhibitors, all of which show close atomic contact between the 5′-hydroxyl and H257:Nδ (8,51). Mutagenesis of H257 to Gly reduces the kcat/Km of the guanosine substrate ∼600-fold compared with the wild-type hPNP (52). Finally, eliminating the interaction between H257 and the 5′-hydroxyl moiety by the use of 5′-deoxy-ImmG decreased binding affinity by 1200-fold (53).
FIGURE 7.

Power spectra of the O5′⋯N4′ distance autocorrelation function and the O5′⋯N4′ and H257:Nδ⋯O5′ distance-distance cross-correlation functions of hPNP·ImmG·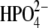 (a) and E:R hPNP·ImmG·
(a) and E:R hPNP·ImmG·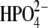 (b).
(b).
The correlated motions of H257:Nδ, O5′ and O4′ are also observed in the complex of hPNP with guanosine substrate, and in E:R hPNP, the spectra are almost superimposable (Fig. 8). Throughout the last 100 ps of the (260 ps) simulation with guanosine, the summation of the two distances O5′⋯O4′ and H257:Nδ⋯O5′ are more compressed in E:R hPNP than in hPNP. This indicates a stronger coupling of promoting vibrations to the oxygen stacking of O5′ and O4′. This compression mode is proposed to play a role in forming the TS. Altering the dynamics of H257 influences the O5′ motion in the catalytically important oxygen stack and thus facilitates dynamic motions important for catalysis.
FIGURE 8.

Power spectra of the O5′⋯O4′ distance autocorrelation function and the O5′⋯O4′ and H257:Nδ⋯O5′ distance-distance cross-correlation functions of guanosine and 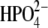 complexed with hPNP and E:R hPNP (a). The plots show the distance O5′⋯O4′, H257:Nδ⋯O5′, and the summation of two distances throughout the last 100 ps of a 260 ps simulation (b).
complexed with hPNP and E:R hPNP (a). The plots show the distance O5′⋯O4′, H257:Nδ⋯O5′, and the summation of two distances throughout the last 100 ps of a 260 ps simulation (b).
Subunit-subunit communication via the interface
In the previous section, we demonstrated that mutations remote from the catalytic sites alter active site dynamics and influence catalytic properties. Here we explore how those remote mutations affect the protein dynamics. We calculated the RMSF of peptide backbone residues to identify the relative displacements of residues in trimer PNP (Fig. 9, a and b). The distribution of RMSF for backbone atoms as a function of residue number indicates that F159 of subunit C, noted as F159C, was found to be the center of the most mobile region (0.8 Å fluctuation) in E:R hPNP but was not observed in hPNP. We applied PCA to identify correlated motions in the subunit of E:R hPNP and hPNP complexed with ImmG and phosphate. The first 10 eigenvectors were analyzed, but only the first few eigenvectors contribute significantly (Fig. S4 in Data S1). Eigenvector 1 of E:R hPNP reveals correlated motions between residues 57–65 of subunit A (the phosphate loop) and residues 155–160 of subunit C (the F159C loop) (Fig. 10). These loops interact through the subunit interface. The F159C loop of subunit C directly affects the active site of subunit A with a contact to ImmG via residue F159C (Figs. 9, c and d, and 11). Residue F159C is the only residue from the neighboring subunit that participates in the catalytic site of an adjacent subunit. The slightly different behavior of the three subunits was observed, and that corresponded well with the H-D exchange experiments, which demonstrated that filling a single catalytic site of the trimer with a TS analog reduces H-D exchange in the peptides of adjacent subunits, demonstrating the cooperative nature of subunit interactions (54).
FIGURE 9.

Residue flexibility for backbone atoms of hPNP (a) and E:R hPNP (b). Ribbon diagrams (c and d) are colored on the basis of the relative amplitudes of fluctuations of individual residue for hPNP and E:R hPNP, respectively. A blue-to-red color spectrum is used to represent different levels of flexibilities, where the smallest motions are in blue and the largest are in red. The phosphate-loop and Phe-159C-loop regions are indicated. Structure of ImmG, His-257A, Phe-200A, Tyr-88A, and Phe-159C are shown.
FIGURE 10.

Eigenvectors 1–3 (a–c) of hPNP and E:R hPNP in complex with ImmG and phosphate. The collective motions of subunit A (black), subunit B (red), and subunit C (green) are indicated.
FIGURE 11.

Details of the ImmG (green) binding site in subunit A of hPNP (a) and E:R hPNP (b). The top panel shows the side chain of Arg-104C (magenta) perturbing the flexibility (dynamics) of the Phe-159C loop (red) to increase its mobility, represented by the color spectrum of backbone fluctuation. The Phe-159C (red) contacting the catalytic site alters the dynamics of the residues His-257A, Phe-200A, and Tyr-88A (orange), which form a hydrophobic cluster. Those hydrophobic residues pack in a more favorable conformation in E:R hPNP. The yellow arrows (b) indicate the vector of conformational perturbation. The average structure of the last 100 ps of a total 260 ps dynamic simulation is shown.
In E:R hPNP, the His104Arg mutation, where the side chain of Arg is within 4 Å of the F159C loop, perturbs the dynamics of the F159C loop of subunit C, enhancing mobility as indicated by the collective motions in PCA and high RMSF (Figs. 9 and 10). Interestingly, F159C is the key residue in a cluster of hydrophobic residues (F159C, H257A, Y88A, and F200A) covering ImmG in the catalytic pocket (Fig. 11) from bulk solvent. Enhancing the motion of the F159C loop affects conformational packing of the hydrophobic cluster and, consequently, increases cluster stability and also interactions with the ImmG TS inhibitor. To further test this hypothesis, we calculated the nonbonded interactions, including van der Waals and electrostatic terms, between ImmG and neighboring residues. A total of 1000 configurations (0.1 ps sampling) during the last 100 ps of 260-ps simulations were collected and averaged (Fig. 12). The distances between the center-of-mass and backbone residues indicate that, in E:R hPNP, the changes in dynamics arising from F159C contacts alter the geometry of the hydrophobic cluster.
FIGURE 12.

Average distances between residues (a) and pairwise nonbonded interaction energies (b) averaged over 1000 structures during the last 100 ps of a total 260 ps dynamic simulation (0.1 ps configurations sampling). The error bars show the standard deviations.
Although two residues were selected for mutation to alter the structure of hPNP toward bvPNP, we found that the K22E mutation plays no significant role in the altered dynamics and catalysis. The K22E mutation is a spectator that arose from experimental attempts to crystallize the protein by altering the electrostatic properties of hPNP toward the bvPNP surface. From the trimeric PNP structure, the K22E mutation is more distant from the neighboring active site (45 Å) and does not participate in the connectivity observed for the H104R mutation.
Temperature-dependent kinetic study
Computational support for stronger coupling of the promoting vibrations in E:R hPNP indicates that E:R hPNP is a better catalyst than hPNP. This hypothesis was tested by measuring the temperature dependence of the catalytic step (k3) for both enzymes (Fig. 4). Single-turnover kinetics established the chemical step by monitoring the increase of fluorescence intensity from the formation of PNP·guanine·R1-P complexes (55,56). The turnover numbers for both native and E:R hPNP increased monotonically with increasing temperature. However, E:R hPNP showed a higher rate (k3) at all temperatures, and the difference increased at physiological temperature. For example, the turnover numbers for E:R hPNP were 214 s−1 and 840 s−1 at 25°C and 37°C, respectively, compared with 154 s−1 and 540 s−1 for native hPNP. The full experimental data of the turnover rate as the function of temperature are given in the Supplementary Material (Table S1 in Data S1).
CONCLUSIONS
Computational and experimental results indicate that E:R hPNP exhibits stronger coupling of the promoting vibrations to the reaction coordinate than found in native hPNP. Mutations remote from, but linked by dynamic motion to, the catalytic site of hPNP alter at least one promoting vibration and the catalytic properties of this enzyme. Power spectra comparing native and E:R hPNP show a correlation between the vibrations of O5′⋯N4′ and H257:Nδ⋯ImmG:O5′ consistent with a coupling of these motions. These modes are linked to the protein promoting vibrations. Stronger coupling of motions to the reaction coordinate increases the possibility of reaching the TS and also the activation free energy. Coincident with the approach to the TS, the sum of the distances of O4′⋯O5′⋯H257:Nδ becomes smaller, which is implicated in stabilizing the oxacarbenium ion. Combined results from crystallography, mutational analysis, chemical kinetics, and computational analysis are consistent with dynamic compression playing a significant role in forming the TS. Stronger coupling of these pairs is observed in the catalytically enhanced mutant E:R hPNP. That motion and catalysis are enhanced by mutations remote from the catalytic site implicates dynamic motion coupling through the protein architecture as a component of catalysis in hPNP.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank the Minnesota Supercomputer Center and Prof. Jiali Gao for a generous allotment of supercomputer time.
The authors acknowledge the support of the National Institutes of Health, grant GM068036.
Editor: Arthur G. Palmer 3rd.
References
- 1.Garcia-Viloca, M., J. Gao, M. Karplus, and D. G. Truhlar. 2004. How enzymes work: analysis by modern rate theory and computer simulations. Science. 303:186–195. [DOI] [PubMed] [Google Scholar]
- 2.Thorpe, I. F., and C. L. Brooks. 2005. Conformational substates modulate hydride transfer in dihydrofolate reductase. J. Am. Chem. Soc. 127:12997–13006. [DOI] [PubMed] [Google Scholar]
- 3.Bruice, T. C. 2006. Computational approaches: reaction trajectories, structures, and atomic motions. Enzyme reactions and proficiency. Chem. Rev. 106:3119–3139. [DOI] [PubMed] [Google Scholar]
- 4.Pu, J. Z., J. Gao, and D. G. Truhlar. 2006. Multidimensional tunneling, recrossing, and the transmission coefficient for enzymatic reactions. Chem. Rev. 106:3140–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antikainen, N. M., R. D. Smiley, S. J. Benkovic, and G. G. Hammes. 2005. Conformation coupled enzyme catalysis: single-molecule and transient kinetics investigation of dihydrofolate reductase. Biochemistry. 44:16835–16843. [DOI] [PubMed] [Google Scholar]
- 6.Hammes, G. G. 2002. Multiple conformational changes in enzyme catalysis. Biochemistry. 41:8221–8228. [DOI] [PubMed] [Google Scholar]
- 7.Eisenmesser, E. Z., O. Millet, W. Labeikovsky, D. M. Korzhnev, M. Wolf-Watz, D. A. Bosco, J. J. Skalicky, L. E. Kay, and D. Kern. 2005. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 438:117–121. [DOI] [PubMed] [Google Scholar]
- 8.Schramm, V. L. 2005. Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch. Biochem. Biophys. 433:13–26. [DOI] [PubMed] [Google Scholar]
- 9.Antoniou, D., S. Caratzoulas, C. Kalyanaraman, J. S. Mincer, and S. D. Schwartz. 2002. Barrier passage and protein dynamics in enzymatically catalyzed reactions. Eur. J. Biochem. 269:3103–3112. [DOI] [PubMed] [Google Scholar]
- 10.Mincer, J. S., and S. D. Schwartz. 2004. Rate-promoting vibrations and coupled hydrogen-electron transfer reactions in the condensed phase: a model for enzymatic catalysis. J. Chem. Phys. 120:7755–7760. [DOI] [PubMed] [Google Scholar]
- 11.Basner, J. E., and S. D. Schwartz. 2004. Donor-acceptor distance and protein promoting vibration coupling to hydride transfer: a possible mechanism for kinetic control in isozymes of human lactate dehydrogenase. J. Phys. Chem. B. 108:444–451. [Google Scholar]
- 12.Basner, J. E., and S. D. Schwartz. 2005. How enzyme dynamics helps catalyze a reaction in atomic detail: a transition path sampling study. J. Am. Chem. Soc. 127:13822–13831. [DOI] [PubMed] [Google Scholar]
- 13.Nunez, S., D. Antoniou, V. L. Schramm, and S. D. Schwartz. 2004. Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J. Am. Chem. Soc. 126:15720–15729. [DOI] [PubMed] [Google Scholar]
- 14.Antoniou, D., J. Basner, S. Nunez, and S. D. Schwartz. 2006. Computational and theoretical methods to explore the relation between enzyme dynamics and catalysis. Chem. Rev. 106:3170–3187. [DOI] [PubMed] [Google Scholar]
- 15.Masgrau, L., A. Roujeinikova, L. O. Johannissen, P. Hothi, J. Basran, K. E. Ranaghan, A. J. Mulholland, M. J. Sutcliffe, N. S. Scrutton, and D. Leys. 2006. Atomic description of an enzyme reaction dominated by proton tunneling. Science. 312:237–241. [DOI] [PubMed] [Google Scholar]
- 16.Hammes-Schiffer, S., and S. J. Benkovic. 2006. Relating protein motion to catalysis. Annu. Rev. Biochem. 75:519–541. [DOI] [PubMed] [Google Scholar]
- 17.Benkovic, S. J., and S. Hammes-Schiffer. 2003. A perspective on enzyme catalysis. Science. 301:1196–1202. [DOI] [PubMed] [Google Scholar]
- 18.Stoeckler, J. D., C. Cambor, and R. E. Jr. Parks. 1980. Human erythrocytic purine nucleoside phosphorylase: reaction with sugar-modified nucleoside substrates. Biochemistry. 19:102–107. [DOI] [PubMed] [Google Scholar]
- 19.Friedkin, M., and H. M. Kalckar. 1950. Desoxyribose-1-phosphate: I. The phosphorolysis and resynthesis of purine desoxyribose nucleoside. J. Biol. Chem. 184:437–448. [PubMed] [Google Scholar]
- 20.Erion, M. D., K. Takabayashi, H. B. Smith, J. Kessi, S. Wagner, S. Honger, S. L. Shames, and S. E. Ealick. 1997. Purine nucleoside phosphorylase. 1. Structure-function studies. Biochemistry. 36:11725–11734. [DOI] [PubMed] [Google Scholar]
- 21.Krenitsky, T. A., J. V. Tuttle, G. W. Koszalka, I. S. Chen, L. M. Beacham 3rd, J. L. Rideout, and G. B. Elion. 1976. Deoxycytidine kinase from calf thymus. Substrate and inhibitor specificity. J. Biol. Chem. 251:4055–4061. [PubMed] [Google Scholar]
- 22.Carson, D. A., J. Kaye, and J. E. Seegmiller. 1977. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: possible role of nucleoside kinase(s). Proc. Natl. Acad. Sci. USA. 74:5677–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell, B. S., E. Mejias, P. E. Daddona, and W. N. Kelley. 1978. Purinogenic immunodeficiency diseases: selective toxicity of deoxyribonucleosides for T cells. Proc. Natl. Acad. Sci. USA. 75:5011–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ullman, B., L. J. Gudas, S. M. Clift, and D. W. Jr. Martin. 1979. Isolation and characterization of purine-nucleoside phosphorylase-deficient T-lymphoma cells and secondary mutants with altered ribonucleotide reductase: genetic model for immunodeficiency disease. Proc. Natl. Acad. Sci. USA. 76:1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ealick, S. E., Y. S. Babu, C. E. Bugg, M. D. Erion, W. C. Guida, J. A. Montgomery, and J. A. Secrist 3rd. 1991. Application of crystallographic and modeling methods in the design of purine nucleoside phosphorylase inhibitors. Proc. Natl. Acad. Sci. USA. 88:11540–11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schramm, V. L. 2002. Development of transition state analogues of purine nucleoside phosphorylase as anti-T-cell agents. Biochim. Biophys. Acta. 1587:107–117. [DOI] [PubMed] [Google Scholar]
- 27.Kline, P. C., and V. L. Schramm. 1993. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 32:13212–13219. [DOI] [PubMed] [Google Scholar]
- 28.Lewandowicz, A., and V. L. Schramm. 2004. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 43:1458–1468. [DOI] [PubMed] [Google Scholar]
- 29.Schramm, V. L. 1998. Enzymatic transition states and transition state analog design. Annu. Rev. Biochem. 67:693–720. [DOI] [PubMed] [Google Scholar]
- 30.Jencks, W. P. 1975. Binding energy, specificity, and enzymic catalysis: the circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43:219–410. [DOI] [PubMed] [Google Scholar]
- 31.Wolfenden, R. 1969. Transition state analogues for enzyme catalysis. Nature. 223:704–705. [DOI] [PubMed] [Google Scholar]
- 32.Wolfenden, R. 2003. Thermodynamic and extrathermodynamic requirements of enzyme catalysis. Biophys. Chem. 105:559–572. [DOI] [PubMed] [Google Scholar]
- 33.Schramm, V. L. 2005. Enzymatic transition states and transition state analogues. Curr. Opin. Struct. Biol. 15:604–613. [DOI] [PubMed] [Google Scholar]
- 34.Kline, P. C., and V. L. Schramm. 1992. Purine nucleoside phosphorylase. Inosine hydrolysis, tight binding of the hypoxanthine intermediate, and third-the-sites reactivity. Biochemistry. 31:5964–5973. [DOI] [PubMed] [Google Scholar]
- 35.Kline, P. C., and V. L. Schramm. 1995. Pre-steady-state transition-state analysis of the hydrolytic reaction catalyzed by purine nucleoside phosphorylase. Biochemistry. 34:1153–1162. [DOI] [PubMed] [Google Scholar]
- 36.Miles, R. W., P. C. Tyler, R. H. Furneaux, C. K. Bagdassarian, and V. L. Schramm. 1998. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. 37:8615–8621. [DOI] [PubMed] [Google Scholar]
- 37.Ghanem, M., L. Li, C. Wing, and V. L. Schramm. 2008. Thermodynamics from remote mutations altering human toward bovine purine nucleoside phosphorylase. Biochemistry. 47:2559–2564. [DOI] [PubMed] [Google Scholar]
- 38.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 39.Bzowska, A., G. Koellner, B. Wielgus-Kutrowska, A. Stroh, G. Raszewski, A. Holy, T. Steiner, and J. Frank. 2004. Crystal structure of calf spleen purine nucleoside phosphorylase with two full trimers in the asymmetric unit: important implications for the mechanism of catalysis. J. Mol. Biol. 342:1015–1032. [DOI] [PubMed] [Google Scholar]
- 40.Erion, M. D., J. D. Stoeckler, W. C. Guida, R. L. Walter, and S. E. Ealick. 1997. Purine nucleoside phosphorylase. 2. Catalytic mechanism. Biochemistry. 36:11735–11748. [DOI] [PubMed] [Google Scholar]
- 41.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 42.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 43.Sauve, A. A., S. M. Cahill, S. G. Zech, L. A. Basso, A. Lewandowicz, D. S. Santos, C. Grubmeyer, G. B. Evans, R. H. Furneaux, P. C. Tyler, A. McDermott, M. E. Girvin, and V. L. Schramm. 2003. Ionic states of substrates and transition state analogues at the catalytic sites of N-ribosyltransferases. Biochemistry. 42:5694–5705. [DOI] [PubMed] [Google Scholar]
- 44.Allen, M. P., and D. J. Tildesley. 1989. Computer Simulation of Liquids. Clarendon Press, Oxford University Press, Oxford.
- 45.Caves, L. S. D., J. D. Evanseck, and M. Karplus. 1998. Locally accessible conformations of proteins: Multiple molecular dynamics simulations of crambin. Protein Sci. 7:649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia, A. E. 1992. Large-amplitude nonlinear motions in protein. Phys. Rev. Lett. 68:2696–2699. [DOI] [PubMed] [Google Scholar]
- 47.Amadei, A. L., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 48.Nunez, S., C. Wing, D. Antoniou, V. L. Schramm, and S. D. Schwartz. 2006. Insight into catalytically relevant correlated motions in human purine nucleoside phosphorylase. J. Phys. Chem. A. 110:463–472. [DOI] [PubMed] [Google Scholar]
- 49.Kim, B. K., S. Cha, and R. E. Jr. Parks. 1968. Purine nucleoside phosphorylase from human erythroyctes. II. Kinetic analysis and substrate-binding studies. J. Biol. Chem. 243:1771–1776. [PubMed] [Google Scholar]
- 50.Lewandowicz, A., W. Shi, G. B. Evans, P. C. Tyler, R. H. Furneaux, L. A. Basso, D. S. Santos, S. C. Almo, and V. L. Schramm. 2003. Over-the-barrier transition state analogues and crystal structure with Mycobacterium tuberculosis purine nucleoside phosphorylase. Biochemistry. 42:6057–6066. [DOI] [PubMed] [Google Scholar]
- 51.Fedorov, A., W. Shi, G. Kicska, E. Fedorov, P. C. Tyler, R. H. Furneaux, J. C. Hanson, G. J. Gainsford, J. Z. Larese, V. L. Schramm, and S. C. Almo. 2001. Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry. 40:853–860. [DOI] [PubMed] [Google Scholar]
- 52.Murkin, A., M. R. Brick, A. Rinaldo-Matthis, W. Shi, E. A. Taylor, and V. L. Schramm. 2007. Neighboring group participation in the transition state of human purine nucleoside phosphorylase. Biochemistry. 46:5038–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kicska, G. A., P. C. Tyler, G. B. Evans, R. H. Furneaux, W. Shi, A. Fedorov, A. Lewandowicz, S. M. Cahill, S. C. Almo, and V. L. Schramm. 2002. Atomic dissection of the hydrogen bond network for transition-state analogue binding to purine nucleoside phosphorylase. Biochemistry. 41:14489–14498. [DOI] [PubMed] [Google Scholar]
- 54.Wang, F., R. W. Miles, G. Kicska, E. Nieves, V. L. Schramm, and R. H. Angeletti. 2000. Immucillin-H binding to purine nucleoside phosphorylase reduces dynamic solvent exchange. Protein Sci. 9:1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wielgus-Kutrowska, B., A. Bzowska, J. Tebbe, G. Koellner, and D. Shugar. 2002. Purine nucleoside phosphorylase from Cellulomonas sp.: physicochemical properties and binding of substrates determined by ligand-dependent enhancement of enzyme intrinsic fluorescence, and by protective effects of ligands on thermal inactivation of the enzyme. Biochim. Biophys. Acta. 1597:320–334. [DOI] [PubMed] [Google Scholar]
- 56.Dlugosz, M., A. Bzowska, and J. M. Antosiewicz. 2005. Stopped-flow studies of guanine binding by calf spleen purine nucleoside phosphorylase. Biophys. Chem. 115:67–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.