

Abstract
Based on curvature energy considerations, nonbilayer phase-forming phospholipids in excess water should form stable bicontinuous inverted cubic (QII) phases at temperatures between the lamellar (Lα) and inverted hexagonal (HII) phase regions. However, the phosphatidylethanolamines (PEs), which are a common class of biomembrane phospholipids, typically display direct Lα/HII phase transitions and may form intermediate QII phases only after the temperature is cycled repeatedly across the Lα/HII phase transition temperature, TH, or when the HII phases are cooled from T > TH. This raises the question of whether models of inverted phase stability, which are based on curvature energy alone, accurately predict the relative free energy of these phases. Here we demonstrate the important role of a noncurvature energy contribution, the unbinding energy of the Lα phase bilayers, gu, that serves to stabilize the Lα phase relative to the nonlamellar phases. The planar Lα phase bilayers must separate for a QII phase to form and it turns out that the work of their unbinding can be larger than the curvature energy reduction on formation of QII phase from Lα at temperatures near the Lα/QII transition temperature (TQ). Using gu and elastic constant values typical of unsaturated PEs, we show that gu is sufficient to make TQ > TH for the latter lipids. Such systems would display direct Lα → HII transitions, and a QII phase might only form as a metastable phase upon cooling of the HII phase. The gu values for methylated PEs and PE/phosphatidylcholine mixtures are significantly smaller than those for PEs and increase TQ by only a few degrees, consistent with observations of these systems. This influence of gu also rationalizes the effect of some aqueous solutes to increase the rate of QII formation during temperature cycling of lipid dispersions. Finally, the results are relevant to protocols for determining the Gaussian curvature modulus, which substantially affects the energy of intermediates in membrane fusion and fission. Recently, two such methods were proposed based on measuring TQ and on measuring QII phase unit cell dimensions, respectively. In view of the effect of gu on TQ that we describe here, the latter method, which does not depend on the value of gu, is preferable.
INTRODUCTION
Although their basic building elements are liquid crystalline lipid bilayers, biomembranes also contain a large fraction of lipids able to form inverted nonlamellar phases under physiological conditions (1,2). For example, the phosphatidylethanolamines (PEs), which are a common, widely spread membrane lipid class, easily transform from lamellar, Lα, into inverted hexagonal, HII, or, occasionally, into inverted bicontinuous cubic, QII, phase in aqueous dispersions (3). Many glycolipids can also form nonlamellar phases (4). Under specific conditions, such as low pH or the presence of divalent cations, inverted nonlamellar phases can also be induced in phosphatidylserines, cardiolipins, and phosphatidic acids. Moreover, it has been shown recently that total and polar lipid extracts and membrane-mimicking lipid compositions also readily form nonlamellar phases at close to physiological temperatures (5). Several different types of inverted nonlamellar phases have been found to form in fully hydrated dispersions of membrane lipids, all of them appearing at temperatures above the existence range of the lamellar liquid crystalline phase Lα. In a hypothetical “full” phase sequence, these phases arrange in the following order with increase of temperature:
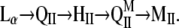 |
L, Q, H, and M refer to lamellar, cubic, hexagonal, and micellar phases, respectively. QII denotes a set of three inverted bicontinuous (bilayer) cubic phases, Im3m, Pn3m, and Ia3d, each of which may appear depending on lipid type and specific conditions.
In this work we address a problem related to the relative stabilities of the Lα and QII phases. It is known from a number of studies that dispersions of double-chain nonlamellar membrane lipids most frequently display a direct lamellar-inverted hexagonal, Lα → HII, phase transition, omitting the intermediate QII phase. The Lα → QII transitions, followed by HII formation at higher temperature, are relatively rare, and they are typically observed for short-chain PEs and glycolipids (6,7). Similarly, mixtures of saturated phosphatidylcholines (PCs) and fatty acids with shorter lauric and myristic acyl chains can form intermediate QII phase, whereas mixtures with longer chains (palmitic, stearic) again display direct Lα → HII transitions (8). However, it was shown for several PEs, which display direct Lα → HII phase transitions, that they can be converted into the intermediate QII phase when the temperature is cycled repeatedly across the Lα/HII phase transition (9–13). In these cases, traces of QII phase were found to form on cooling from the HII range and to gradually replace the initial lamellar Lα phase over the whole range of its existence (11). These observations suggest that the bilayer QII phases are metastable with respect to Lα in PEs with intermediate chain lengths of 16–18 carbon atoms such as dielaidoyl PE (DEPE), dipalmitoyl PE (DPPE), dihexadecyl PE (DHPE), dipalmitoleoyl PE (DPoPE), and dioleoyl PE (DOPE).
On the other hand, the Lα → QII → HII sequence of phases can be obtained theoretically if the free energy differences between these phases are assumed to be due only to differences in their curvature energies (e.g., Siegel and Kozlov (14) and Siegel (15)). Thus the absence of QII phase on the heating of most PEs, in particular those with intermediate and longer chains, raises the question of to what extent theoretical descriptions of the curvature energies of QII and HII phases are sufficient to predict the relative stability of these phases, especially the thermodynamic stability of a QII phase with respect to the neighboring Lα and HII phases.
Here we show that the apparent discrepancy between experimental observations and curvature energy considerations can be resolved by taking into account a noncurvature energy contribution, the unbinding energy, gu, of the Lα phase, which stabilizes the lamellar phase relative to the QII and HII phases. The planar Lα phase bilayers must separate for QII phase to form and it turns out that the work of their unbinding can be larger than the curvature energy reduction upon formation of QII from the Lα phase. Using gu values and values of elastic constants typical of unsaturated PEs, we show that gu is sufficiently large to shift the temperature TQ of the Lα → QII transition to values above the temperature TH of the Lα → HII transition. Such systems with TQ > TH would display direct Lα →HII transitions, and a QII phase might only form as a metastable phase upon cooling from the HII phase region.
These results have important implications for the choice of methods that can be used to determine the Gaussian curvature elasticity modulus. This modulus has a substantial effect on the energy of intermediates in the process of membrane fusion and fission, and the contribution of the Gaussian curvature elastic energy to the energy of fusion intermediates is on the same order of magnitude as the contribution of the bending energy (16).
THEORY
Curvature energy differences between lamellar and nonlamellar phases
If the free energy differences between lamellar and nonlamellar phases are assumed to be due only to curvature energy, one would obtain that a lipid system exhibits an Lα → QII → HII sequence of phases with increasing temperature (14,15). This is because the spontaneous curvature of lipid monolayers, Js, becomes more negative with increasing temperature. Respectively, the Lα phase should give way to a QII phase where the monolayers can adopt more negative mean curvatures while adopting slightly unfavorable Gaussian curvature. At still higher T, the HII phase has a more negative mean curvature energy than QII. In the HII phase, the monolayer curvature becomes ≈Js, although at the penalty of overcoming unfavorable interstice-packing energies at the boundaries between the HII phase cylinders.
As is known, the quadratic terms in the Helfrich phenomenological membrane curvature energy equation are insufficient to explain the bicontinuous QII phase stability (Discussion). We therefore use here a previously derived expression for the curvature energy per lipid molecule in the QII phase with respect to planar bilayers, which is accurate to fourth order in curvature (15). Here we delineate its derivation. The model in Siegel (15) is based on the Helfrich model for the curvature energy (17), complemented with the third- and fourth-order curvature terms as formulated by Mitov (18). The curvature energy of the lipid monolayers is then given by
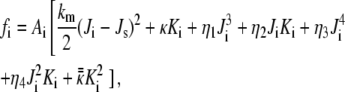 |
(1) |
where the subscripts i are labels for the two monolayers of the bilayer, and Ai is the area of each monolayer; km and κ are the bending (splay) modulus and the Gaussian (saddle splay) modulus of the lipid monolayers, respectively; Ji, Js, and Ki are the mean curvature at the monolayer neutral plane, the spontaneous curvature at the neutral plane, and the Gaussian curvature of the monolayers, respectively; and finally, ηj and 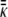 are the elastic moduli of the respective third- and fourth-order curvature terms.
are the elastic moduli of the respective third- and fourth-order curvature terms.
Assuming a constant displacement δ of the monolayer neutral planes from the bilayer midplanes, and using a mathematical relationship between the curvatures of parallel surfaces (19), Ji, Ki, and Ai of the monolayers can be expressed in terms of mean curvature, Gaussian curvature, and area (J, K, and A, respectively) of the bilayer midplane. Since the bilayer midplanes of bicontinuous QII phases lie on minimal surfaces, J = 0. It is found that the values of Ji and Ki for the two monolayers are equivalent, and
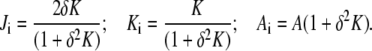 |
(2) |
The curvature energy per unit area of the bilayer can then be expressed in terms of fi. Expressing (1 + δ2K)−1 as a series of powers of δ2K and collecting terms with the same powers of K, the curvature energy per unit area of bilayer to fourth order in curvature (K2) is
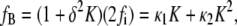 |
(3) |
where
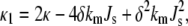 |
(4) |
and
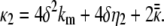 |
(5) |
The area average of Eq. 3 is the energy per unit area of the bilayer in the QII phase. The integrals of KN over the unit cells of the different QII phases are
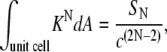 |
(6) |
where c is the cell constant of the QII phase and the coefficients SN are geometric constants that have been calculated numerically for each of the infinite periodic minimal surfaces corresponding to the different QII phases (20,21). The area-averaged curvature energy is then
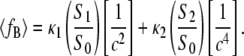 |
(7) |
We find the equilibrium value of c in excess water, ceq, by minimizing Eq. 7 with respect to c at constant Js (constant temperature). This yields
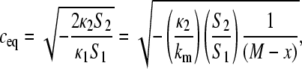 |
(8) |
where, in the second equality, we have inserted Eq. 4 and simplified notation by making the substitutions
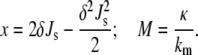 |
(9) |
The result of Eq. 8 is a real number because S1 < 0, and κ1, κ2, and S2 are all positive in the range of application of this expression (see below). Substituting Eqs. 8 and 9 into Eq. 7 and dividing by the number of lipid molecules per unit area of bilayer yields the equilibrium curvature energy per lipid molecule in the QII phase in excess water:
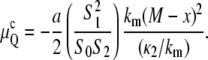 |
(10) |
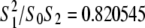 for all three Bonnet-related QII phases (Im3m, Pn3m, and Ia3d). δ is ∼1.3 nm for phospholipids with oleoyl chains, and a is the area per lipid molecule at the neutral surface. The values of M and the ratio κ2/km can be determined experimentally from the temperature dependence of the QII unit cell constant in excess water using Eqs. 8 and 9 if the values of δ, Js, and km are known over the relevant range of temperature (see below). For N-monomethylated dioleoylphosphatidylethanolamine (DOPE-Me), using the data in (22), M = − 0.90 ± 0.09 and κ2/km = 2.4 ± 0.3 nm2 (15).
for all three Bonnet-related QII phases (Im3m, Pn3m, and Ia3d). δ is ∼1.3 nm for phospholipids with oleoyl chains, and a is the area per lipid molecule at the neutral surface. The values of M and the ratio κ2/km can be determined experimentally from the temperature dependence of the QII unit cell constant in excess water using Eqs. 8 and 9 if the values of δ, Js, and km are known over the relevant range of temperature (see below). For N-monomethylated dioleoylphosphatidylethanolamine (DOPE-Me), using the data in (22), M = − 0.90 ± 0.09 and κ2/km = 2.4 ± 0.3 nm2 (15).
An important feature of the QII curvature energy model in Eq. 10 is that it predicts the existence of a threshold temperature, TK, limiting from below the range of QII stability. Js decreases with increasing temperature, and TK is the temperature at which x = M (Eq. 9). Above TK, the curvature energy is always negative (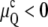 ), rendering the QII phase more stable than Lα. At TK,
), rendering the QII phase more stable than Lα. At TK, 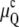 becomes equal to 0 and the QII phase unit cell constant diverges to infinity. No finite equilibrium values for the QII unit cell constant exist below that temperature (15). TK is the lowest temperature at which formation of stable structures with negative Gaussian curvature, like QII phases, is still possible in the given lipid system. The value of TK is determined by the balance of elastic constants and spontaneous curvature of the lipid membrane, through the temperature dependence of Js and Eq. 9. For supercooled QII phases at T < TK, the curvature energy is positive and is not given by Eq. 10. Its value and the nonequilibrium value of the QII unit cell constant depend on the temperature history of the lipid sample and will not be discussed further here.
becomes equal to 0 and the QII phase unit cell constant diverges to infinity. No finite equilibrium values for the QII unit cell constant exist below that temperature (15). TK is the lowest temperature at which formation of stable structures with negative Gaussian curvature, like QII phases, is still possible in the given lipid system. The value of TK is determined by the balance of elastic constants and spontaneous curvature of the lipid membrane, through the temperature dependence of Js and Eq. 9. For supercooled QII phases at T < TK, the curvature energy is positive and is not given by Eq. 10. Its value and the nonequilibrium value of the QII unit cell constant depend on the temperature history of the lipid sample and will not be discussed further here.
The curvature energy per lipid molecule in the HII phase in excess water with respect to planar bilayers is (14)
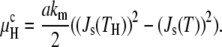 |
(11) |
Here Js(TH) is the value of Js at the observed Lα/HII transition temperature, TH.
In the absence of other contributions, the free energy differences between the Lα phase and the QII and HII phases are given by the curvature energies 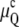 and
and 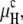 respectively. In Fig. 1 A,
respectively. In Fig. 1 A, 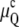 and
and 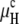 are plotted as a function of temperature using parameters determined for DOPE-Me (15). The values for km and δ are 10 kBT and 1.3 nm, respectively, and a = 0.6 nm2. The values of Js(T) are taken from Siegel and Kozlov (14), and the calculated threshold temperature, TK, is 50°C (15). The observed value of TH for DOPE-Me is 62°C (22). The curvature energy plots in Fig. 1 A should be representative also for DOPE and mixtures of DOPE with 5–30 mol % DOPC at temperatures near the respective TH, because of similar HII phase lattice constants as a function of temperature (23,24) and similar values of km for DOPE and DOPC (25). This means that Js in the temperature range of the bilayer/nonbilayer phase transitions and km of these lipids should be similar to those of DOPE-Me. The same can also be said for PEs with effective chain lengths of 18 carbons and a variety of acyl chain structures including cis- and trans-monounsaturated chains and saturated chains with methyl, dimethyl, ethyl, or cylcohexyl substitution near the methyl terminals (23,26–28). Based on curvature energy considerations alone, lipids with these values of the elastic constants should form stable QII phases in a temperature range below TH, except for lipids with a value of κ/km that is very close to −1 (14,15). However, the few measurements of κ to date indicate that κ is significantly less negative (closer to zero) than this (14,15,29).
are plotted as a function of temperature using parameters determined for DOPE-Me (15). The values for km and δ are 10 kBT and 1.3 nm, respectively, and a = 0.6 nm2. The values of Js(T) are taken from Siegel and Kozlov (14), and the calculated threshold temperature, TK, is 50°C (15). The observed value of TH for DOPE-Me is 62°C (22). The curvature energy plots in Fig. 1 A should be representative also for DOPE and mixtures of DOPE with 5–30 mol % DOPC at temperatures near the respective TH, because of similar HII phase lattice constants as a function of temperature (23,24) and similar values of km for DOPE and DOPC (25). This means that Js in the temperature range of the bilayer/nonbilayer phase transitions and km of these lipids should be similar to those of DOPE-Me. The same can also be said for PEs with effective chain lengths of 18 carbons and a variety of acyl chain structures including cis- and trans-monounsaturated chains and saturated chains with methyl, dimethyl, ethyl, or cylcohexyl substitution near the methyl terminals (23,26–28). Based on curvature energy considerations alone, lipids with these values of the elastic constants should form stable QII phases in a temperature range below TH, except for lipids with a value of κ/km that is very close to −1 (14,15). However, the few measurements of κ to date indicate that κ is significantly less negative (closer to zero) than this (14,15,29).
FIGURE 1.

Free energy per lipid molecule of the QII (μQ) and HII (μH) phases relative to the Lα phase as functions of the temperature for a lipid with the curvature energy parameters of DOPE-Me for different values of the Lα phase unbinding energy, gu. (A) gu = 0 (i.e., μQ and μH are determined only by curvature elastic energy via Eqs. 10 and 11, respectively). (B) gu = 0.01 erg/cm2, a value typical of methylated PEs and mixtures of PEs with small mole fractions of PC (Table 1). (C) gu = 0.15 erg/cm2, a value typical of pure PEs (Table 1). The curvature elastic energy parameters for DOPE-Me are from Siegel and Kozlov (14) and Siegel (15).
Unbinding energy of lamellar phases
Bilayers in lamellar phases interact across the interlamellar water spaces by a combination of attractive and repulsive forces (van der Waals, hydration, and electrostatic and bilayer undulation forces; see Nagle and Tristram-Nagle (30) for a recent review). In addition, PE bilayers may bind to each other by formation of hydrogen bonds between phospholipid headgroups in adjacent interfaces at separations of <1 nm (31). The interplay of these forces generates an interbilayer potential typically with a minimum at a given interbilayer distance (water layer thickness). For lamellar phases in excess water, the bilayers adopt the interbilayer separation corresponding to this minimum. The unbinding energy, gu, is the work per unit area required to separate these bilayers to infinite separation from the equilibrium interbilayer separation. In separating the bilayers, most of gu is expended in increasing the interbilayer separation by the first several nanometers.
The unbinding energy can be evaluated through analysis of x-ray diffraction experiments on osmotically stressed multilamellar liposomes (32) or determined more directly by optically measuring the deformation of adhering giant unilamellar vesicles as a function of membrane tension applied by pipette aspiration (33,34). Values of the unbinding energy for a number of lipid systems are summarized in Table 1 (32,35,36).
TABLE 1.
Unbinding energies of the lamellar Lα phase of lipid systems
| Lipid system | gu (erg/cm2) | Reference |
|---|---|---|
| POPE | >0.15 | (36) |
| POPE (30°C) | 0.14 | (32) |
| Egg-PE | 0.14 | (32) |
| N-monomethylated egg-PE | 0.01 | (32) |
| POPE/SOPC = 9/1 | 0.06 | (36) |
| POPE/SOPC = 4/1 | 0.04 | (36) |
| DOPE/DOPC = 3/1 | 0.03 | (32) |
| SOPC, egg-PC, and DMPC | 0.01–0.015 | (34) |
| Egg-PC/cholesterol = 1/1 | 0.003 | (32) |
Unless otherwise noted, the measurement temperature was 22°C.
Contributions of the unbinding energy to the free energies of the QII and HII phases relative to the Lα phase
The bilayers in a lamellar phase must separate by several nanometers to form a bicontinuous QII phase. This is necessary because the typical distances between lipid/water interfaces in bicontinuous QII phases, which are composed of labyrinths of bilayers, are significantly greater than those in the Lα phase. The average separation is given by twice the average water channel radius, rw (37):
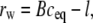 |
(12) |
where B is a geometric constant specific to the space group of each QII phase, and l is the lipid monolayer thickness, typically ∼2 nm. B is 0.305 for the Im3m, 0.391 for the Pn3m, and 0.248 for the Ia3d cubic phases. Since the QII unit cell constants for PEs can be >20–25 nm (11), the average bilayer separations in these QII phases exceed 10–15 nm and are far larger than the typical Lα interlamellar separations of ∼2 nm or less.
Since the only attractive interaction at bilayer separations above 1 nm is the van der Waals interaction, the bilayer interaction energy in a QII phase can be approximated with the van der Waals term for flat bilayers at the average interbilayer separation. The van der Waals energy of two parallel bilayers is given by (30)
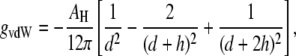 |
(13) |
where AH is the Hamaker constant, d is the distance between the bilayers, and h is the bilayer thickness. The distance dependence of gvdW is more complicated in QII phases because the bilayers are bent into complex shapes and it is not straightforward to calculate their van der Waals energy. However, in nonplanar geometries the potential between objects like slabs, cylinders, and spheres is still generally proportional to d−n, with n > 1 (38). With QII interbilayer separations five or more times larger than the separation between bilayers in the Lα phase, the van der Waals attraction in the QII phase must be at least 5–10 times smaller than gu of the Lα phase. To a good approximation, we can assume that all of gu is expended in separating the bilayers to the distances within QII phases. The sum of the curvature and unbinding energy difference per lipid molecule between the QII and Lα phases, μQ, would then become
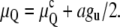 |
(14) |
In Eq. 14, 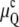 is given by Eq. 10, a is the area per lipid molecule at the neutral planes of the monolayers, and the factor of 1/2 accounts for the presence of two monolayers per adhering bilayer.
is given by Eq. 10, a is the area per lipid molecule at the neutral planes of the monolayers, and the factor of 1/2 accounts for the presence of two monolayers per adhering bilayer.
In HII phases of PEs and methylated PEs in excess water conditions, the interfacial separations, approximated as the HII aqueous tube diameter, are typically 3.5–4 nm (23,26), ∼2–3 times larger than the typical Lα interlamellar separation. This implies that the residual van der Waals attraction in the HII phase is again significantly smaller than that in the Lα phase. We have not found expressions for van der Waals interactions in an HII phase, but fortunately our argument concerning the relative effects of the unbinding energy gu on TQ and TH turns out to be rather insensitive to the actual magnitude of the residual van der Waals term in the HII phase. As an approximation, we assume that there is no residual interaction of the lipid monolayers in the HII phase, similarly to our assumption for the QII phase. Then we can write an expression for the total energy difference between HII and Lα per lipid molecule, μH, which is analogous to Eq. 14:
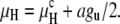 |
(15) |
However, as shown below, the final result would be nearly the same even if we assumed that the van der Waals interaction energy per lipid molecule in the HII phase has not changed and has remained equal to that of the Lα phase, so that Eq. 15 reduces to 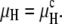
Effects of the unbinding energy on TQ and TH
As is evident from Fig. 1 A, the curvature energy 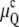 initially decreases very slowly with increasing temperature, whereas the decline of
initially decreases very slowly with increasing temperature, whereas the decline of 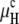 is much steeper. If there is an additional positive free energy term besides the curvature energies that translates the free energy versus temperature curves upward, it is obvious that TQ will increase by much more than TH, because of the different slopes of μQ and μH. Below we consider the role of unbinding energy gu, and Fig. 1, B and C, illustrates the effects on TQ and TH of gu values typical of phospholipids.
is much steeper. If there is an additional positive free energy term besides the curvature energies that translates the free energy versus temperature curves upward, it is obvious that TQ will increase by much more than TH, because of the different slopes of μQ and μH. Below we consider the role of unbinding energy gu, and Fig. 1, B and C, illustrates the effects on TQ and TH of gu values typical of phospholipids.
In Fig. 1 B, μQ and μH are plotted using the same curvature energy parameters, but with gu = 0.01 erg/cm2. This is a relatively small value, typical for methylated PEs, PCs, and PC-cholesterol mixtures (Table 1). Relative to Fig. 1 A, where gu = 0, TQ increases by ∼6 K to 56°C, whereas TH increases by <0.3 K. TH is still higher than TQ, but the temperature range between Lα and HII phases, where QII is the stable phase, has decreased from 12 K to only 6 K.
In Fig. 1 C, gu is set at 0.15 erg/cm2, which is typical of PEs in the Lα phase at room temperature (Table 1). Such a value results in a 25 K increase in TQ to ∼75°C, whereas TH increases by only 4 K to 66°C. Notably, TQ is now substantially higher than TH and in such a system the HII phase would form before the QII phase. Indeed, Fig. 1 C indicates that the QII phase would not become stable at any temperature. In summary, Fig. 1, A–C, shows that unbinding energies typically found in PEs can make TQ ≥ TH. Elevating TQ to above TH effectively eliminates the QII phase from the sequence Lα → QII → HII and reduces it to a direct Lα → HII phase transition. This effect explains the apparent absence of QII phases upon direct heating of PEs, whereas QII can be observed upon heating of monomethylated PEs (22) and PE mixtures with small amounts of PCs (39–42), which have much smaller values of gu (Table 1). In some of these studies the presence of QII phases was inferred from observation of isotropic 31P-NMR resonances and was not demonstrated by x-ray diffraction (39,40,42). However, the presence of isotropic 31P-NMR resonances has been associated with QII phase formation in DOPE-Me (22,23,39), temperature-cycled DEPE (10,11) and DOPE (9,12), and soy PE/egg-PC (41).
A plot of TQ and TH as a function of gu for a lipid with the elastic constants of DOPE-Me is shown in Fig. 2. TQ increases much faster with gu than TH does and becomes greater than TH for gu values >∼0.04 erg/cm2. Since the effect of gu on TH is much smaller than that on TQ, the results are not sensitive to the assumption in Eq. 15 regarding the gu difference between the Lα and HII phases. In the other limiting case, i.e., if we assume that gu is the same in the Lα and HII phases so that μH = 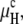 TH would become independent of gu, as indicated by the dotted line in Fig. 2. However, all conclusions of importance regarding the difference between TQ and TH as a functions of gu would remain practically unaffected. The data in Table 1 and Fig. 2 suggest that addition of small amounts of PC to PE can reduce gu to a level where there will be a direct Lα → QII transition.
TH would become independent of gu, as indicated by the dotted line in Fig. 2. However, all conclusions of importance regarding the difference between TQ and TH as a functions of gu would remain practically unaffected. The data in Table 1 and Fig. 2 suggest that addition of small amounts of PC to PE can reduce gu to a level where there will be a direct Lα → QII transition.
FIGURE 2.

Dependence of the transition temperatures TQ and TH on the unbinding energy gu for a lipid with the curvature elastic constants of DOPE-Me. The dashed horizontal line indicates the value of TH if we assume that gu in the HII phase has remained equal to that of the Lα phase (see text).
The gu values in Table 1, which we used for the calculations shown in Fig. 1, B and C, have been determined at room temperature. They should be somewhat smaller at higher temperatures due primarily to an increase in the strength of repulsive undulation forces, since short-range hydration repulsion and van der Waals forces do not change appreciably with temperature (38,43). However, we are not aware of direct measurements of gu in the temperature range above 40°C–50°C, where lamellar/nonlamellar phase transitions most often take place. A qualitative comparison of the unbinding energies of different lipids can sometimes be made on the basis of their lamellar spacings. For example, the lamellar spacing of DOPE-Me increases from 6.2 nm at 20°C to 6.4 nm at 65°C, whereas, in contrast, the lamellar spacing of DEPE decreases from 5.5 nm at 40°C to 5.2 nm at 60°C (44). Since the lamellar spacings in the methylated versus unmethylated PE are much larger at both low and high temperatures, and given the monotonic dependence of the attractive van der Waals forces between bilayers on interbilayer spacing, the unbinding energy of the PEs probably remains much larger than in methylated PE also at 60°C, similarly to the case at room temperature (Table 1). Thus, at least for temperatures up to 60°C, the temperature dependence of gu should not affect our conclusions concerning the QII phase-forming propensities of these lipids.
DISCUSSION
Effect of unbinding energy on the inverted bicontinuous cubic phase stability
In this work we developed a simple argument that explains why dispersions of membrane lipids frequently display direct Lα → HII transitions, omitting the intermediate bicontinuous QII cubic phase. This argument makes use of the circumstance that the lipid Lα phase is stabilized relative to the QII phase by a noncurvature energy contribution, the unbinding energy gu. Taking gu into account shifts the temperature TQ of the Lα → QII transition upward. Our principal result, shown in Fig. 2, is that TQ increases much faster with gu and may readily become higher than the temperature TH of a potential direct, higher temperature Lα → HII transition. Although an Lα → QII → HII sequence of phases can be expected on theoretical grounds if one considers only the curvature energy differences between the lamellar and nonlamellar phases, at least for the range of PE elastic constants measured to date, we show that values of gu that are typical of PEs with intermediate chain lengths of 16–18 carbon atoms are sufficiently large to prevent the QII phase from appearing as a stable phase between the Lα and HII phases.
As can be seen in Fig. 2, for gu values exceeding ∼0.04 erg/cm2, TQ becomes greater than TH, and QII becomes metastable with respect to the Lα and HII phases. Such lipids would display direct Lα → HII transitions and eventually form some (trace) amount of metastable QII phase on cooling from the HII range. Many PEs such as DEPE, DOPE, DPoPE, DPPE, and DHPE appear to conform to this type of phase behavior (Introduction). Moreover, dispersions of these lipids can be fully converted into QII phase by means of temperature cycling across the Lα/HII phase transition. In all cases studied, amounts of QII phase that increase with increasing numbers of cycles were invariably found to form only during the cooling stage of the temperature cycles (11). On the other hand, it is known that short-chain PEs and glycolipids exhibit the sequences of phases Lα → QII → HII (3,4,6,7). In the framework of this model, it may be that these lipids have smaller values of gu than longer chain lipids and/or a less negative value of M (Eqs. 9 and 10) that would allow a stable intermediate QII phase to form. It would be interesting to measure these quantities and see if this prediction is borne out.
However, lipids and lipid mixtures with values of gu smaller than ∼0.04 erg/cm2 (Table 1) and with appropriate spontaneous curvatures and bending elastic moduli would still be able to form intermediate QII phases upon heating. The best studied example of this kind, considered here in more detail, is DOPE-Me, whose behavior is consistent with the gu value for N-monomethylated egg-PE (Table 1). Using the experimentally determined DOPE-Me elastic constants and substituting the latter gu value in Eq. 14, one calculates an Lα → QII transition temperature TQ of 56°C. That is very close to the temperature of 55°C at which QII phase is first observed in DOPE-Me dispersions (22) (see also Fig. 3). Thus, the observed TQ in DOPE-Me is consistent with the model for the QII curvature elastic energy in Eq. 10 and with the effect of gu postulated in Eq. 14.
FIGURE 3.

Direct transformation of the DOPE-Me lamellar Lα phase into bilayer cubic Im3m phase upon incubation at 55°C. The final Im3m lattice constant is 33.0 nm. Consecutive diffraction frames of 1 s exposure were recorded every 15 min. The sample was a 16.7% (w/v) DOPE-Me dispersion in PBS pH 7.2.
Fourth-order curvature energy expression for the QII phase
The reason for the use of higher order curvature terms in the expression for the QII phase curvature energy (Eq. 10) is treated in detail in the derivation of this model (15). If one uses only quadratic terms in the expression for the QII phase curvature energy, one obtains the unphysical result that the QII phase unit cell should contract indefinitely, which is incompatible with experiment (45). Obviously something stops that contraction of the unit cell size. For QII phases with very small lattice constants, one might expect that their contraction could be stopped by steric/hydration repulsion, but not for the PE QII phases considered here, which have lattice constants typically of more than 20 nm. Similarly to previous authors (e.g., Seddon and Templer (45) and Ljunggren and Eriksson (46)), we suggest that higher order curvature energy terms explain the stabilization of QII phases with large values of the equilibrium unit cell constant. The close correspondence noted above (within 1°) between observed and calculated values of TQ indicates that the model developed in Siegel (15) is fairly accurate, at least in the temperature interval between TK and TQ. The QII phase is not the only system whose theoretical description requires a fourth-order curvature model. Such a model is also necessary to describe the prefission constriction of Golgi membrane tubules (47), for similar reasons.
Mechanisms of inverted bicontinuous cubic phase formation
Our low-angle x-ray diffraction measurements show that QII phases can form by two different pathways in DOPE-Me dispersions (Figs. 3 and 4). These measurements were made at the Advanced Photon Source, Argonne, using a low-angle x-ray setup described in detail elsewhere (48). The first pathway is the direct Lα → QII transition at constant temperature incubation (Fig. 3). It involves disordering (unbinding) of the Lα phase with concomitant formation of QII phase (Im3m under the conditions here). The lattice parameter of the Im3m phase gradually decreases with time and reaches a final value of 33.0 nm after 5–6 h of incubation. A slow disordering of the DOPE-Me Lα phase preceding the formation of the cubic phase in this lipid was also reported in previous work (23,49). Such a direct Lα → QII transition may also take place at very slow rates of temperature increase (22).
FIGURE 4.

Complete conversion of the lamellar Lα phase into Im3m cubic phase in DOPE-Me dispersion in a single heating-cooling cycle Lα → HII → Im3m. The Im3m lattice constant is 40.3 nm. Heating rate 1°C/min; cooling rate 5°C/min. Diffraction frames of 1 s exposure were recorded every 1 min. Lα → HII transition onset at 67.5°C. HII → Im3m transition onset ∼55°C. The sample was a 10% (w/v) dispersion of DOPE-Me in PBS pH 7.2.
If the incubation is long enough or the heating rate slow enough, one will observe QII phase formation at T = TQ, and the value of gu should strongly affect this value, as shown above (Fig. 2). The second pathway is indirect conversion in a heating-cooling cycle Lα → HII → QII (Fig. 4). The heating rate of 1°C/min used in this cycle is obviously not slow enough to allow QII formation in the heating direction, and the dispersion first undergoes the Lα → HII transition. This is a clear indication for the existence of kinetic barriers that significantly slow down the formation of the QII phase even though it is the thermodynamically stable phase in the respective temperature range. On the other hand, an HII → QII transition in cooling turns out to be very fast, 1–2 orders of magnitude faster than the Lα → QII transition, and the DOPE-Me dispersion fully converts into QII phase (Im3m phase with lattice parameter of ∼40 nm in Fig. 4) without any traces of reappearing Lα phase.
Cubic phase formation in PEs
Although DOPE-Me readily forms QII phase in a single heating-cooling cycle, this is not the case for PE dispersions, which typically require several tens of cycles for complete conversion into the QII phase (11). We speculate that only a fraction of the lipid in the HII phase enters the QII phase on each cooling cycle in PEs because the unbinding energy gu is ∼15 times larger for PEs than for monomethylated PEs (Table 1). This would strongly favor recovery of the Lα phase on cooling from the HII range at the expense of QII formation. However, the QII fraction, once it forms, is stable upon supercooling into the Lα phase region, even down to the chain-melting temperature of the dispersion (9,11), so QII phase accumulates during successive cycles until it completely replaces the Lα phase.
In earlier work we found that salts (NaSCN, NaCl, NaH2PO4, Na2SO4) as well as sugars (sucrose, trehalose) substantially reduced the number of cycles necessary to convert PE dispersions into the QII phase (11). The relative efficacy of the Na salts in promoting QII phase formation did not appear to correlate with salt ordering in the lyotropic series. The data, rather, appeared to indicate that the accelerating solutes destabilize to some extent the Lα phase and maintain some portion of the sample in a state of uncorrelated, single bilayers. We considered these disordered bilayers a more likely source for formation of cubic phases on the grounds elaborated in this work, i.e., that the attractive interactions in a well-stacked lamellar phase, which could be expected to impede the formation of a bicontinuous cubic phase, would be much weaker between bilayers separated by larger spaces (11). Recently it has been shown that monovalent salts and small carbohydrate molecules decrease the van der Waals attraction between electrostatically neutral bilayers in the Lα phase (50,51). Therefore, it appears that these solutes are effective in accelerating QII phase formation because they decrease the unbinding energy gu and thus facilitate the bilayer separation.
Physiological relevance
Because of the effect of gu, for lipid bilayers with elastic constants such that TK < TH and with gu > 0, there is a range of temperatures in which the QII phase is metastable with respect to the Lα phase, but represents the stable configuration with respect to unbound, disordered bilayers for which gu = 0. Consider two bilayers composed of lipid which is in this temperature range of metastable QII phase under the ambient conditions. The bilayers are maintained in the unbound state by some external factor, such as the presence of hydrated macromolecules between them. If the two bilayer interfaces are opposed over a small area, the lipids in this region might then be able to spontaneously form structures like fusion pores, which may be considered QII phase precursors (14,15). In these cases where unbinding factors are present, lipid compositions might be more fusogenic than indicated by their bulk phase behavior in the absence of such factors.
PCs are examples of membrane lipids that are effective in reducing gu (Table 1). In model systems, lipid molecules with bulky headgroups such as polyethylene glycol (PEG)-conjugated phospholipids can be particularly effective in this. In previous work we have shown that 5 mol % of PEG-conjugated dimyristoyl PE (DMPE-PEG510) disorders the Lα phase and promotes formation of stable intermediate QII phase in a temperature range between the Lα and HII phases in DEPE dispersions (44,52). Even at low concentrations, the PEG lipids appear to create steric hindrances that prevent the bilayer stacking into Lα phase and thus favor formation of QII phases.
Protocols for measuring the Gaussian (saddle splay) curvature elastic modulus
The big effect of the unbinding energy gu on the temperature TQ of the Lα/QII transition has important implications for the choice of protocols for measuring the Gaussian curvature elastic modulus κ of membranes. Recently two such protocols were suggested based on observations of QII phase behavior (14,15). The method in Siegel and Kozlov (14) relies on a measurement of TQ. The method in Siegel (15) is based on measurements of the QII phase unit cell constant as a function of temperature. The results of the latter protocol do not depend on the value of TQ and, since TQ is strongly affected by gu, it is clear that the utility of the technique in Siegel and Kozlov (14) is more limited than the technique in Siegel (15). The techniques in Siegel and Kozlov (14) and Siegel (15) will yield measurements of similar accuracy limited by the accuracy of values of δ, km, Js, and their temperature dependences. However, for comparative measurements on a host lipid system with minor fractions of lipid or peptide additives, the precision of comparisons by the technique in Siegel (15) will be superior because of the lack of a gu effect. This is significant for studies of the mechanism of membrane fusion and fission because the Gaussian curvature modulus has almost as large an effect on the curvature energy of the stalk fusion intermediate as the monolayer bending modulus (16). It would be important to measure the effects of lipid composition and of moieties of fusion- or fission-catalyzing proteins on this modulus.
Acknowledgments
Use of the advanced photon source was supported by the U.S. Department of Energy under contract No. W-31-102-Eng-38. DuPont-NorthwesternDow Collaborative Access Team is supported by E. I. DuPont de Nemours, Dow Chemical, the National Science Foundation through grant DMR-9304725, and the state of Illinois through the Department of Commerce and the Board of Higher Education grant IBHE HECA NWU 96. BioCAT is a National Institutes of Health (NIH)-supported research center through grant RR08630. B.G.T. acknowledges support from NIH grant GM57305 and from the Center for Cancer Nanotechnology Excellence initiative of the NIH National Cancer Institute under award U54CA119341.
Editor: Thomas J. McIntosh.
References
- 1.Seddon, J. M., and R. H. Templer. 1995. Polymorphism of lipid-water systems. In Handbook of Biological Physics. R. Lipowsky and E. Sackmann, editors. Elsevier Science, Amsterdam. 97–160.
- 2.Lewis, R. N. A. H., D. A. Mannock, and R. N. McElhaney. 1997. Membrane lipid molecular structure and polymorphism. In Lipid Polymorphism and Membrane Properties (Current Topics in Membranes, Vol. 44). R. Epand, editor. Academic Press, San Diego, CA. 25–102.
- 3.Seddon, J. M., G. Cevc, R. D. Kaye, and D. Marsh. 1984. X-ray-diffraction study of the polymorphism of hydrated diacylphosphatidylethanolamine and dialkylphosphatidylethanolamine. Biochemistry. 23:2634–2644. [DOI] [PubMed] [Google Scholar]
- 4.Hinz, H. J., H. Kuttenreich, R. Meyer, M. Renner, R. Frund, R. Koynova, A. I. Boyanov, and B. G. Tenchov. 1991. Stereochemistry and size of sugar head groups determine structure and phase-behavior of glycolipid membranes—densitometric, calorimetric, and x-ray studies. Biochemistry. 30:5125–5138. [DOI] [PubMed] [Google Scholar]
- 5.Koynova, R., and R. C. MacDonald. 2007. Natural lipid extracts and biomembrane-mimicking lipid compositions are disposed to form nonlamellar phases, and they release DNA from lipoplexes most efficiently. Biochim. Biophys. Acta. 1768:2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koynova, R., and M. Caffrey. 1994. Phases and phase-transitions of the hydrated phosphatidylethanolamines. Chem. Phys. Lipids. 69:1–34. [DOI] [PubMed] [Google Scholar]
- 7.Koynova, R., and M. Caffrey. 1994. Phases and phase-transitions of the glycoglycerolipids. Chem. Phys. Lipids. 69:181–207. [DOI] [PubMed] [Google Scholar]
- 8.Koynova, R., B. Tenchov, and G. Rapp. 1997. Mixing behavior of saturated short-chain phosphatidylcholines and fatty acids—eutectic points, liquid and solid phase immiscibility, non-lamellar phases. Chem. Phys. Lipids. 88:45–61. [Google Scholar]
- 9.Shyamsunder, E., S. M. Gruner, M. W. Tate, D. C. Turner, P. T. C. So, and C. P. S. Tilcock. 1988. Observation of inverted cubic phase in hydrated dioleoylphosphatidylethanolamine membranes. Biochemistry. 27:2332–2336. [DOI] [PubMed] [Google Scholar]
- 10.Veiro, J. A., R. G. Khalifah, and E. S. Rowe. 1990. P-31 nuclear-magnetic-resonance studies of the appearance of an isotropic component in dielaidoylphosphatidylethanolamine. Biophys. J. 57:637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tenchov, B., R. Koynova, and G. Rapp. 1998. Accelerated formation of cubic phases in phosphatidylethanolamine dispersions. Biophys. J. 75:853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erbes, J., C. Czeslik, W. Hahn, R. Winter, M. Rappolt, and G. Rapp. 1994. On the existence of bicontinuous cubic phases in dioleoylphosphatidylethanolamine. Ber. Bunsenges. Phys. Chem. 98:1287–1293. [Google Scholar]
- 13.Jordanova, A., Z. Lalchev, and B. Tenchov. 2003. Formation of monolayers and bilayer foam films from lamellar, inverted hexagonal and cubic lipid phases. Eur Biophys J. 31:626–632. [DOI] [PubMed] [Google Scholar]
- 14.Siegel, D. P., and M. M. Kozlov. 2004. The Gaussian curvature elastic modulus of N-monomethylated dioleoylphosphatidylethanolamine: relevance to membrane fusion and lipid phase behavior. Biophys. J. 87:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel, D. P. 2006. Determining the ratio of the Gaussian curvature and bending elastic moduli of phospholipids from Q(II) phase unit cell dimensions. Biophys. J. 91:608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozlovsky, Y., A. Efrat, D. A. Siegel, and M. M. Kozlov. 2004. Stalk phase formation: effects of dehydration and saddle splay modulus. Biophys. J. 87:2508–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helfrich, W. 1973. Elastic properties of lipid bilayers—theory and possible experiments. Z Naturforsch [C] 28:693–703. [DOI] [PubMed] [Google Scholar]
- 18.Mitov, M. D. 1978. 3rd and 4th order curvature elasticity of lipid bilayers. Comptes Rendus de l'Academie Bulgare des Sciences. 31:513–515. [Google Scholar]
- 19.do Carmo, M. P. 1976. Differential Geometry of Curves and Surfaces. Prentice-Hall, Englewood Cliffs, NJ. 212.
- 20.Anderson, D. M., S. M. Gruner, and S. Leibler. 1988. Geometrical aspects of the frustration in the cubic phases of lyotropic liquid-crystals. Proc. Natl. Acad. Sci. USA. 85:5364–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz, U. S., and G. Gompper. 2001. Bending frustration of lipid-water mesophases based on cubic minimal surfaces. Langmuir. 17:2084–2096. [Google Scholar]
- 22.Cherezov, V., D. P. Siegel, W. Shaw, S. W. Burgess, and M. Caffrey. 2003. The kinetics of non-lamellar phase formation in DOPE-Me: relevance to biomembrane fusion. J. Membr. Biol. 195:165–182. [DOI] [PubMed] [Google Scholar]
- 23.Gruner, S. M., M. W. Tate, G. L. Kirk, P. T. C. So, D. C. Turner, D. T. Keane, C. P. S. Tilcock, and P. R. Cullis. 1988. X-ray-diffraction study of the polymorphic behavior of N-methylated dioleoylphosphatidylethanolamine. Biochemistry. 27:2853–2866. [DOI] [PubMed] [Google Scholar]
- 24.Tate, M. W., and S. M. Gruner. 1989. Temperature-dependence of the structural dimensions of the inverted hexagonal (HII) phase of phosphatidylethanolamine-containing membranes. Biochemistry. 28:4245–4253. [DOI] [PubMed] [Google Scholar]
- 25.Fuller, N., and R. P. Rand. 2001. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 81:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harper, P. E., D. A. Mannock, R. N. A. H. Lewis, R. N. McElhaney, and S. M. Gruner. 2001. X-ray diffraction structures of some phosphatidylethanolamine lamellar and inverted hexagonal phases. Biophys. J. 81:2693–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis, R. N. A. H., D. A. Mannock, R. N. McElhaney, D. C. Turner, and S. M. Gruner. 1989. Effect of fatty acyl chain-length and structure on the lamellar gel to liquid-crystalline and lamellar to reversed hexagonal phase-transitions of aqueous phosphatidylethanolamine dispersions. Biochemistry. 28:541–548. [DOI] [PubMed] [Google Scholar]
- 28.Epand, R. M., N. Fuller, and R. P. Rand. 1996. Role of the position of unsaturation on the phase behavior and intrinsic curvature of phosphatidylethanolamines. Biophys. J. 71:1806–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Templer, R. H., B. J. Khoo, and J. M. Seddon. 1998. Gaussian curvature modulus of an amphiphilic monolayer. Langmuir. 14:7427–7434. [Google Scholar]
- 30.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIntosh, T. J., and S. A. Simon. 1996. Adhesion between phosphatidylethanolamine bilayers. Langmuir. 12:1622–1630. [Google Scholar]
- 32.Rand, R. P., and V. A. Parsegian. 1989. Hydration forces between phospholipid-bilayers. Biochim. Biophys. Acta. 988:351–376. [Google Scholar]
- 33.Needham, D. 1993. Measurement of interbilayer adhesion energies. Methods Enzymol. 220:111–129. [DOI] [PubMed] [Google Scholar]
- 34.Needham, D., and D. V. Zhelev. 2000. Use of micropipette manipulation techniques to measure the properties of giant lipid vesicles. In Giant Vesicles. P. L. Luisi and P. Walde, editors. John Wiley & Sons, New York. 103–147.
- 35.Evans, E., and D. Needham. 1987. Physical properties of surfactant bilayer-membranes—thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J. Phys. Chem. 91:4219–4228. [Google Scholar]
- 36.Evans, E., and D. Needham. 1986. Giant vesicle bilayers composed of mixtures of lipids, cholesterol and polypeptides—thermomechanical and (mutual) adherence properties. Faraday Discuss. Chem. Soc. 81:267–280. [DOI] [PubMed] [Google Scholar]
- 37.Kraineva, J., R. A. Narayanan, E. Kondrashkina, P. Thiyagarajan, and R. Winter. 2005. Kinetics of lamellar-to-cubic and intercubic phase transitions of pure and cytochrome c containing monoolein dispersions monitored by time-resolved small-angle x-ray diffraction. Langmuir. 21:3559–3571. [DOI] [PubMed] [Google Scholar]
- 38.Parsegian, V. A. 2006. Van der Waals Forces. Cambridge University Press, Cambridge, UK.
- 39.Ellens, H., D. P. Siegel, D. Alford, P. L. Yeagle, L. Boni, L. J. Lis, P. J. Quinn, and J. Bentz. 1989. Membrane-fusion and inverted phases. Biochemistry. 28:3692–3703. [DOI] [PubMed] [Google Scholar]
- 40.Tilcock, C. P. S., M. B. Bally, S. B. Farren, and P. R. Cullis. 1982. Influence of cholesterol on the structural preferences of dioleoylphosphatidylethanolamine dioleoylphosphatidylcholine systems—A P-31 and deuterium nuclear magnetic-resonance study. Biochemistry. 21:4596–4601. [DOI] [PubMed] [Google Scholar]
- 41.Tilcock, C. P. S., P. R. Cullis, and S. M. Gruner. 1986. On the validity of P-31-NMR determinations of phospholipid polymorphic phase-behavior. Chem. Phys. Lipids. 40:47–56. [Google Scholar]
- 42.Separovic, F., and K. Gawrisch. 1996. Effect of unsaturation on the chain order of phosphatidylcholines in a dioleoylphosphatidylethanolamine matrix. Biophys. J. 71:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon, S. A., S. Advani, and T. J. McIntosh. 1995. Temperature-dependence of the repulsive pressure between phosphatidylcholine bilayers. Biophys. J. 69:1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koynova, R., B. Tenchov, and G. Rapp. 1997. Low amounts of PEG-lipid induce cubic phase in phosphatidylethanolamine dispersions. Biochim. Biophys. Acta. 1326:167–170. [DOI] [PubMed] [Google Scholar]
- 45.Seddon, J. M., and R. H. Templer. 1993. Cubic phases of self-assembled amphiphilic aggregates. Philos. Trans. R. Soc. Lond. A. 344:377–401. [Google Scholar]
- 46.Ljunggren, S., and J. C. Eriksson. 1992. Minimal-surfaces and Winsor III microemulsions. Langmuir. 8:1300–1306. [Google Scholar]
- 47.Shemesh, T., A. Luini, V. Malhotra, K. N. J. Burger, and M. M. Kozlov. 2003. Prefission constriction of Golgi tubular carriers driven by local lipid metabolism: a theoretical model. Biophys. J. 85:3813–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tenchov, B. G., R. C. MacDonald, and D. P. Siegel. 2006. Cubic phases in phosphatidylcholine-cholesterol mixtures: cholesterol as membrane “fusogen”. Biophys. J. 91:2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siegel, D. P., and J. L. Banschbach. 1990. Lamellar/inverted cubic (L alpha/QII) phase transition in N-methylated dioleoylphosphatidylethanolamine. Biochemistry. 29:5975–5981. [DOI] [PubMed] [Google Scholar]
- 50.Deme, B., M. Dubois, and T. Zemb. 2002. Swelling of a lecithin lamellar phase induced by small carbohydrate solutes. Biophys. J. 82:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petrache, H. I., S. Tristram-Nagle, D. Harries, N. Kucerka, J. F. Nagle, and V. A. Parsegian. 2006. Swelling of phospholipids by monovalent salt. J. Lipid Res. 47:302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koynova, R., B. Tenchov, and G. Rapp. 1999. Effect of PEG-lipid conjugates on the phase behavior of phosphatidylethanolamine dispersions. Colloids Surf. A Physicochem. Eng. Aspect. 149:571–575. [Google Scholar]