

Abstract
It is well known, although not well understood, that smoking and eating just do not go together. Smoking is associated with decreased food intake and lower body weight. Nicotine, administered either by smoking or by smokeless routes, is considered the major appetite-suppressing component of tobacco. Perhaps the most renowned example of nicotine's influence on appetite and feeding behavior is the significant weight gain associated with smoking cessation. This article presents an overview of the literature at, or near, the interface of nicotinic receptors and appetite regulation. We first consider some of the possible sites of nicotine's action along the complex network of neural and non-neural regulators of feeding. We then present the hypothesis that the lateral hypothalamus is a particularly important locus of the anorectic effects of nicotine. Finally, we discuss the potential role of endogenous cholinergic systems in motivational feeding, focusing on cholinergic pathways in the lateral hypothalamus.
Keywords: orexin, M-H, lateral hypothalamus, acetylcholine
INTRODUCTION
Nicotine, the major addictive component of tobacco, reduces appetite and alters feeding patterns typically resulting in reduced body weight (Grunberg, 1986; Grunberg et al., 1986; Miyata et al., 1999). Combined epidemiologic studies of more than a quarter of a million subjects converge on a strong inverse relationship between cigarette smoking and body weight, with smokers weighing significantly less than non-smokers of the same age and sex (Albanes et al., 1987; Levin et al., 1987). Particularly striking is the hyperphagia and resultant weight gain that accompanies smoking cessation of 70–80% of people who quit smoking, with women being most affected (Grunberg et al., 1986; Klesges et al., 1989; Pomerleau, 1999; Pomerleau et al., 2000). Withdrawal of nicotine leads to increases in body weight of sufficient magnitude to confound maintained abstinence, significantly altering the outcomes of smoking cessation programs as manifest in weight-gain associated relapse (Nordstrom et al., 1999; Pomerleau, 1999).
The effects of chronic nicotine administration on appetite suppression, decreased food intake, and leanness are not confined to human subjects (e.g., Blaha et al., 1998; Bray, 2000; Zhang et al., 2001). Likewise, nicotine withdrawal from laboratory animals previously subjected to chronic drug administration induces hyperphagia and weight gain that are at least qualitatively similar to the effects elicited in humans by smoking cessation without nicotine replacement therapy (Grunberg et al., 1986; Levin et al., 1987). Such findings indicate that postcessation weight-gain results from a fundamental physiologic response, rather than just from socio-cultural factors, and underscore the importance of understanding the mechanisms that underlie nicotine and nicotine withdrawal-induced changes in appetite and food intake.
POSSIBLE MECHANISMS FOR THE EFFECTS OF NICOTINE ON FEEDING BEHAVIOR
Even a simplified schematic of the basic neural and non-neural regulators of energy balance cannot hide the mechanistic complexity of feeding behavior (see Fig. 1 and Woods et al., 1998; Kalra et al., 1999; Schwartz et al., 2000; Chiesi et al., 2001; Speigelman and Flier, 2001, for recent reviews). Peripheral signals of adiposity and satiety—in the form of both chemical and mechanical indicators of sufficient metabolic stores and food intake—arise from adipose, liver, pancreas, and gastrointestinal tissues. These multiple signals assure accurate feedback in the regulation of “start-feeding” versus “stop-feeding” behaviors. These peripheral signals are carried by both humoral and neural routes, and include sugars, peptides (e.g., leptin, insulin, cholecystokinin, TNFα), eicosanoids, fatty acids, and other lipid-derived molecules (e.g., prostaglandins, triglycerides, LDL). Peripheral neuronal reporters of satiety, originating with mechanical and chemical signals from the gut and liver, are primarily conveyed via vagal afferents to the nucleus of the solitary tract (NTS) resulting in the activation of both ascending and descending circuits involved in food intake. Humoral indicators of adequate fat storage (such as leptin) act directly on central neurons to regulate autonomic and limbic circuits controlling motivational aspects of feeding (see Fig. 1 and Woods et al., 1998; Kalra et al., 1999; Schwartz et al., 2000; Chiesi et al., 2001; Speigelman and Flier, 2001, for recent reviews).
Figure 1.
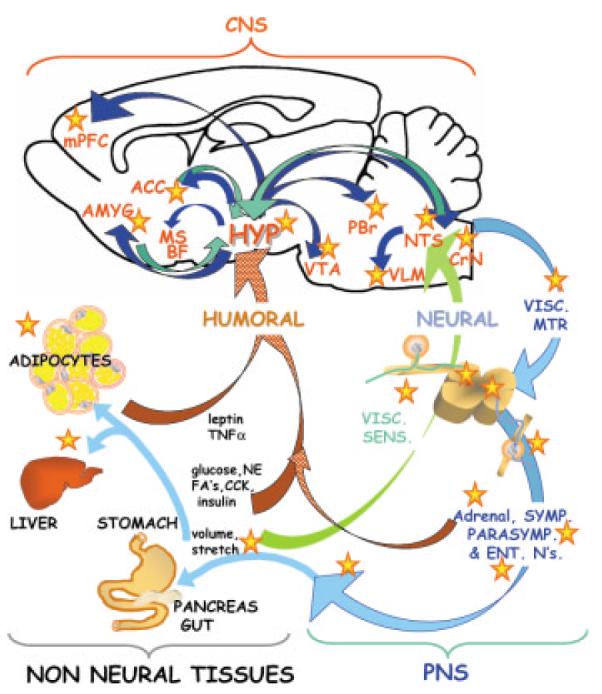
Possible loci of nicotine effects on feeding behavior. The brain receives a multitude of signals from the periphery reporting on adequacy of food intake and energy balance. These include humoral signals (red ribbon arrow) such as hormones and cytokines (e.g., leptin, TNFα, insulin, cholescystokinin, norepinephrine) and metabolites (e.g., glucose and fatty acids) as well as neural signals (green ribbon arrow). Within the hypothalamus (HYP) and the nucleus of the tractus solitarius (NTS) this information is integrated and transmitted to multiple brain regions (dark blue arrows), and the appropriate behavior is elicited. In addition to these central responses, the PNS (light blue) neurons including sympathetic, parasympathetic, and enteric, innervate the gastrointestinal tract, adipose depots, and endocrine organs. Possible sites at which nicotine might modify feeding behavior or energy balance are indicated with stars. A number of non-neural tissues express nAChRs, and could repsond directly to nicotine. In the CNS, nicotine could act within the hypothalamus, the NTS, and in the regions throughout the neuroaxis to which these structures project (mPFC, medial septal, and basal forebrain nuclei; VTA, ventral tegmental area; NTS, nucleus tractus solitarius; PBr, parabrachial nucleus; VLM, ventrolateral medullary nucleus; CrN, cranial nerve nuclei; VISC. SENS and VISC MTR, visceral sensory and motor neurons; NE, norephnephrine, ENT, N's, enteric neurons; CCK, cholecystokinin; FAs, fatty acids).
This review, and the diagram in Figure 1, emphasizes the key integrative role of hypothalamic neurons in the relay of feeding-linked information to and from limbic and autonomic structures throughout the neuraxis. Thus, the hypothalamus coordinates motivation and emotion-related features of feeding behavior (via direct interactions with medial prefrontal; mPFC and cingulate cortex, basal forebrain and medial septal nuclei; MS/BF) with more fundamental aspects of appetitive and aversive responses (via interaction with nucleus accumbens, ACC, amygdala AMYG, ventral tegmental area; VTA, substantia nigra and raphe; e.g., Bittencourt and Elias, 1998; Broberger et al., 1998; Peyron et al., 1998; Stratford and Kelley, 1999; Pajolla et al., 2001; Fadel and Deutch, 2002). In this manner, it is the interactions of the hypothalamus that convey the emotional salience of feeding “context” from the cortex to brainstem autonomic nuclei (e.g., the nucleus tractus solitarius, NTS, parabrachial, PBr, and ventrolateral medullary nuclei, VLM) and ultimately to the basic visceral motor and sensory mediators of biting, chewing, and swallowing (e.g., cranial nerve nuclei 7, 9 and 10; CrN, see Fig. 1).
The first challenge, for those of us interested in understanding the mechanism of nicotine-dependent hypo- versus. hyperphagia, is to find where in this complex sequence of sites nicotine can act. The answers from the literature, as outlined below, include several possible loci of the nicotinic regulation of food intake (indicated by red stars in Fig. 1). From these possible sites of action, some of which we touch on below, our discussion focuses on nicotine effects on the excitability of lateral hypothalamic projection neurons and potentially important contributions of endogenous cholinergic systems in the control of normal feeding behavior.
DOES NICOTINE AFFECT FEEDING VIA INTERACTIONS AT PERIPHERAL (NON-NEURAL) SITES OF ENERGY METABOLISM?
Many reports implicate nicotine as the appetite suppressing component of smoking (Grunberg, 1986; Blaha et al., 1998; Li et al., 2000; Zhang et al., 2001). Studies of feeding behavior reveal that smoking or nicotine administration decreases meal size, measured as the amount of food intake per meal, without substantial changes in meal number (Blaha et al., 1998; Miyata et al., 2001). Although studies of nicotine-induced changes in overall metabolic rate are more variable (Stamford et al., 1986; Sztalryd et al., 1996), nicotine administration has been shown to alter metabolic processing in humans, intact animals and in both hepatocytes and adipocytes, in vitro (Sztalryd et al., 1996; Ashakumary and Vijayammal, 1997; Arai et al., 2001). Nicotine decreases lipolysis by inhibiting lipoprotein lipase activity, decreasing triglyceride uptake and hence lessening net storage in adipose tissue (Sztalryd et al., 1996). Activation of nicotinic receptors also induces the expression of uncoupling protein 1 (UCP1) in both white and brown adipose tissue (Arai et al., 2001). As UCP1 shifts the balance of energy metabolism from the generation of ATP to the generation of heat, this represents a shift in metabolic efficiency compatible with the average lower body-weight of smokers (Klesges et al., 1989). Overall, the data on the effects of nicotine on energy metabolism in peripheral, non-neural tissue are reasonably compelling and certainly consistent with a significant contribution of these mechanisms to nicotine's anorectic activity.
Could the Anorectic Effects of Nicotine Involve Regulation of “Adiposity” Signaling by Leptin?
Leptin is a peptide hormone subject to regulated synthesis and release by adipocytes during the well-fed state (Zhang et al., 1994; Ahima et al., 2000). Leptin–receptor activation in the CNS and PNS decreases appetite and increases energy expenditure, thus functioning as a key afferent component of the negative feedback loops that stabilize adipose tissue mass. Intravenous leptin increases norepinephrine release from the adrenal gland and increases sympathetic nerve activity to both thermogenic and non-thermogenic tissues (e.g., brown adipose vs. adrenal gland; see Fig. 1). Administration of exogenous leptin to ob/ob mice, which have a phenotype of massive over-eating and obesity due to the lack of leptin production, markedly decreased food intake and body weight (Campfield et al., 1995; Halaas et al., 1995; Pelleymounter et al., 1995). Animals of the db/db genotype are similarly obese, but in this case it is due to the lack of expression of leptin receptors and, as such, their weight is unaffected by exogenous leptin administration (Caro et al., 1996). Thus, regulated synthesis of leptin and its cytokine receptor are necessary components in the maintenance of normal body weight and energy homeostasis (see Flier and Maratos-Flier, 1998; Ahima et al., 2000; Schwartz et al., 2000; Elmquist, 2001, for reviews).
Nicotine, as an appetite suppressant, may decrease feeding by increasing leptin levels and/or by enhancing step(s) along the leptin–receptor-mediated signaling cascade. Several laboratories have attempted to test these hypotheses; overall, it appears that nicotine-induced changes in leptin levels—measured either as leptin mRNA in adipocytes or as plasma leptin concentration—are highly dependent on experimental protocol. Some studies indicate that chronic smokers have significantly higher serum leptin concentrations than nonsmokers, consistent with nicotine's hypophagic effects (Hodge et al., 1997; Wei et al., 1997; Eliasson and Smith, 1999; Nicklas et al., 1999). Other reports find no correlation (either positive or negative) between chronic nicotine exposure and serum leptin concentrations (Yoshinari et al., 1998; Donahue et al., 1999). Nevertheless, the consensus remains that the anorectic affects of nicotine are somehow linked to altered leptin signaling. Even investigators who found that chronic cigarette smokers had significantly reduced plasma leptin levels propose that the low leptin levels reflect nicotine-induced enhancement of leptin–receptor binding or increased sensitivity of downstream transduction cascades (Hodge et al., 1997; Wei et al., 1997). Recent analyses of nicotine affects on adipocytes in vitro—i.e., under relatively controlled conditions—revealed that the changes in leptin release depended on the duration of nicotine treatment and were not necessarily correlated with the degree of nicotine-induced decreases in food intake in vivo (Arai et al., 2001).
It remains to be tested whether nicotine can increase the efficacy of leptin–receptor signaling, as proposed. In principle, nicotine could enhance or augment leptin–receptor signaling by several transcriptionally independent mechanisms—acting in concert with reported electrophysiologic effects of leptin on specific populations of CNS and PNS neurons (Harvey et al., 1997; Spanswick et al., 1997; Niijima, 1998; Liu et al., 1999; Sha and Szurszewski, 1999; Shiraishi et al., 2000; Spanswick et al., 2000; Cowley et al., 2001). Alternatively, nicotine may act by enhancing leptin receptor-mediated JAK-STAT signal transduction or the transcriptional regulation of one or more of the identified target genes of leptin receptor signaling (Tartaglia, 1997; Elias et al., 1999; Ahima et al., 2000; Elias et al., 2000). Nevertheless, although nicotine and leptin elicit similar responses (whether at the level of excitability or transcriptional regulation) in certain hypothalamic, vagal afferent and sympathetic efferent neurons, their sites of action are often distinct. Thus, the anorexigenic actions of nicotine parallel those of leptin and appear to involve both common and distinct cellular targets.
DOES NICOTINE AFFECT APPETITE VIA INTERACTION WITH PERIPHERAL AUTONOMIC, ENTERIC, OR SENSORY NEURONS?
Nicotine—like many other anorexigenic substances, including leptin, CCK, and insulin—generally exerts opposite effects on the regulation of sympathetic outflow and appetite (Haynes et al., 1997; Yettefti et al., 1997; Niijima, 1998; Krisufek et al., 1999; Sha and Szurszewski, 1999; Bray, 2000). Thus, the net effects of nicotine include elevated blood pressure, heart rate, and gastric motility while eliciting a sustained decrease in food intake. Autonomic, sensory, and enteric neurons each constitute potentially important loci for nicotine-mediated changes in feeding behavior. Peripheral tissues involved in the regulation of food intake receive input from sympathetic and/or para-sympathetic neurons that are themselves controlled by cholinergic transmission. All autonomic, many enteric, and even a subset of sensory neurons are cholinoceptive, expressing a variety of nAChR subunits and nAChR subtypes that are potently activated by nicotine (e.g., Rust et al., 1994; Conroy and Berg, 1995; De Biasi et al., 2000; Roth et al., 2000; Cooper, 2001; Picciotto et al., 2001; Shoop et al., 2001). Thus, nicotine administration via smoking can evoke significant activation (and sustained inactivation, via desensitization) of many peripheral neuron components of visceral-sensory and visceral-motor pathways involved in the regulation of feeding.
The extent (and duration) of nicotine-elicited alterations in peripheral neuron activity depends on multiple factors, such as the concentration and duration of nicotine exposure, the subtype(s) of nicotinic AChR expressed and the pre- versus postsynaptic distribution of the nAChRs (for recent reviews, see Gotti et al., 1997; McGehee, 1999; De Biasi et al., 2000; Temburni et al., 2000; Cooper, 2001; Dani, 2001; Leonard and Bertrand, 2001; Picciotto et al., 2001, and articles in this volume). In addition to altering neuronal excitability directly by the activation of synaptic and “extra-synaptic” nAChRs, nicotine is an established modulator of transmitter release in the periphery. Nicotine enhances the release of both ACh and norepinephrine (NE) from pre- and postganglionic neurons by interaction with presynaptic nAChRs (e.g., McGehee et al., 1995; Krisufek et al., 1999; De Biasi et al., 2000; Leonard and Bertrand, 2001; Picciotto et al., 2001). Peripheral administration of nicotine results in decreased food intake and increased sympathetic activity to liver, adipose and gut, reminiscent of its effects on cardiovascular parameters (Bray, 2000). Although consistent with direct, nicotine-induced enhancement of norepinephrine release from peripheral nerve terminals, such changes are also due to more indirect routes of controlling food intake and sympathetic outflow (Fig. 1). Systemic administration of nicotine stimulates visceral afferents and activates brain stem autonomic control centers, such as the solitary tract, ventrolateral medullary, and parabrachial nuclei (noted in Fig. 1 as NTS, VLM, and PBr, respectively; Yettefti et al., 1997; Bray, 2000; Cooper, 2001; Tribollet et al., 2001). Nicotine-evoked responses are elicited in less than 25% of the NTS neurons when tested by direct ionotophoretic application, implicating both central and peripheral neuronal circuits in nicotine's effects on feeding (Yettefti et al., 1997). Other studies underscore the contribution of nAChRs on cranial nerve and spinal cord visceral motor neurons in the regulation of sympathetic and parasympathetic outflow (e.g., McGehee et al., 1995; Tribollet et al., 2001). In sum, nicotine's anorexigenic activity is likely due—at least in part—to alterations in the excitability of visceral sensory and motor neurons as well as to modulation of transmitter release at brain stem, spinal cord (preganglionic), and/or peripheral synapses.
Last, but not least, the activation of nicotinic receptors on peripheral neurons has also been linked to altered phospholipid and fatty-acid metabolism (e.g., Vijayaraghavan et al., 1995; Marin et al., 1997; Stella and Piomelli, 2001; Tieman et al., 2001, and references therein). As several of these metabolites affect appetite and/or modulate nAChRs, the reciprocal interaction of nicotinic and lipid-signaling pathways may constitute an important “feedback” (pun intended) mechanism for regulating peripheral signals and circuits controlling food intake (e.g., Vijayaraghavan et al., 1995; Marin et al., 1997; Bray, 2000; Du and Role, 2001; Tieman et al., 2001).
ARE THE ANORECTIC EFFECTS OF NICOTINE DUE TO MODULATION OF CENTRAL AUTONOMIC NEURONS
The Hypothalamus
The hypothalamus is a region of the brain critical for regulation of homeostatic processes such as feeding, thermoregulation, and reproduction (Elmquist et al., 1999). To accomplish these ends, the hypothalamus receives neural, endocrine, and metabolic signals, integrates these inputs, and engages distinct effector pathways, resulting in behavioral, autonomic, and endocrine responses. The essential role of the hypothalamus in appetite regulation was initially suggested by studies utilizing “localized” lesion and stimulation of the hypothalamus. Disruption of the ventromedial hypothalamus produced hyperphagia and obesity, while lesions of the lateral hypothalamus resulted in hypophagia and dramatic weight loss. Although early proposals for a ventromedial “satiety” center and a lateral hypothalamic “appetite” controller were oversimplified (and somewhat confounded by varying disruption of medial forebrain bundle projections), they correctly established the hypothalamus as a key contributor to motivational aspects of feeding (see Flier and Maratos-Flier, 1998; Woods et al., 1998; Elmquist et al., 1999; Kalra et al., 1999; Schwartz et al., 2000; Chiesi et al., 2001; Speigelman and Flier, 2001). The understanding of the chemical signals and neuronal circuitry involved in the regulation of appetite has grown substantially during the past decade. As such, it is particularly surprising that we know so little about the role of nicotinic receptors in the hypothalamic control of food intake in smokers, ex-smokers and nonsmokers. In Figure 2, and in the discussion below, we consider the key relays to, from and within the hypothalamus that may be involved in the anorexigenic actions of nicotine.
Figure 2.
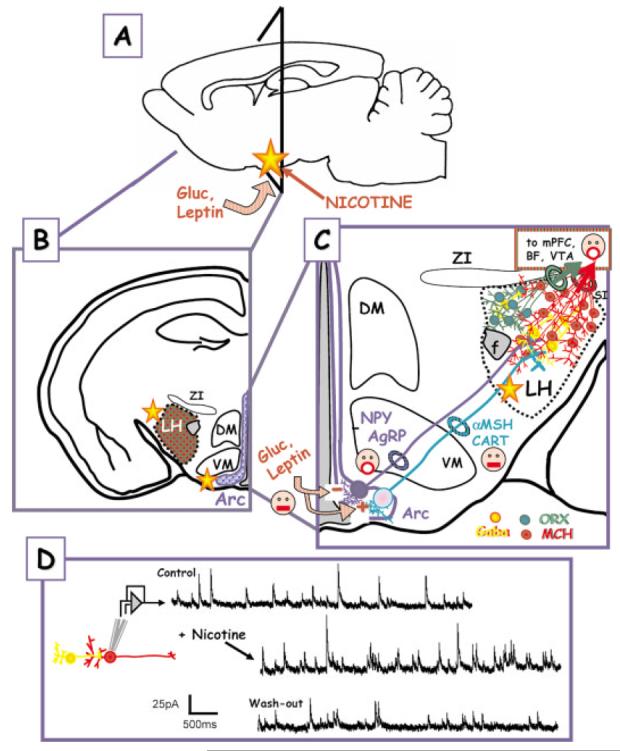
CNS sites of glucose, leptin, and (possibly) nicotine effects—up close and personal. (A) Glucose (Gluc), leptin, and nicotine signaling converge on the arcuate nucleus within the hypothalamus. The triangle shows the region from which the coronal sections in (B) and (C) are taken. (B) Higher magnification, coronal section showing the spatial relationship of the lateral hypothalamus (LH), the dorsal medial, and ventromedial hypothalamus (DM, VM) and the arcuate nuclus (Arc). ZI—zona inserta. (C) Glucose and leptin responsive neurons in the arcuate project to the LH. The first group of neurons, which are inhibited by the anorexigenic leptin signal (face with closed mouth), express the orexigenic peptides NPY and AgRP (face with open mouth). Leptin also excites a second group of neurons that contain the anorexigenic peptides, αMSH and CART. Both classes of Arc neurons project to the LH interacting with neurons and interneurons that express GABA (yellow), orexin (green), or MCH (red). Orexin and MCH neurons are orexigenic and project to other brain regions such as the mPFC, BF, VTA, etc. Key relays implicated in the regulation of feeding within the hypothalamic nuclei. The yellow stars represent possible loci of nicotine effects. (D) Nicotine treatment of LH neurons in vitro enhances neurotransmitter (GABA) release. The nicotine target is believed to be preynaptic nAChRs on GABAergic interneurons. The figure was modified from Jo and Role, 2002b, in press. (NPY, neuropeptide Y; AgRP, agouti-related peptide; αMSH, α-melanocyte-stiimulating hormone; CART, cocaine- and amphetamine-regulated transcript; ORX, orexin, MCH, melanin-concentrating hormone; f, fornix).
First and foremost, nicotinic receptors with the pharmacology of both α7-containing (α7*) and α/β-like pharmacology are detected throughout the hypothalamus (Harfstrand et al., 1988; Pabreza et al., 1991; Britto et al., 1992; Okuda et al., 1993; Shioda et al., 1997; O'Hara et al., 1998; Davila-Garcia et al., 1999; Hatton and Yang, 2002). Application of nicotine to in vitro preparations of hypothalamus elicits the usual panoply of neurophysiologic responses, including direct depolarization as well as alterations in GABA, glutamate, norepinephrine, and serotonin release (Pabreza et al., 1991; Meguid et al., 2000; Hatton and Yang, 2002). These findings are consistent with localization of nAChRs at both pre- and postsynaptic sites (Schwartz et al., 1984; Li and Pan, 2001). In situ hybridization with nAChR-subunit probes reveals moderate to high levels of expression of α4, α7, and β2-mRNAs in hypothalamus, with particularly prominent expression in the supraoptic and suprachiasmatic nuclei. Examination of the nAChR distribution within the subnuclei involved in the regulation of ingestive behavior further guided our search for candidate sites of nicotine's anorectic activity to focus on the lateral hypothalamus (Jo and Role, 2002, and unpublished observations).
Numerous studies have emphasized the contributions of the arcuate, ventromedial, dorsomedial, paraventricular, and/or lateral hypothalamic nuclei in appetite regulation. In view of the stated emphasis of this volume we will confine our discussion to the first and last of these regions as of particular interest to the nicotinologist.
The Arcuate Nucleus
The arcuate nucleus is located at the base of the hypothalamus on either side of the third cerebral ventricle (Arc; Fig 2), extending rostrocaudally from the posterior borders of the optic chiasm to the mamillary bodies. Two relatively recent observations have placed the arcuate nucleus in a position of prominence among the sites that are associated with the detection and integration of orexigenic versus anorexigenic signals [indicated by the open-mouthed vs. closed-mouth faces, respectively, in Fig. 2(C)]. First, the arcuate nucleus contains a high density of neurons that produce the orexigenic peptides, neuropeptide Y (NPY; Erickson et al., 1996), opioids, dynorphin (Neal and Newman, 1989), β-endorphin (Lantos et al., 1995), and galanin (Merchenthaler et al., 1993). The agouti-related peptide, a selective antagonist of αMSH receptors (see below), is coexpressed with NPY mRNA in the arcuate nucleus (Broberger et al., 1998). These two peptides also coexist in nerve terminals surrounding and in close apposition to perikarya and processes of both orexin (ORX) and melanin-concentrating hormone (MCH)-immunoreactive cells in the LH [Fig 2(C); Broberger et al., 1998; Horvath et al., 1999]. Thus, ORX and MCH expressing neurons within the lateral hypothalamus are considered likely downstream targets of NPY-positive projections from the arcuate nucleus and, as such, are important mediators of NPY-induced feeding signals.
Melanocortins such as α-melanocyte-stimulating hormone (αMSH) are peptides that are cleaved from the pro-opiomelanocortin (POMC) precursor molecule and exert their effects by binding to members of the family of melanocortin receptors (Lawrence et al., 1999). A synthetic agonist of these receptors suppresses food intake, whereas a synthetic antagonist has the opposite effect (Fan et al., 1997). The report that mice lacking the MC4-type of αMSH receptor are hyperphagic and very obese indicates that under normal conditions tonic signaling by MC4 receptors limits food intake and body fat mass (Huszar et al., 1997).
The cocaine- and amphetamine-regulated transcript (CART) was originally identified as an mRNA involved in CNS responses to drugs of abuse. Intracerebroventricular (i.c.v.) administration of CART encoded peptides inhibits normal, starvation-, and NPY-induced feeding in rats (Kristensen et al., 1998; Lambert et al., 1998), whereas i.c.v. administration of neutralizing anti-CART sera increases night-time feeding (Kristensen et al., 1998). These results support a role for CART peptides as endogenous inhibitors of feeding. Interestingly, POMC and CART are colocalized in a subset of Arc neurons distinct from the NPY/AgRP coexpressing neurons (Elias et al., 1998). Taken together, these studies of the expression patterns and the distinct downstream effects of NPY/AgRP vs. αMSH/CART on feeding indicate that circuits originating in the Arc have highly specialized roles in energy homeostasis.
In addition, due to the absence of a blood–brain barrier, the arcuate nucleus is strategically positioned to be in direct communication with peripheral signals such as leptin and insulin (reviewed in Kalra et al., 1999). A majority of both NPY/AgRP and POMC/CART neurons express leptin receptors (Cowley et al., 2001). Of particular importance in this regard are studies demonstrating that leptin differentially regulates the NPY/AgRP versus POMC/CART neurons projecting from the arcuate to the LH nuclei [indicated by + vs. − in Fig. 2(C); Elias et al., 1999; Cowley et al., 2001]. In addition, the presence of direct glucose-sensing neurons within the arcuate (as well as within the VMN) underscores their likely contribution to direct monitoring and transduction of primary metabolic signals to control food intake (e.g., Spanswick et al., 2000). The arcuate nucleus is thus considered to play a key integrative role between the initial afferent signals from the periphery (e.g., leptins and glucose) and CNS responses in the regulation of appetite.
The Lateral Hypothalamus
The lateral hypothalamus is comprised of a band of cells contiguous with the dorsomedial, and lateral to the ventromedial, nuclei of the hypothalamus [Fig. 2(B) and (C)], extending rostrally from the mesencephalic tegmentum to the lateral preoptic area. It is the site of passage for the medial forebrain bundle, a major conduit of projections connecting forebrain and midbrain structures with each other and with several hypothalamic sites. Lesions in the LH produced temporary aphagia, adipsia, and loss in body weight consistent with an important contribution of LH to appetite regulation. The demonstration that lateral hypothalamic neurons are essentially unique in their expression of the orexigenic neuropeptides, melanin-concentrating hormone (MCH) (Qu et al., 1996), and orexin (ORX) (Sakurai et al., 1998) (also called hypocretin; de Lecea et al., 1998) reaffirmed the essential role of the LH in motivational aspects of feeding. Starvation sharply increases the levels of mRNA encoding both of these orexigenic signals (Qu et al., 1996; Sakurai et al., 1998), when leptin levels rapidly fall (Ahima et al., 1996).
Cells expressing MCH and ORX comprise distinct populations of neurons within the LH. ORX cells are located primarily in the perifornical LH, whereas MCH cells are more broadly distributed within the LH, with the most anterior MCH neurons lining the borders of the LH, by the zona incerta and substantia innominata. More posterior MCH neurons are in the ventral lateral portion of the perifornical area and surround the ORX population (Broberger et al., 1998; Fadel and Deutch, 2002). Interestingly, CART-positive cell bodies in the LH coexpress MCH and glutamic acid decarboxylase mRNA (Elias et al., 2001). ORX-immunoreactive terminals originating from the LH make direct synaptic contact with neurons of the arcuate nucleus that express NPY and contain leptin receptors (Horvath et al., 1999). ORX-containing neurons also express leptin receptor immunoreactivity (Horvath et al., 1999). The excitatory actions of ORX could increase NPY release, resulting in enhanced feeding behavior, whereas leptin, released from adipose tissue as an indicator of fat stores, would have the opposite effect on the same neurons, leading to a decrease in NPY and NPY-mediated hypothalamic functions.
Why is the LH an important candidate locus of nicotine-elicited appetite suppression? First, nicotine administration into the LH significantly decreases food intake (Miyata et al., 1999; Yang et al., 1999; Miyata et al., 2001). Second, nicotine treatment elicits both short- and long-term changes in the release of a variety of transmitters in LH (Miyata et al., 1999; Meguid et al., 2000; Li and Pan, 2001; Zhang et al., 2001). Third, nicotine alters the expression of feeding-related neuropeptides and their receptors within the LH (Frankish et al., 1995; Kane et al., 2000; Li et al., 2000a, Li et al., 2000b; Kane et al., 2001). In particular, nicotine regulates dopamine and serotonin release from extrinsic projections to the LH (Miyata et al., 1999; Meguid et al., 2000) and it modulates GABA and glutamate transmission within the LH [Fig. 2(D) and Y. Jo and L. Role, unpublished observations]. Likewise, nicotine regulates the levels of expression of NPY, orexin and an orexin receptor. The complexity arises in assessing which of the many changes that are elicited by nicotine in the lateral hypothalamus underlie the nicotine-induced hypophagia and, perhaps even more importantly, the persistent hyperphagia accompanying nicotine cessation (Grunberg et al., 1986; Levin et al., 1987; Nordstrom et al., 1999; Pomerleau et al., 2000).
Does Nicotine Affect Appetite via Regulation of Release from Dopaminergic or Serotoninergic Projections to the Lateral Hypothalamus?
First let's consider the effects of nicotine on the release of dopamine and serotonin, two critical food intake-related neurotransmitters. The absence of dopamine production (in mice that cannot express tyrosine hydroxylase (Szczypka et al., 2000) results in an inability to initiate feeding, although the basic motor ability to eat is unaffected. In the hypothalamus, dopamine release is associated with the duration of meal consumption, an important determinant of feeding pattern. As dopamine is required to initiate meals, decreased dopamine is associated with reduced meal number and meal duration (reviewed in Meguid et al., 2000). In contrast, serotonin is implicated in the inhibition of food intake (see Meguid et al., 2000). Serotonin release in the hypothalamus is enhanced during feeding (D.H. Schwartz et al., 1989) and is thought to promote satiety. Mice lacking serotinergic receptors display food intake and body weight-related abnormalities (Nonogaki et al., 1998).
Studies of nicotine infusion and amine release in hypothalamus support the idea that activation of nAChRs on serotinergic projections to LH may contribute to the anorectic effects of nicotine. The role of dopamine release in nAChR-induced appetite suppression—as opposed to meal initiation—is less clear (Miyata et al., 1999; Yang et al., 1999; Meguid et al., 2000). Continuous nicotine administration into the LH induces long-lasting increases in 5-HT release that parallel the sustained suppression of food intake in the same animals. If enhanced serotinergic transmission in hypothalamus signals satiety as proposed (Schwartz et al., 1989; Nonogaki et al., 1998), nicotine may act as a false indicator of the well-fed state, resulting in early meal termination (i.e., decreasing meal size and duration, as previously reported; Miyata et al., 2001) due to nAChR-mediated presynaptic facilitation of serotonin release.
Does Nicotine Affect Appetite via Regulation of NPY Neurons that Project to the Lateral Hypothalamus?
NPY, a 36-amino acid neuropeptide, is abundant in the brain of rats, and is highly concentrated in the hypothalamus. NPY administration is potently orexigenic and expression of NPY mRNA in the arcuate nucleus is increased in response to fasting or chronic, but moderate food restriction (see Woods et al., 1998; Kalra et al., 1999; Lawrence et al., 1999; M.W. Schwartz et al., 2000, for reviews). A simple prediction is that if NPY signaling is a target for nicotine's anorectic action, then nicotine treatment might suppress the expression of NPY or NPY receptors. In the short term, this appears to be true. Frankish and collaborators found that acute (24-h) nicotine administration, reduced food intake by 30%, and lowered NPY and NPY mRNA levels in the Arc nucleus by 35% (Frankish et al., 1995). The situation following chronic nicotine administration is murky; however, as 2 weeks of nicotine treatment, lowers food intake but significantly increases levels of NPY mRNA. Li and colleagues (Li et al., 2000) found that after 14 days nicotine suppressed food intake and elicited a dose-dependent increase in hypothalamic NPY mRNA levels (by 20–50% depending on the dose of nicotine) and increased NPY immunoreactivity by 24–69% (the magnitude of the increase varied in different regions of the hypothalamus). In a valiant attempt to resolve the conflict between nicotine-induced hypophagia and the observed induction of a renowned orexigenic peptide, the authors propose that nicotine somehow modifies the sensitivity of NPY and/or nicotinic acetylcholine receptors; consequently, modulating NPY synthesis and food intake. A long-term increase in NPY levels might desensitize the NPY receptor systems. We provide an alternative explanation, based on initial studies of nicotine effects on LH circuits, below.
Does Nicotine Affect Appetite via Regulation of Orexin Neurons in the Lateral Hypothalamus?
Orexin A and B (also called hypocretins) are two peptides derived from a single 131-residue precursor peptide (prepro-orexin), that are produced exclusively in the LH (Sakurai, 1999; Shiraishi et al., 2000; Willie et al., 2001; Kotz et al., 2002). In view of the orexigenic activity of these peptides, the simplest predicted link between nicotine-induced hypophagia and orexins would be that nicotine depresses orexin signaling. No such luck.
Kane and colleagues (Kane et al., 2000) showed that chronic nicotine treatment (14 days) elicited a dose-dependent increase in the expression of prepro-orexin mRNA expression in hypothalamus. This increase in prepro-orexin mRNA in nicotine-treated animals is expected to yield increased levels of orexin protein in the LH and/or at sites of projection of orexin-producing neurons. This increase might lead to downregulation of orexin and/or NPY receptor expression. Indeed, the same authors demonstrated that hypothalamic orexin-A binding sites were downregulated by chronic nicotine treatment (Kane et al., 2001). It should be kept in mind that orexin may regulate sleep/wakefulness states, rather than feeding behavior (Hungs and Mignot, 2001; Sutcliffe and De Lecea, 2002). Mice lacking orexin show a phenotype similar to human narcolepsy, including behavioral arrests and premature entry into rapid eye movement (REM) sleep (Chemelli et al., 1999). Hara and colleagues (Hara et al., 2001) observed similar results in mice lacking orexin neurons. Thus, effects of nicotine on orexin signaling might be manifested as alterations in sleep/wake states rather than feeding behavior.
Does Nicotine Affect Appetite via Regulation of MCH Neurons in Hypothalamus?
MCH, another peptide uniquely expressed in lateral hypothalamic neurons, is also orexigenic (see discussion above and Fig. 2). Fasting animals upregulate hypothalamic MCH mRNA levels and MCH receptor antagonists block NPY, β-endorphin, and fasting-induced increases in food intake. Mice lacking MCH have lower body weights and enhanced leanness, associated with marked hypophagia, compared with control animals (Shimada et al., 1998). Recent studies showing that MCH receptor-deficient mice, like those lacking MCH expression, are hypophagic, further supporting the importance of MCH in appetite regulation (Marsh et al., 2002).
The activity of MCH-expressing neurons is elaborately regulated by interneurons within the LH, inputs from other hypothalamic nuclei and by projections from other brain regions (e.g., Qu et al., 1996; Bayer et al., 1999; Fig. 2; see below, and Fig. 3). Orexigenic, NPY-positive inputs from the Arc strongly enhance the activity of MCH neurons (Flier and Maratos-Flier, 1998). AgRP neurons have the same net effect as costored NPY on the activity of MCH neurons but the mechanisms are distinct. AgRP is an antagonist of receptors for the anorexigenic peptide, αMSH, and thereby inhibits the inhibition (!) normally mediated by αMSH–MC4 receptor interactions. Decreased activation of MCH neurons results from enhanced inhibitory GABAergic input and by inhibitory influences of CART/αMSH projections (Fan et al., 1997). Finally, MCH projections may mediate a positive feedback control by inhibition of GABAergic inputs (Gao and van den Pol, 2001).
Figure 3.

Acetylcholine containing neurons are found throughout the lateral hypothalamus. (A) ACh-positive neurons (yellow/green circles) and ACh-positive projections (green, yellow/green shading; green represents highest density) are distributed throughout the LH. Some ACh-positive neurons are also found in the Arc. (B) ACh-positive neurons identified by VAChT (vesicular ACh transport) immunoreactivity and MCH-positive neurons (red—MCH immunoreactivity) are interspersed. (C) VAChT-positive bouton-like structures (red/brown immunostaining) are present in the perifornical region of the LH.
Recent work investigates the possibility that nicotinic receptor activation regulates the excitability of MCH neurons in LH [Fig. 2(D); Y. Jo and L. Role, unpublished observations]. In principle, nicotine could suppress appetite either by decreasing any of the multiple inputs that normally activate MCH neurons or by increasing the overall inhibition of MCH neurons. We propose that both mechanisms are involved in nicotine's actions [Fig. 2(C) and (D)].
GABA is the primary inhibitory transmitter in the hypothalamus (Elias et al., 2001) and a prominent component of synaptic input in in vitro preparations of rat, chick, and mouse lateral hypothalamus (Gao and van den Pol, 2001; Jo and Role, 2002a; Jo and Role, unpublished). Activation of nicotinic receptors by treatment with nicotine [Fig. 2(D)] or by increasing ACh release (not shown) strongly facilitates GABAergic transmission to LH neurons (Jo and Role, 2002b; Jo and Role, unpublished data). Initial efforts in mouse brain slices to identify the post synaptic LH neurons under study are consistent with our proposal that MCH-positive neurons receive GABAergic input and that GABA release is potently enhanced by presynaptic nAChRs. In this scenario, activation of presynaptic nAChRs on GABAergic interneurons would offset the pro-appetite, orexigenic signaling of NYP/AgRP inputs, favoring a net inhibition of MCH-positive neurons. This sort of synaptic tuning of LH neurons could explain, at least in part, nicotine's anorexigenic activity.
The model can also resolve the apparent conflict between elevated NPY expression and the sustained hypophagia that characterizes chronic nicotine exposure. Under these conditions, we would predict that the nAChR-mediated enhancement of GABA release is maintained and results in an elevated set point for steady-state inhibition of orexigenic outflow via MCH projections. Increased expression of NPY peptide (due to diminished negative regulation by leptin, as leptin levels fall with chronic nicotine exposure) cannot elicit sufficient activation of MCH neurons to override the elevated GABAergic inhibition. Obviously, this model can (and must) be tested. Whether correct in detail or not, such studies would provide a systematic foray into the effects of nicotine and nicotine withdrawal in appetite regulation.
How Might Endogenous Cholinergic Circuits Be Involved in the Normal Regulation of Appetite?
The effects of neuronal nAChR activation on hypothalamic activity, food intake, and body weight regulation beg the question of how central cholinergic systems might participate in normal appetite control. The role of cholinergic and/or cholinoceptive neurons in the hypothalamus has received relatively little attention. Several recent studies, however, implicate acetylcholine effects within the hypothalamus in the control of both neuroendocrine and behavioral functions, including food intake and arousal (Hara et al., 2001; Saper et al., 2001).
Cholinergic projections to neurons within the hypothalamus arise from both extrinsic and intrinsic sources; approximate areas of cholinergic input are indicated in Figure 3 by green (high level) and yellow (lower level) shading (Tago et al., 1987; Risold et al., 1989; Woolf, 1991; Schafer et al., 1998; Bayer et al., 1999). Cholinergic neurons intrinsic to the hypothalamus, identified by their positive immunoreactivity for choline acetyltransferase (ChAT), or the vesicular transporter for ACh (VAChT) are present in the arcuate nucleus and scattered through the paraventricular, periventricular, and posterior nuclei (Fig. 3; adapted from Tago et al., 1987; Risold et al., 1989; Woolf, 1991; Schafer et al., 1998; Bayer et al., 1999). Higher densities of both large and small ChAT-stained somata are found in the lateral hypothalamic area and in the adjacent substantia innominata (Tago et al., 1987). Extrinsic cholinergic projections to the hypothalamus arise primarily from the ponto-mesencephalic cell groups (i.e., the pedunculopontine and laterodorsal tegmental nucleus; Woolf, 1991).
The density of cholinergic fibers (whether intrinsic or extrinsic in origin) within the LH ranges from moderate to strong (see especially Tago et al., 1987; Bayer et al., 1999). LH areas with substantial numbers of ACh-positive projections include the perifornical area, the dorsal border of the LH (adjacent to the zona incerta) and, at a more anterior level than illustrated here, along the lateral border of the LH and substantia innominata. The overlap between MCH and cholinergic neurons and fibers is particularly striking, especially with respect to proposed mechanisms of nicotine effects in LH. In the zona incerta and perifornical regions of the rat LH, many choline acetyltransferase (ChAT) fibers are detected in the immediate vicinity of MCH-positive perikarya and their proximal dendrites [Bayer et al., 1999; and Fig. 3(B) and (C)].
Of course, new levels of complexity are added by changing the question from “how does nicotine affect appetite?” to an analysis of how central cholinergic circuits might participate in feeding regulation. An obvious, and substantial wrinkle, is that ACh (as opposed to nicotine) interacts with multiple subtypes of muscarinic as well as nicotinic AChRs. Several subtypes of both nicotinic and muscarinic AChRs (mAChRs) are expressed within the LH including α4, α7, and β2 type nAChRs, and m1 and m2 type mAChRs (Ehlert and Tran, 1990; Britto et al., 1992; Okuda et al., 1993; J. Wei et al., 1994).
So, what happens when cholinergic neurons or cholinergic projections in the lateral hypothalamus are activated? Little is known. Initial attempts, using changes in GABAergic transmission as an assay of elevated ACh release in LH slice, appear to confirm in vitro results demonstrating corelease of GABA and ATP, but differential modulation of GABAergic and purinergic transmission by nicotinic versus muscarinic receptors (Jo and Role 2002a, 2002b, and Y. Jo and L. Role, unpublished). The net effect of nicotinic receptor activation in hypothalamus—even with ATP and glutamate transmission added to the analysis—is increased inhibition. Muscarinic receptors mediate exactly the opposite effects: GABAergic transmission is depressed and purinergic transmission is enhanced.
Similar pharmacologic approaches to assessing the role of cholinergic signaling in LH examine the effects of carbachol treatment on expression of MCH mRNA in hypothalamic slices in vitro. Carbachol induces a rapid increase in MCH mRNA, which is abolished both by atropine, a muscarinic antagonist, and hexamethonium, a nicotinic antagonist (Bayer et al., 1999). This finding is consistent with studies examining the regulation of MCH mRNA in animals lacking specific muscarinic signaling pathways. MCH mRNA is increased in WT, fasted animals, thereby inducing a compensatory increase in food intake. However, MCH mRNA levels are significantly reduced, even with fasting, in certain types of muscarinic receptor knock-out mice (Yamada et al., 2001).
Taken together, the data are consistent with endogenous cholinergic control of hypothalamic circuits involved in food intake. Basal levels of cholinergic activity in the LH appear to elicit a low level of tonic inhibition that involves nicotinic receptor-mediated enhancement of GABAergic transmission distinct from its effects in other hypothalamic nuclei (see D.P. Li and Pan, 2001; D.P. Li et al., 2001). Increased ACh levels appear to activate M3 receptor-mediated stimulation of MCH mediated-orexigenic pathways (Shimada et al., 1998; Yamada et al., 2001; Marsh et al., 2002). The muscarinic induction of MCH signaling must occur along the LH circuit past the point of leptin activation of the αMSH/CART system, but before the MCH neuron. In view of in vitro findings and prior proposals (Flier and Maratos-Flier, 1998; Elmquist et al., 1999; Jo and Role, 2002b; Y Jo and L. Role, unpublished), it is tempting to speculate that cholinoceptive GABAergic neurons are the missing link between the arcuate projections to the LH and the MCH neurons that coordinate cortical and brainstem regions involved in motivational feeding (Fadel and Deutch, 2002; Qu et al., 1996; Bittencourt and Elias, 1998; Elias et al., 2001; Marsh et al., 2002).
SUMMARY, CONCLUSIONS, AND FUTURE STUDIES
The literature is clear with respect to the effects of nicotine and nicotinic receptors in the regulation of appetite. Smokers are leaner and smoking cessation in the absence of nicotine replacement therapy typically results in significant and sustained hyperphagia and weight gain.
Identifying the cellular substrates of nicotine action is no small task. Despite numerous reports on the seriousness of nicotine-cessation related weight gain, considerable progress in our understanding of nAChRs (to wit this volume) and staggering discoveries in the identification of the molecular scaffolds of feeding behavior, the interface between the fields of smoking and eating remains sketchy, at best. We are still lacking systematic studies aimed at identifying the major regulatory nodes of where nicotine effects and appetite control converge. Our knowledge of the role of central cholinergic circuits in appetite is embryonic, despite impressive efforts in the neuroanatomy and immunocytochemistry of cholinergic neurons and their diffuse terminal fields. In sum, it appears that the physiologists—from systems to synaptic-analysts—have not (ahem) towed their weight in addressing these fundamental issues in the neural regulation of feeding behavior. There's lots of work to do.
Acknowledgments
Contract grant sponsor: National Institutes of Health; contract grant numbers: NS29071, NS22061, and CA79737.
REFERENCES
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front Neuroendocrinol. 2000;21:263–307. doi: 10.1006/frne.2000.0197. [DOI] [PubMed] [Google Scholar]
- Albanes D, Jones DY, Micozzi MS, Mattson ME. Associations between smoking and body weight in the US population: analysis of NHANES II. Am J Public Health. 1987;77:439–444. doi: 10.2105/ajph.77.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Kim K, Kaneko K, Iketani M, Otagiri A, Yamauchi N, Shibasaki T. Nicotine infusion alters leptin and un-coupling protein 1 mRNA expression in adipose tissues of rats. Am J Physiol Endocrinol Metab. 2001;280:E867–E876. doi: 10.1152/ajpendo.2001.280.6.E867. [DOI] [PubMed] [Google Scholar]
- Ashakumary L, Vijayammal PL. Effect of nicotine on lipoprotein metabolism in rats. Lipids. 1997;32:311–315. doi: 10.1007/s11745-997-0038-8. [DOI] [PubMed] [Google Scholar]
- Bayer L, Risold PY, Griffond B, Fellmann D. Rat diencephalic neurons producing melanin-concentrating hormone are influenced by ascending cholinergic projections. Neuroscience. 1999;91:1087–1101. doi: 10.1016/s0306-4522(98)00678-2. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Elias CF. Melanin-concentrating hormone and neuropeptide EI projections from the lateral hypothalamic area and zona incerta to the medial septal nucleus and spinal cord: a study using multiple neuronal tracers. Brain Res. 1998;805:1–19. doi: 10.1016/s0006-8993(98)00598-8. [DOI] [PubMed] [Google Scholar]
- Blaha V, Yang ZJ, Meguid M, Chai JK, Zadak Z. Systemic nicotine administration suppresses food intake via reduced meal sizes in both male and female rats. Acta Med. 1998;41:167–173. [PubMed] [Google Scholar]
- Bray GA. Reciprocal relation of food intake and sympathetic activity: experimental observations and clinical implications. Int J Obes Relat Metab Disord. 2000;24(Suppl 2):S8–S17. doi: 10.1038/sj.ijo.0801269. [DOI] [PubMed] [Google Scholar]
- Britto LR, Keyser KT, Lindstrom JM, Karten HJ. Immunohistochemical localization of nicotinic acetylcholine receptor subunits in the mesencephalon and diencephalon of the chick (Gallus gallus) J Comp Neurol. 1992;317:325–340. doi: 10.1002/cne.903170402. [DOI] [PubMed] [Google Scholar]
- Broberger C, De Lecea L, Sutcliffe JG, Hokfelt T. Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol. 1998;402:460–474. [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chiesi M, Huppertz C, Hofbauer KG. Pharmacotherapy of obesity: targets and perspectives. Trends Pharmacol Sci. 2001;22:247–254. doi: 10.1016/s0165-6147(00)01664-3. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Berg DK. Neurons can maintain multiple classes of nicotinic acetylcholine receptors distinguished by different subunit compositions. J Biol Chem. 1995;270:4424–4431. doi: 10.1074/jbc.270.9.4424. [DOI] [PubMed] [Google Scholar]
- Cooper E. Nicotinic acetylcholine receptors on vagal afferent neurons. Ann N Y Acad Sci. 2001;940:110–118. doi: 10.1111/j.1749-6632.2001.tb03670.x. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Dani JA. Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry. 2001;49:166–174. doi: 10.1016/s0006-3223(00)01011-8. [DOI] [PubMed] [Google Scholar]
- Davila-Garcia MI, Houghtling RA, Qasba SS, Kellar KJ. Nicotinic receptor binding sites in rat primary neuronal cells in culture: characterization and their regulation by chronic nicotine. Brain Res Mol Brain Res. 1999;66:14–23. doi: 10.1016/s0169-328x(98)00344-1. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Nigro F, Xu W. Nicotinic acetylcholine receptors in the autonomic control of bladder function. Eur J Pharmacol. 2000;393:137–140. doi: 10.1016/s0014-2999(00)00008-x. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue RP, Zimmet P, Bean JA, Decourten M, DeCarlo Donahue RA, Collier G, Goldberg RB, Prineas RJ, Skyler J, Schneiderman N. Cigarette smoking, alcohol use, and physical activity in relation to serum leptin levels in a multiethnic population: The Miami Community Health Study. Ann Epidemiol. 1999;9:108–113. doi: 10.1016/s1047-2797(98)00037-4. [DOI] [PubMed] [Google Scholar]
- Du C, Role LW. Differential modulation of nicotinic acetylcholine receptor subtypes and synaptic transmission in chick sympathetic ganglia by PGE(2) J Neurophysiol. 2001;85:2498–2508. doi: 10.1152/jn.2001.85.6.2498. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ, Tran LP. Regional distribution of M1, M2 and non-M1, non-M2 subtypes of muscarinic binding sites in rat brain. J Pharmacol Exp Ther. 1990;255:1148–1157. [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21:1375–1385. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- Elias CF, Lee CE, Kelly JF, Ahima RS, Kuhar M, Saper CB, Elmquist JK. Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol. 2001;432:1–19. doi: 10.1002/cne.1085. [DOI] [PubMed] [Google Scholar]
- Eliasson B, Smith U. Leptin levels in smokers and long-term users of nicotine gum. Eur J Clin Invest. 1999;29:145–152. doi: 10.1046/j.1365-2362.1999.00420.x. [DOI] [PubMed] [Google Scholar]
- Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic and behavioral effects of leptin. Int J Obes Relat Metab Disord. 2001;25:S78–S82. doi: 10.1038/sj.ijo.0801918. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Hollopeter G, Palmiter RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science. 1996;274:1704–1707. doi: 10.1126/science.274.5293.1704. [DOI] [PubMed] [Google Scholar]
- Fadel J, Deutch AY. Anatomical substrates of orexindopamine interactions: lateral hypothalamic projections to the ventral tegmental area. Neuroscience. 2002;111:379–387. doi: 10.1016/s0306-4522(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- Flier JS, Maratos-Flier E. Obesity and the hypothalamus: novel peptides for new pathways. Cell. 1998;92:437–440. doi: 10.1016/s0092-8674(00)80937-x. [DOI] [PubMed] [Google Scholar]
- Frankish HM, Dryden S, Wang Q, Bing C, MacFarlane IA, Williams G. Nicotine administration reduces neuropeptide Y and neuropeptide Y mRNA concentrations in the rat hypothalamus: NPY may mediate nicotine's effects on energy balance. Brain Res. 1995;694:139–146. doi: 10.1016/0006-8993(95)00834-d. [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol. 2001;533:237–252. doi: 10.1111/j.1469-7793.2001.0237b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- Grunberg NE. Nicotine as a psychoactive drug: appetite regulation. Psychopharmacol Bull. 1986;22:875–881. [PubMed] [Google Scholar]
- Grunberg NE, Bowen DJ, Winders SE. Effects of nicotine on body weight and food consumption in female rats. Psychopharmacology (Berlin) 1986;90:101–105. doi: 10.1007/BF00172879. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Hara, Beuckmann Ct, Nambu, Willie Jt, Chemelli Rm, Sinton Cm, Sugiyama, Yagami, Goto, Yanagisawa, Sakurai Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Harfstrand A, Adem A, Fuxe K, Agnati L, Andersson K, Nordberg A. Distribution of nicotinic cholinergic receptors in the rat tel- and diencephalon: a quantitative receptor autoradiographical study using [3H]-acetylcholine, [alpha-125I]bungarotoxin and [3H]nicotine. Acta Physiol Scand. 1988;132:1–14. doi: 10.1111/j.1748-1716.1988.tb08291.x. [DOI] [PubMed] [Google Scholar]
- Harvey J, McKenna F, Herson PS, Spanswick D, Ashford ML. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. J Physiol. 1997;504(Pt 3):527–535. doi: 10.1111/j.1469-7793.1997.527bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI, Yang QZ. Synaptic potentials mediated by alpha 7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci. 2002;22:29–37. doi: 10.1523/JNEUROSCI.22-01-00029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–278. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge AM, Westerman RA, de Courten MP, Collier GR, Zimmet PZ, Alberti KG. Is leptin sensitivity the link between smoking cessation and weight gain? Int J Obes Relat Metab Disord. 1997;21:50–53. doi: 10.1038/sj.ijo.0800362. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, van den Pol AN. Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. J Neurosci. 1999;19:1072–1087. doi: 10.1523/JNEUROSCI.19-03-01072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungs M, Mignot E. Hypocretin/orexin, sleep and narcolepsy. Bioessays. 2001;23:397–408. doi: 10.1002/bies.1058. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Jacob RJ, Dziura J, Medwick MB, Leone P, Caprio S, During M, Shulman GI, Sherwin RS. The effect of leptin is enhanced by microinjection into the ventromedial hypothalamus. Diabetes. 1997;46:150–152. doi: 10.2337/diab.46.1.150. [DOI] [PubMed] [Google Scholar]
- Jo YH, Role LW. Coordinate release of ATP and GABA at in vitro synapses of lateral hypothalamic neurons. J. Neurosci. 2002a;22:4794–4804. doi: 10.1523/JNEUROSCI.22-12-04794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Role LW. Differential modulation of synaptic transmission by nicotinic and muscarinic-receptor mediated pathways in lateral hypothalamus. J Neurophysiol. 2002b [Google Scholar]
- Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100. doi: 10.1210/edrv.20.1.0357. [DOI] [PubMed] [Google Scholar]
- Kane JK, Parker SL, Li MD. Hypothalamic orexin-A binding sites are downregulated by chronic nicotine treatment in the rat. Neurosci Lett. 2001;298:1–4. doi: 10.1016/s0304-3940(00)01730-4. [DOI] [PubMed] [Google Scholar]
- Kane JK, Parker SL, Matta SG, Fu Y, Sharp BM, Li MD. Nicotine up-regulates expression of orexin and its receptors in rat brain. Endocrinology. 2000;141:3623–3629. doi: 10.1210/endo.141.10.7707. [DOI] [PubMed] [Google Scholar]
- Klesges RC, Meyers AW, Klesges LM, La Vasque ME. Smoking, body weight, and their effects on smoking behavior: a comprehensive review of the literature. Psychol Bull. 1989;106:204–230. doi: 10.1037/0033-2909.106.2.204. [DOI] [PubMed] [Google Scholar]
- Kotz CM, Teske JA, Levine JA, Wang C. Feeding and activity induced by orexin A in the lateral hypothalamus in rats. Regul Pept. 2002;104:27–32. doi: 10.1016/s0167-0115(01)00346-9. [DOI] [PubMed] [Google Scholar]
- Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N, Larsen PJ, Hastrup S. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature. 1998;393:72–76. doi: 10.1038/29993. [DOI] [PubMed] [Google Scholar]
- Krisufek D, Stocker E, Boehm S, Huck S. Somatic and prejunctional nicotinic receptors in cultured rat sympathetic neurons show different agonist profiles. J Physiol. 1999;516(Pt 3):739–756. doi: 10.1111/j.1469-7793.1999.0739u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PD, Couceyro PR, McGirr KM, Dall Vechia SE, Smith Y, Kuhar MJ. CART peptides in the central control of feeding and interactions with neuropeptide Y. Synapse. 1998;29:293–298. doi: 10.1002/(SICI)1098-2396(199808)29:4<293::AID-SYN1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Lantos TA, Gorcs TJ, Palkovits M. Immunohistochemical mapping of neuropeptides in the premamillary region of the hypothalamus in rats. Brain Res Brain Res Rev. 1995;20:209–249. doi: 10.1016/0165-0173(94)00013-f. [DOI] [PubMed] [Google Scholar]
- Lawrence CB, Turnbull AV, Rothwell NJ. Hypothalamic control of feeding. Curr Opin Neurobiol. 1999;9:778–783. doi: 10.1016/s0959-4388(99)00032-x. [DOI] [PubMed] [Google Scholar]
- Leonard S, Bertrand D. Neuronal nicotinic receptors: from structure to function. Nicotine Tob Res. 2001;3:203–223. doi: 10.1080/14622200110050213. [DOI] [PubMed] [Google Scholar]
- Levin ED, Morgan MM, Galvez C, Ellison GD. Chronic nicotine and withdrawal effects on body weight and food and water consumption in female rats. Physiol Behav. 1987;39:441–444. doi: 10.1016/0031-9384(87)90370-2. [DOI] [PubMed] [Google Scholar]
- Li DP, Pan HL. Potentiation of glutamatergic synaptic input to supraoptic neurons by presynaptic nicotinic receptors. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1105–R1113. doi: 10.1152/ajpregu.2001.281.4.R1105. [DOI] [PubMed] [Google Scholar]
- Li DP, Pan YZ, Pan HL. Acetylcholine attenuates synaptic GABA release to supraoptic neurons through presynaptic nicotinic receptors. Brain Res. 2001;920:151–158. doi: 10.1016/s0006-8993(01)03055-4. [DOI] [PubMed] [Google Scholar]
- Li MD, Kane JK, Parker SL, McAllen K, Matta SG, Sharp BM. Nicotine administration enhances NPY expression in the rat hypothalamus. Brain Res. 2000a;867:157–164. doi: 10.1016/s0006-8993(00)02283-6. [DOI] [PubMed] [Google Scholar]
- Li MD, Parker SL, Kane JK. Regulation of feeding-associated peptides and receptors by nicotine. Mol Neurobiol. 2000b;22:143–165. doi: 10.1385/MN:22:1-3:143. [DOI] [PubMed] [Google Scholar]
- Liu M, Seino S, Kirchgessner AL. Identification and characterization of glucoresponsive neurons in the enteric nervous system. J Neurosci. 1999;19:10305–10317. doi: 10.1523/JNEUROSCI.19-23-10305.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin P, Hamon B, Glowinski J, Premont J. Nicotine-induced inhibition of neuronal phospholipase A2. J Pharmacol Exp Ther. 1997;280:1277–1283. [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, Van der Ploeg LH, Qian S. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci USA. 2002;99:3240–3245. doi: 10.1073/pnas.052706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS. Molecular diversity of neuronal nicotinic acetylcholine receptors. Ann NY Acad Sci. 1999;868:565–577. doi: 10.1111/j.1749-6632.1999.tb11330.x. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Meguid MM, Fetissov SO, Varma M, Sato T, Zhang L, Laviano A, Rossi-Fanelli F. Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition. 2000;16:843–857. doi: 10.1016/s0899-9007(00)00449-4. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Lopez FJ, Negro-Vilar A. Anatomy and physiology of central galanin-containing pathways. Prog Neurobiol. 1993;40:711–769. doi: 10.1016/0301-0082(93)90012-h. [DOI] [PubMed] [Google Scholar]
- Miyata G, Meguid MM, Fetissov SO, Torelli GF, Kim HJ. Nicotine's effect on hypothalamic neurotransmitters and appetite regulation. Surgery. 1999;126:255–263. [PubMed] [Google Scholar]
- Miyata G, Meguid MM, Varma M, Fetissov SO, Kim HJ. Nicotine alters the usual reciprocity between meal size and meal number in female rat. Physiol Behav. 2001;74:169–176. doi: 10.1016/s0031-9384(01)00540-6. [DOI] [PubMed] [Google Scholar]
- Neal CR, Jr, Newman SW. Prodynorphin peptide distribution in the forebrain of the Syrian hamster and rat: a comparative study with antisera against dynorphin A, dynorphin B, and the C-terminus of the prodynorphin precursor molecule. J Comp Neurol. 1989;288:353–386. doi: 10.1002/cne.902880302. [DOI] [PubMed] [Google Scholar]
- Nicklas BJ, Tomoyasu N, Muir J, Goldberg AP. Effects of cigarette smoking and its cessation on body weight and plasma leptin levels. Metabolism. 1999;48:804–808. doi: 10.1016/s0026-0495(99)90183-x. [DOI] [PubMed] [Google Scholar]
- Niijima A. Afferent signals from leptin sensors in the white adipose tissue of the epididymis, and their reflex effect in the rat. J Auton Nerv Syst. 1998;73:19–25. doi: 10.1016/s0165-1838(98)00109-x. [DOI] [PubMed] [Google Scholar]
- Nonogaki K, Strack AM, Dallman MF, Tecott LH. Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nat Med. 1998;4:1152–1156. doi: 10.1038/2647. [DOI] [PubMed] [Google Scholar]
- Nordstrom BL, Kinnunen T, Utman CH, Garvey AJ. Long-term effects of nicotine gum on weight gain after smoking cessation. Nicotine Tob Res. 1999;1:259–268. doi: 10.1080/14622299050011381. [DOI] [PubMed] [Google Scholar]
- O'Hara BF, Edgar DM, Cao VH, Wiler SW, Heller HC, Kilduff TS, Miller JD. Nicotine and nicotinic receptors in the circadian system. Psychoneuroendocrinology. 1998;23:161–173. doi: 10.1016/s0306-4530(97)00077-2. [DOI] [PubMed] [Google Scholar]
- Okuda H, Shioda S, Nakai Y, Nakayama H, Okamoto M, Nakashima T. Immunocytochemical localization of nicotinic acetylcholine receptor in rat hypothalamus. Brain Res. 1993;625:145–151. doi: 10.1016/0006-8993(93)90147-f. [DOI] [PubMed] [Google Scholar]
- Pabreza LA, Dhawan S, Kellar KJ. [3H]cytisine binding to nicotinic cholinergic receptors in brain. Mol Pharmacol. 1991;39:9–12. [PubMed] [Google Scholar]
- Pajolla GP, Crippa GE, Correa SA, Moreira KB, Tavares RF, Correa FM. The lateral hypothalamus is involved in the pathway mediating the hypotensive response to cingulate cortex-cholinergic stimulation. Cell Mol Neurobiol. 2001;21:341–356. doi: 10.1023/A:1012650021137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Caldarone BJ, Brunzell DH, Zachariou V, Stevens TR, King SL. Neuronal nicotinic acetylcholine receptor subunit knockout mice: physiological and behavioral phenotypes and possible clinical implications. Pharmacol Ther. 2001;92:89–108. doi: 10.1016/s0163-7258(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Pomerleau CS. Issues for women who wish to stop smoking. In: Seidman DF, Covey LS, editors. Helping the hard-core smoker. Lawrence Erlbaum; London: 1999. pp. 73–91. 1999. [Google Scholar]
- Pomerleau CS, Pomerleau OF, Namenek RJ, Mehringer AM. Short-term weight gain in abstaining women smokers. J Subst Abuse Treat. 2000;18:339–342. doi: 10.1016/s0740-5472(99)00085-9. [DOI] [PubMed] [Google Scholar]
- Pu S, Jain MR, Horvath TL, Diano S, Kalra PS, Kalra SP. Interactions between neuropeptide Y and gamma-aminobutyric acid in stimulation of feeding: a morphological and pharmacological analysis. Endocrinology. 1999;140:933–940. doi: 10.1210/endo.140.2.6495. [DOI] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- Risold PY, Fellmann D, Lenys D, Bugnon C. Coexistence of acetylcholinesterase-, human growth hormone-releasing factor(1–37)-, alpha-melanotropin- and melanin-concentrating hormone-like immunoreactivities in neurons of the rat hypothalamus: a light and electron microscope study. Neurosci Lett. 1989;100:23–28. doi: 10.1016/0304-3940(89)90654-x. [DOI] [PubMed] [Google Scholar]
- Roth AL, Shoop RD, Berg DK. Targeting alpha7-containing nicotinic receptors on neurons to distal locations. Eur J Pharmacol. 2000;393:105–112. doi: 10.1016/s0014-2999(00)00092-3. [DOI] [PubMed] [Google Scholar]
- Rust G, Burgunder JM, Lauterburg TE, Cachelin AB. Expression of neuronal nicotinic acetylcholine receptor subunit genes in the rat autonomic nervous system. Eur J Neurosci. 1994;6:478–485. doi: 10.1111/j.1460-9568.1994.tb00290.x. [DOI] [PubMed] [Google Scholar]
- Sakurai T. Orexins and orexin receptors: implication in feeding behavior. Regul Pept. 1999;85:25–30. doi: 10.1016/s0167-0115(99)00076-2. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richarson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–731. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Eiden LE, Weihe E. Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. I. Central nervous system. Neuroscience. 1998;84:331–359. doi: 10.1016/s0306-4522(97)00516-2. [DOI] [PubMed] [Google Scholar]
- Schick RR, Samsami S, Zimmermann JP, Eberl T, Endres C, Schusdziarra V, Classen M. Effect of galanin on food intake in rats: involvement of lateral and ventromedial hypothalamic sites. Am J Physiol. 1993;264:R355–R361. doi: 10.1152/ajpregu.1993.264.2.R355. [DOI] [PubMed] [Google Scholar]
- Schwartz DH, McClane S, Hernandez L, Hoebel BG. Feeding increases extracellular serotonin in the lateral hypothalamus of the rat as measured by microdialysis. Brain Res. 1989;479:349–354. doi: 10.1016/0006-8993(89)91639-9. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Lehmann J, Kellar KJ. Presynaptic nicotinic cholinergic receptors labeled by [3H]acetylcholine on catecholamine and serotonin axons in brain. J Neurochem. 1984;42:1495–1498. doi: 10.1111/j.1471-4159.1984.tb02818.x. [DOI] [PubMed] [Google Scholar]
- Sha L, Szurszewski JH. Leptin modulates fast synaptic transmission in dog pancreatic ganglia. Neurosci Lett. 1999;263:93–96. doi: 10.1016/s0304-3940(99)00122-6. [DOI] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- Shioda S, Nakajo S, Hirabayashi T, Nakayama H, Nakaya K, Matsuda K, Nakai Y. Neuronal nicotinic acetylcholine receptor in the hypothalamus: morphological diversity and neuroendocrine regulations. Brain Res Mol Brain Res. 1997;49:45–54. doi: 10.1016/s0169-328x(97)00122-8. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Oomura Y, Sasaki K, Wayner MJ. Effects of leptin and orexin-A on food intake and feeding related hypothalamic neurons. Physiol Behav. 2000;71:251–261. doi: 10.1016/s0031-9384(00)00341-3. [DOI] [PubMed] [Google Scholar]
- Shoop RD, Chang KT, Ellisman MH, Berg DK. Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci. 2001;21:771–781. doi: 10.1523/JNEUROSCI.21-03-00771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- Speigelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Stamford BA, Matter S, Fell RD, Papanek P. Effects of smoking cessation on weight gain, metabolic rate, caloric consumption, and blood lipids. Am J Clin Nutr. 1986;43:486–494. doi: 10.1093/ajcn/43.4.486. [DOI] [PubMed] [Google Scholar]
- Stanley BG, Chin AS, Leibowitz SF. Feeding and drinking elicited by central injection of neuropeptide Y: evidence for a hypothalamic site(s) of action. Brain Res Bull. 1985;14:521–524. doi: 10.1016/0361-9230(85)90100-5. [DOI] [PubMed] [Google Scholar]
- Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- Stratford TR, Kelley AE. Evidence of a functional relationship between the nucleus accumbens shell and lateral hypothalamus subserving the control of feeding behavior. J Neurosci. 1999;19:11040–11048. doi: 10.1523/JNEUROSCI.19-24-11040.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JG, De Lecea L. The hypocretins: setting the arousal threshold. Nat Rev Neurosci. 2002;3:339–349. doi: 10.1038/nrn808. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM, Palmiter RD. Feeding behavior in dopamine-deficient mice. Proc Natl Acad Sci USA. 1999;96:12138–12143. doi: 10.1073/pnas.96.21.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypka MS, Rainey MA, Palmiter RD. Dopamine is required for hyperphagia in Lep(ob/ob) mice. Nat Genet. 2000;25:102–104. doi: 10.1038/75484. [DOI] [PubMed] [Google Scholar]
- Sztalryd C, Hamilton J, Horowitz BA, Johnson P, Kraemer FB. Alterations of lipolysis and lipoprotein lipase in chronically nicotine-treated rats. Am J Physiol. 1996;270:E215–E223. doi: 10.1152/ajpendo.1996.270.2.E215. [DOI] [PubMed] [Google Scholar]
- Tago H, McGeer PL, Bruce G, Hersh LB. Distribution of choline acetyltransferase-containing neurons of the hypothalamus. Brain Res. 1987;415:49–62. doi: 10.1016/0006-8993(87)90268-x. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- Temburni MK, Blitzblau RC, Jacob MH. Receptor targeting and heterogeneity at interneuronal nicotinic cholinergic synapses in vivo. J Physiol. 2000;525(Pt 1):21–29. doi: 10.1111/j.1469-7793.2000.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieman TL, Steel DJ, Gor Y, Kehoe J, Schwartz JH, Feinmark SJ. A pertussis toxin-sensitive 8-lipoxygenase pathway is activated by a nicotinic acetylcholine receptor in Aplysia neurons. J Neurophysiol. 2001;85:2150–2158. doi: 10.1152/jn.2001.85.5.2150. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Bertrand D, Raggenbass M. Role of neuronal nicotinic receptors in the transmission and processing of information in neurons of the central nervous system. Pharmacol Biochem Behav. 2001;70:457–466. doi: 10.1016/s0091-3057(01)00700-6. [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan S, Huang B, Blumenthal EM, Berg DK. Arachidonic acid as a possible negative feedback inhibitor of nicotinic acetylcholine receptors on neurons. J Neurosci. 1995;15:3679–3687. doi: 10.1523/JNEUROSCI.15-05-03679.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Walton EA, Milici A, Buccafusco JJ. m1–m5 muscarinic receptor distribution in rat CNS by RT-PCR and HPLC. J Neurochem. 1994;63:815–821. doi: 10.1046/j.1471-4159.1994.63030815.x. [DOI] [PubMed] [Google Scholar]
- Wei M, Stern MP, Haffner SM. Serum leptin levels in Mexican Americans and non-Hispanic whites: association with body mass index and cigarette smoking. Ann Epidemiol. 1997;7:81–86. doi: 10.1016/s1047-2797(96)00114-7. [DOI] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci. 2001;24:429–458. doi: 10.1146/annurev.neuro.24.1.429. [DOI] [PubMed] [Google Scholar]
- Woods S, Seeley C, Porte RJ, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1478–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ, Xia B, Crawley JN, Felder CC, Deng CX, Wess J. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature. 2001;410:207–212. doi: 10.1038/35065604. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Blaha V, Meguid MM, Oler A, Miyata G. Infusion of nicotine into the LHA enhances dopamine and 5-HT release and suppresses food intake. Pharmacol Biochem Behav. 1999;64:155–159. doi: 10.1016/s0091-3057(99)00111-2. [DOI] [PubMed] [Google Scholar]
- Yettefti K, Orsini JC, Perrin J. Neuronal responses to systemic nicotine in the solitary tract nucleus: origin and possible relation with nutritional effects of nicotine. Pharmacol Biochem Behav. 1997;58:529–535. doi: 10.1016/s0091-3057(97)00247-5. [DOI] [PubMed] [Google Scholar]
- Yoshinari M, Wakisaka M, Fujishima M. Serum leptin levels in smokers with type 2 diabetes. Diabetes Care. 1998;21:516–517. doi: 10.2337/diacare.21.4.516. [DOI] [PubMed] [Google Scholar]
- Zhang L, Meguid MM, Miyata G, Varma M, Fetissov SO. Role of hypothalamic monoamines in nicotine-induced anorexia in menopausal rats. Surgery. 2001;130:133–142. doi: 10.1067/msy.2001.115513. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]