

Abstract
There is an accumulating amount of evidence indicating that lipid oxidation is depressed in the skeletal muscle of obese individuals. Decrements in fatty acid oxidation (FAO) have been reported with obesity in models ranging from whole body measurements to isolated skeletal muscle preparations as well as in myotubes raised in culture. This reduction appears to be associated with a depression in the activities of enzymes involved in various steps of lipid oxidation, which subsequently partitions lipid entering the cell toward storage. The defect in FAO in skeletal muscle may be critical in relation to health, as a reduction in the capacity for lipid oxidation could directly or indirectly contribute to the insulin resistance commonly evident with obesity. Although less characterized, a decrement in FAO has also been linked with weight gain, which suggests that this characteristic may be an integral aspect leading to the obese state. In terms of intervention, weight loss does not seem to correct the defect in FAO with obesity. This review will provide evidence supporting a reduction in muscle FAO with obesity.
Keywords: skeletal muscle, metabolism, insulin action
THE PREVALENCE OF OBESITY is increasing dramatically in the United States and other industrialized nations (33, 51). Obesity is associated with a variety of health-related risks, such as coronary heart disease, hypertension, and type 2 diabetes, all of which may center around insulin resistance (i.e., the metabolic syndrome) (39). It is thus essential to identify characteristics that predispose individuals toward obesity or negative conditions that occur during the development of the obese state, which subsequently alter metabolism and health status in a negative manner.
Skeletal muscle is a metabolically active tissue that is critical to maintaining whole body homeostasis due to its energy needs as well as its relatively large mass. In terms of carbohydrate metabolism, a major portion of an ingested glucose load is either stored as glycogen or oxidized in skeletal muscle (14). Skeletal muscle also plays an important role in fatty acid oxidation (FAO). During the fasted state, FAO is the predominant metabolic activity of skeletal muscle (13, 51). At rest, the oxidation of lipid contributes significantly to overall energy needs with most of the energy requirements of muscle being obtained via FAO (10, 13). Such data indicates that skeletal muscle is quantitatively important in relation to the fate of lipid. Factors that elicit a decrement in the ability to oxidize lipid in skeletal muscle would thus be anticipated to evoke profound changes in whole body homeostasis. The intent of this paper is to review some of the evidence indicating a decrement in skeletal muscle lipid oxidation with obesity.
Is There a Decrement in Fat Oxidation with Obesity?
Determining the mechanisms that are linked with obesity is problematic in that when studying the obese individual, it is difficult to discern whether the findings obtained are due to 1) an inherent predisposition to obesity, 2) conditions that developed during years of gaining body mass from positive energy balance, or 3) both. In relation to the ability to utilize lipid, there are limited prospective data indicating that a propensity for weight gain is associated with substrate utilization, which favors carbohydrate over lipid oxidation. When discussing whole body substrate utilization the terms respiratory quotient (RQ) and respiratory exchange ration (RER) are used interchangeably; regardless of terminology a higher RQ-RER is indicative of a reduced capacity to oxidize lipid. In nondiabetic Pima Indians, Zurlo et al. (54) reported a familial influence on RQ and that a low capacity for fat oxidation was associated with an increased risk for weight gain. Similar findings were obtained by Marra et al. (31) who reported that a higher whole body RQ predicted weight gain in lean women. Other work has supported the premise that an elevated whole body RQ-RER is predictive of weight gain in both lean and obese individuals (32, 43), although this is not always the case (25). While these findings are somewhat sparse, the data suggests that weight gain is linked with an inability to utilize lipid as measured at the whole body level.
Another experimental method for discerning mechanisms potentially linked with obesity is to examine obese subjects after weight loss. This approach theoretically provides a glimpse of the “preobese” state, as it has already been demonstrated that the subjects have a propensity toward obesity. A weight loss model that our laboratory has utilized is studying extremely obese subjects (body mass index ≥ 40 kg/m2) before and ~1 yr or longer after gastric bypass surgery (36). This intervention induces a long-term weight loss of ~50 kg with patients becoming weight stable at ~12 mo after the surgery (36); subjects are thus tested in a relatively stable condition when confounding factors, such as energy imbalance, are minimized. Using this model, we observed that previously extremely obese women of mixed ethnicities after weight loss still exhibited a preference for carbohydrate over fat oxidation compared with weight-matched individuals who were never extremely obese; substrate utilization was determined at the whole body level with indirect calorimetry during submaximal exercise (17). This observation was supported by tracking the oxidation of [13C] palmitate at rest and during submaximal exercise in lean, extremely obese, and extremely obese women after weight loss (48). The relative percentage of fat oxidation was similar in both the extremely obese and extremely obese women after weight loss and significantly reduced compared with the lean subjects (Fig. 1); these findings suggest that a reduced capacity for lipid oxidation is linked with a propensity toward extreme obesity.
Fig. 1.

Percentage of plasma free fatty acid (FFA) uptake oxidized during basal and exercise (50% maximum O2 consumption). *Significantly decreased in extremely obese and extremely obese subjects after weight reduction (weight-reduced) compared with lean (P < 0.05). From Thyfault et al. (48).
Other studies have examined the effect of weight loss on substrate utilization in obese individuals at the whole body level and obtained similar results. Larson et al. (30) studied previously obese individuals who lost a mean of 57 kg via energy restriction and found that fat oxidation (as determined with indirect calorimetry) was significantly depressed in the weight-loss (postobese) group compared with weight-matched controls. Similarly, Kelley et al. (26) reported no change in resting RQ when determining substrate utilization across a skeletal muscle bed in the legs of obese individuals before and after weight loss. In postobese individuals, a decrement in fat oxidation during submaximal exercise as well as at rest has also been found (for reviews, see Ref. 4–6).
Another approach to discerning whether there is a defect in fat oxidation with obesity at the whole body level has been to examine the responses to dietary lipid in obese or obese individuals after weight loss. Although the mechanistic link between a reduced capacity for fat oxidation and obesity has not been characterized, one possibility is that an inability to increase fat oxidation when faced with the consumption of dietary fat contributes to positive fat balance, the accumulation of adipose tissue, and eventually weight gain and obesity (16, 38). In support of this hypothesis, obese individuals displayed an impaired capacity to oxidize lipid in response to an increase in dietary fat compared with lean subjects (7, 16, 41). Individuals examined after weight loss also failed to increase the ratio of fat to carbohydrate oxidation when exposed to a high-fat diet that resulted in positive fat balance (1). Together, these findings imply that a reduced capacity for fat oxidation is evident at the whole body level with obesity. There are conflicting data, however, which indicate either no difference (46, 47, 50) or an elevated (15, 21, 24) rate of lipid oxidation in obese compared with lean individuals. An explanation for this disparity may involve factors such as the degree of adiposity in the subjects examined, gender differences, nutritive status during the measurement of lipid oxidation (i.e., fasting or insulin-stimulated), how whole body fatty acid oxidation is expressed (i.e., per kilogram of body or fat mass), and, in exercise studies, the intensity of the workload selected (48); however, it is difficult to definitively explain why these differences exist between studies. The intent of the current review is to present and summarize findings indicating lipid oxidation in skeletal muscle is impaired with obesity and allow the reader to use these data to formulate their own hypotheses.
Is There a Decrement in the Ability of Skeletal Muscle to Oxidize Lipid with Obesity?
The whole body measurements discussed above are indicative of a decrement in FAO with obesity, which implies at least a partial involvement of skeletal muscle; in support, direct measurements made in this tissue indicate a decrement in the ability to oxidize lipid. In our laboratory we have observed a reduction in FAO in the skeletal muscle of extremely obese individuals under several different experimental conditions. In one of our first studies, muscle strip preparations were obtained from the rectus abdominus of lean, obese, and extremely obese individuals and incubated with labeled palmitate (22). In muscle strips from extremely obese individuals there was a significant decrease in FAO and increased partitioning of lipid toward storage (22). The finding of a reduction in FAO with obesity was validated in a subsequent study of another muscle group, the vastus lateralis, by obtaining needle biopsies from lean and extremely obese individuals and measuring labeled palmitate oxidation in muscle homogenates (28). The integration of these findings (22, 28) provides relatively convincing evidence for a reduction in FAO in the skeletal muscle of extremely obese individuals, as FAO was reduced in two separate muscle groups in different anatomical areas. Validation in distinct muscle groups is critical as skeletal muscle is a heterogeneous tissue that can vary widely in respect to fiber composition and contractile activity.
Another approach to study metabolism in skeletal muscle is the use of primary cell cultures. Briefly, this method involves isolating satellite cells from a muscle biopsy and treating the cells so that they proliferate into myoblasts and ultimately differentiate into myotubes (3). The myotubes possess many of the qualities of intact muscle fibers and are suitable for examining substrate utilization. Using this human skeletal muscle cell culture method (HSkMC), we compared the capacity for lipid oxidation in cells derived from lean and extremely obese donors and observed that FAO (measured with [l-14C] palmitate) was depressed in cells from obese subjects (23). The magnitude of the decrement in FAO was similar to our in vivo (17, 48) and in vitro (22, 28) findings comparing lean and extremely obese individuals (Fig. 2). The HSkMC technique involves culturing the cells ex vivo for ~4–6 wk; under these conditions it is believed that any expressed phenotype is largely the result of genetic or epigenetic origins (3). If this is the case, our HSkMC data (23) implies that extremely obese individuals may have a predisposition toward a reduction in FAO in skeletal muscle. Another study reported that the ability to oxidize lipid in response to an increased lipid load in HSkMC mirrored metabolic traits determined in vivo in lean individuals, indicating the potential utility of HSkMC in studying lipid oxidation (49).
Fig. 2.
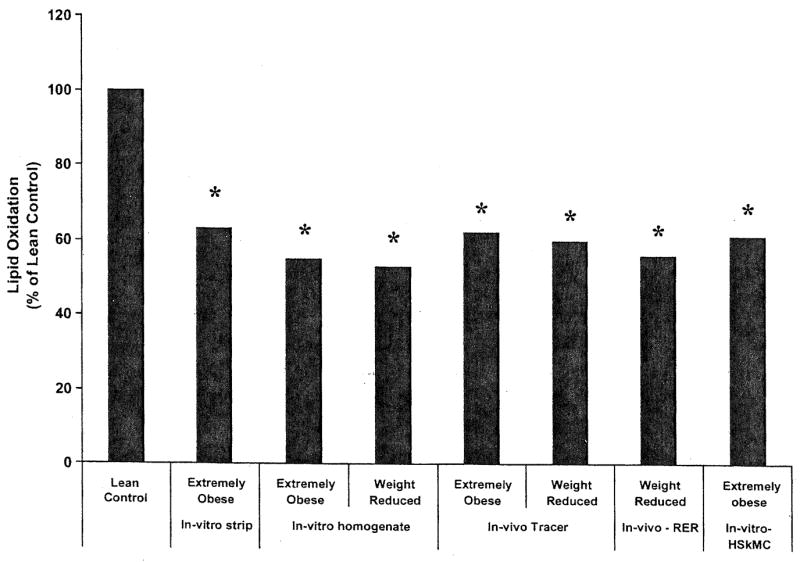
Decrement in fatty acid oxidation (FAO) with obesity as indicated by various methodologies used in our laboratory. Data are redrawn from Refs. 17, 22, 23, 28, 48. *Significantly different (P ≤ 0.05) from lean controls for the listed method.
Other work has indicated a defect in FAO specific to skeletal muscle with obesity. Colberg et al. (11) examined the ability of skeletal muscle to utilize free fatty acids (FFA) with the leg balance technique in lean and obese premenopausal women. These authors (11) reported that women with visceral obesity had a reduced rate of FFA uptake and oxidation in muscle under fasting conditions. Using similar methodology (leg balance), this group confirmed their earlier findings by reporting an elevated leg RQ in obese subjects (26). Utilizing an in vitro muscle preparation, Kirkwood et al. (29) reported a statistically significant and negative relationship between maximally stimulated muscle respiration and body fat content.
The reduction in FAO in the skeletal muscle of obese individuals may be linked, at least in part, with a decreased activity of enzymes involved in lipid oxidation. Although enzymatic activities or protein content may not necessarily reflect substrate flux, Simoneau et al. (45) reported that the activities of carnitine palmitoyl transferase (CPT-1), citrate synthase (CS), and cytochrome c oxidase were significantly depressed in skeletal muscle samples (vastus lateralis) obtained from obese compared with lean subjects. These data are significant in that they suggest defects at several key regulatory steps in FAO, such as lipid transfer into the mitochondria (CPT-1), the Krebs cycle (CS), and the electron transport system (cytochrome c oxidase). Several studies have confirmed reductions in CPT-1, CS, and cytochrome c oxidase in the skeletal muscle of obese individuals (19, 26, 28); similarly, others (27, 40) have reported a reduction in the activity of the respiratory chain in skeletal muscle from obese individuals. Ratios of enzyme activities indicative of glycolytic to oxidative flux, such as glycogen phosphorylase to CS (44) and lipid oxidation to lipid transfer (CPT-1 to fatty acid binding protein) (45) are also supportive of a reduced capacity for lipid oxidation in skeletal muscle with obesity. Zurlo et al. (55) reported a negative relationship between 24-h RQ and (β-hydroxyacl-coenzyme A dehydrogenase activity, which implies that variability exists in the process of β-oxidation, which may influence the capacity for FAO. In support of a defect in β-oxidation, we reported a reduction in (β-hydroxyacl-coenzyme A dehydrogenase activity in the skeletal muscle of extremely obese individuals (28). Together, these data provide evidence for a reduction in the activities of key oxidative enzymes involved in FAO in the skeletal muscle of obese individuals, which is unfortunately not corrected with weight loss (26, 45). While the mechanism(s) responsible for the depression in enzyme activity is not evident, peroxisome-proliferator-activated receptor-γ coactivator-1α, which is an important regulator of mitochondrial biogenesis, has been reported to be lower in the skeletal muscle of obese, insulin-resistant subjects compared with lean controls (19).
In addition to reductions in enzyme activities linked with FAO, there are also alterations in mitochondrial morphology with obesity. Kirwood et al. (29) demonstrated that the central distribution of body fat was associated with a lower mitochondrial density, as determined by electron microscopy. Other work using electron microscopy (27) indicated that skeletal muscle mitochondria were smaller by ~35% in type 2 diabetic and obese subjects compared with lean subjects. The mitoschondria from the obese and diabetic subjects also had a less well-defined inner membrane structure, including a narrower cristae (27). Similar alterations in mitochondrial morphology, such as reduced size, are evident in myopathies in which the function of this organelle is compromised (52). There may also be an alteration in the distribution of mitochondria within the skeletal muscle of obese individuals, as the number of subsarcolemmal mitochondria has been reported to be reduced with obesity and type 2 diabetes along with decreased electron transport chain activity (40). It is believed that one of the primary duties of subsarcolemmal mitochondria is to oxidize lipid (20), the reduction of mass in this fraction coupled with reduced enzymatic activity per unit of mitochondria with obesity (40) may provide an underlying mechanism for the decrement in FAO.
While the above findings indicate a defect in oxidative processes, there also appears to be a congruent increase in the capacity for lipid storage in the skeletal muscle of obese individuals, which would contribute to an accumulation of intramuscular lipid. For example, Simoneau et al. (45) reported elevated levels of fatty acid binding protein in the plasma membrane fraction of muscle from obese individuals. Bonen et al. (8) studied giant sarcolemmal vesicles prepared from skeletal muscle biopsies and reported that long-chain fatty acid transport into skeletal muscle was upregulated by approximately fourfold with obesity. This increase in fatty acid transport was associated with an elevated level of FAT/CD36 (fatty acid translocase) in the sarcolemmal fraction (8). Hulver et al. (23) reported that stearoyl-CoA desaturase-1, a lipogenic gene, was robustly upregulated in the skeletal muscle of extremely obese individuals and that this characteristic was retained in culture (HSkMC). Overexpression of stearoyl-CoA desaturase-1 in myotubes from lean subjects increased fatty acid esterification and depressed FAO, suggesting a potential duel regulation of lipid-handling pathways by stearoyl-CoA desaturase-1. Together, the findings of a depression in FAO coupled with an increased capacity for lipogenesis indicates a global organization of lipid metabolism in obese skeletal muscle that is biased toward storage and esterification (Fig. 3).
Fig. 3.
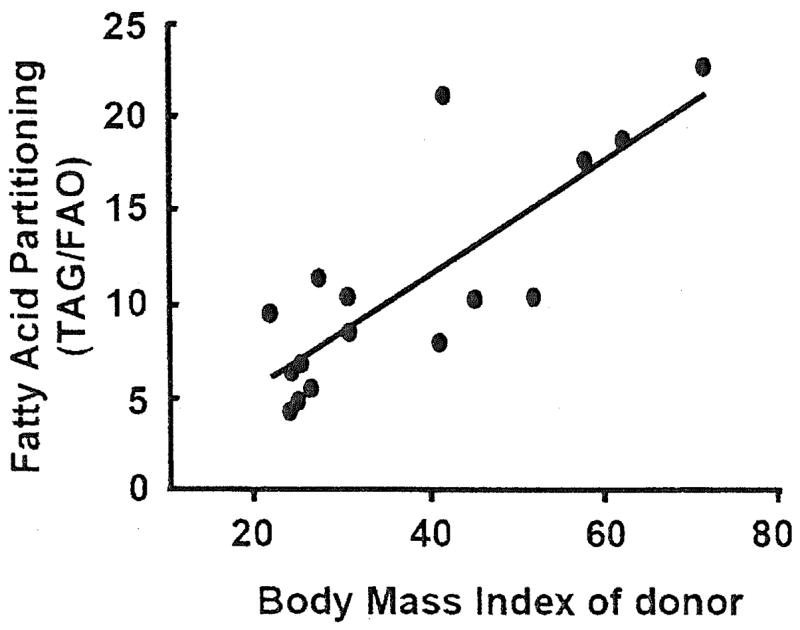
Fatty acid partitioning [labeled oleate incorporation into triacylglycerol (TAG) divided by FAO] in myotubes cultured from skeletal muscle samples obtained from lean and obese donors (23). An increased fatty acid partitioning ratio indicates preferential portioning toward storage within the muscle cell as opposed to oxidation. The positive relationship between the FAO partitioning index of the myotubes and the body mass index of the donor was statistically significant (r2 = 0.66, P < 0.001). HSkMC, human skeletal muscle cell culture method.
Some insight into the mechanisms responsible for the decrement in FAO in skeletal muscle with obesity may be obtained by studying the effects of interventions. Weight loss does not seem to alter fat oxidation in the skeletal muscle of obese individuals (17, 26, 30, 45, 48). On the other hand, contractile activity, i.e., physical activity or exercise training has been reported to enhance the capacity for lipid oxidation in the skeletal muscle of obese subjects. We observed that a short-term endurance-oriented training model, which did not induce weight loss (10 consecutive days, 60 min/day at 75% V̇o2peak) improved fatty acid oxidation in the skeletal muscle of morbidly obese individuals (2). Menshikova et al. (33) reported that in obese individuals moderate physical activity combined with weight loss increased oxidative enzyme activities without a significant increase in mitochondrial DNA, although there was evidence for an increase in mitochondrial cristae. This pattern differs from the classical mitochondrial biogenesis seen with physical activity in lean individuals (33). Together, these findings suggest that some aspect linked with contractile activity either directly overcomes or somehow compensates for the decrement in FAO seen in the skeletal muscle of obese individuals. The exact mechanism(s) responsible, however, remain to be discerned.
Possible Interactions Between Fatty Acid Oxidation and Insulin Action with Obesity
A health concern with obesity is the presence of insulin resistance. The coexistence of insulin resistance, intramuscular lipid accumulation, and a decrement in FAO in skeletal muscle are often simultaneously evident in obese individuals (for reviews, see Refs. 4, 6, 35). However, the mechanistic link between FAO and insulin resistance in the skeletal muscle of obese individuals is not yet apparent.
There is evidence suggesting that the accumulation of lipid within the skeletal muscle of obese individuals induces insulin resistance. Several studies suggest that fatty-acid-derived metabolites, such as long-chain acyl CoA, diacylglycerol, and ceramide accumulate in the cytosol of the skeletal muscle of obese individuals. These intermediates then either directly or indirectly impair insulin signal transduction and/or the activity of enzymes involved in glucose utilization, which in turn induces insulin resistance (for reviews, see Refs. 12, 18, 36). The accumulation of these metabolically active lipid intermediates could be due, at least in part, to the disturbances in mitochondrial function and depression in FAO that may occur with obesity.
There are also findings that suggest the capacity for FAO alone controls insulin action in skeletal muscle. When overexpressing CPT1 in L6 myotubes, Perdomo et al. (35) reported an increase in palmitate and oleate oxidation, which provided a protective effect against fatty-acid-induced insulin resistance. The intriguing finding of this study (35) was that CPT1 overexpression improved insulin action, despite no decrease in intracellular diacylglycerol, long-chain acyl CoA, or ceramide, compared with lipid-treated control cells that were insulin resistant; this observation prompted the hypothesis that accelerating FAO exerts an insulin-sensitizing effect that functions independently of changes in cytosolic lipid content. In support, in vivo human findings indicate that the oxidative capacity of skeletal muscle is an effective predictor of whole body insulin action (9). However, other work utilizing HSkMC reported that CPT1 overexpression was protective against the accumulation of diacylglycerol, ceramide, and insulin resistance (42). While the cellular mechanism(s) responsible for lipid-induced insulin resistance remain to be discerned, the findings remain supportive of the integral role that FAO in skeletal muscle plays in controlling insulin action.
Perspectives and Significance
There is accumulating evidence indicating a defect in the capacity for FAO in the skeletal muscle of obese individuals. The broad implications of this finding is that such a defect may contribute to 1) the insulin resistance often evident with obesity and 2) the weight gain inherent with the obese state. The mechanisms linking the obesity-associated reduction in lipid oxidation with insulin-resistance and weight gain may be a fruitful area for future studies. For example, it is not evident why the skeletal muscle of obese individuals exhibits a depression in the activities of enzymes involved in various steps of lipid oxidation and how this alteration contributes to the partitioning of lipid entering the cell toward storage. It is also not apparent how interventions, such as physical activity, overcome or somehow compensate for the decrement in FAO seen in the skeletal muscle of obese individuals. In conclusion, a reduction in the capacity for lipid oxidation in the skeletal muscle of obese individuals may be an integral component of the obese state.
Acknowledgments
Special thanks to my collaborators Drs. Berggren, Chapman, Dohm, Hulver, Muoio, Pories, Tanner, and Thyfault, for their many contributions.
GRANTS
Work for this article was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-56112.
References
- 1.Astrup A. Dietary composition, substrate balances and body fat in subjects with a predisposition to obesity. Int J Obes. 1993;17(Suppl 3):S32–S36. [PubMed] [Google Scholar]
- 2.Berggren JR, Hulver MW, Dohm GL, Houmard JA. Weight loss and exercise: implications for muscle lipid metabolism and insulin action. Med Sci Sport Exerc. 2004;36:1191–1195. doi: 10.1249/01.mss.0000074670.03001.98. [DOI] [PubMed] [Google Scholar]
- 3.Berggren JR, Tanner CJ, Houmard JA. Primary cell cultures in the study of human muscle metabolism. Exerc Sport Sci Rev. 2007;35:56–61. doi: 10.1249/JES.0b013e31803eae63. [DOI] [PubMed] [Google Scholar]
- 4.Blaak EE. Fatty acid metabolism in obesity and type 2 diabetes mellitus. Proc Nutr Soc. 2003;62:753–760. doi: 10.1079/PNS2003290. [DOI] [PubMed] [Google Scholar]
- 5.Blaak EE. Basic disturbances in skeletal muscle fatty acid metabolism in obesity and type 2 diabetes mellitus. Proc Nutr Soc. 2004;63:323–330. doi: 10.1079/PNS2004361. [DOI] [PubMed] [Google Scholar]
- 6.Blaak EE. Metabolic fluxes in skeletal muscle in relation to obesity and insulin resistance. Best Pract Res Clin Endocrinol Metab. 2005;19:391–403. doi: 10.1016/j.beem.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Blaak EE, Hul G, Verdich C, Stich V, Martinez A, Petersen M, Feskens EFM, Patel K, Oppert JM, Barbe P, Toubro S, Anderson I, Polak J, Astrup A, Macdonald IA, Langin D, Hoist C, Sorensen TI, Saris WHM. Fat oxidation before and after a high fat load in the obese insulin resistant state. J Clin Endocrinol Metab. 2006;91:1462–1469. doi: 10.1210/jc.2005-1598. [DOI] [PubMed] [Google Scholar]
- 8.Bonen A, Parolin ML, Steinberg GR, Calles-Escandon J, Tandon NN, Glatz JFC, Luiken JJFP, Heigenhauser GJF, Dyck DJ. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. 2004;18:1144–1146. doi: 10.1096/fj.03-1065fje. [DOI] [PubMed] [Google Scholar]
- 9.Bruce CR, Anderson MJ, Carey AL, Newman DG, Bonen A, Kriketos AD, Cooney GJ, Hawley JA. Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J Clin Endocrinol Metab. 2003;88:5444–5451. doi: 10.1210/jc.2003-030791. [DOI] [PubMed] [Google Scholar]
- 10.Bulow J. Lipid mobilization and utilization. Principles of exercise biochemistry. Basel: Karger; 140. pp. 163–1988. [Google Scholar]
- 11.Colberg SR, Simoneau JA, Thaete FL, Kelley DE. Skeletal muscle utilization of free fatty acids in women with visceral obesity. J Clin Invest. 1995;95:1846–1853. doi: 10.1172/JCI117864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooney GJ, Thompson AL, Furler SM, Ye J, Kraegen EW. Muscle long-chain acyl CoA esters and insulin resistance. Ann NY Acad Sci. 2002;967:196–207. doi: 10.1111/j.1749-6632.2002.tb04276.x. [DOI] [PubMed] [Google Scholar]
- 13.Dagenais GR, Tancredi RG, Zierlier KL. Free fatty acid oxidation by forearm muscle at rest, and evidence for an intramuscular lipid pool in the human forearm. J Clin Invest. 1976;58:421–431. doi: 10.1172/JCI108486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose: results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 15.Felber JP, Ferrannini E, Golay A, Meyer HU, Theibaud D, Curchod B, Maeder E, Jequier E, DeFronzo RA. Role of lipid oxidation in pathogenesis of insulin resistance of obesity and type II diabetes. Diabetes. 1987;36:1341–1350. doi: 10.2337/diab.36.11.1341. [DOI] [PubMed] [Google Scholar]
- 16.Flatt JP, Ravussin E, Acheson KJ, Jequier E. Effects of dietary fat on postprandial substrate oxidation and on carbohydrate and fat balances. J Clin Invest. 1985;76:1019–1024. doi: 10.1172/JCI112054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guesbeck NR, Hickey MS, MacDonald KG, Pories WJ, Harper I, Ravussin E, Dohm GL, Houmard JA. Substrate utilization during exercise in formerly obese women. J Appl Physiol. 2001;90:1007–1012. doi: 10.1152/jappl.2001.90.3.1007. [DOI] [PubMed] [Google Scholar]
- 18.Hegarty BD, Furier SM, Ye J, Cooney GJ, Kraegen CJ. The role of intramuscular lipid in insulin resistance. Acta Physiol Scand. 2003;178:373–383. doi: 10.1046/j.1365-201X.2003.01162.x. [DOI] [PubMed] [Google Scholar]
- 19.Heilbronn LK, Gan SK, Turner N, Campbell LV, Chisholm DJ. Markers of mitochondrial biogenesis and metabolism are lower in overweight and obese insulin-resistant subjects. J Clin Endocrinol Metab. 2007;92:1467–1473. doi: 10.1210/jc.2006-2210. [DOI] [PubMed] [Google Scholar]
- 20.Hood D. Plasticity in skeletal, cardiac, and smooth muscle: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- 21.Horowitz JF, Klein S. Oxidation of nonplasma fatty acids during exercise is increased in women with abdominal obesity. J Appl Physiol. 2000;89:2276–2282. doi: 10.1152/jappl.2000.89.6.2276. [DOI] [PubMed] [Google Scholar]
- 22.Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003;284:E741–E747. doi: 10.1152/ajpendo.00514.2002. [DOI] [PubMed] [Google Scholar]
- 23.Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm GL, Houmard JA, Muoio DM. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanaley JA, Weatherup-Dentes MM, Alvarado CR, Whitehead G. Substrate oxidation during acute exercise and with exercise training in lean and obese women. Eur J Appl Physiol. 2001;85:68–73. doi: 10.1007/s004210100404. [DOI] [PubMed] [Google Scholar]
- 25.Katzmarzyk PT, Perusse L, Tremblay A, Bouchard C. No association between resting metabolic rate or respiratory exchange ratio and subsequent changes in body mass and fatness: 5′ year follow-up of the Quebec Family Study. Eur J Clin Nutr. 2000;54:610–614. doi: 10.1038/sj.ejcn.1601053. [DOI] [PubMed] [Google Scholar]
- 26.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol Endocrinol Metab. 1999;277:E1130–E1141. doi: 10.1152/ajpendo.1999.277.6.E1130. [DOI] [PubMed] [Google Scholar]
- 27.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 28.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- 29.Kirkwood SP, Zurlo F, Larson K, Ravussin E. Muscle mitochondrial morphology, body composition, and energy expenditure in sedentary individuals. Am J Physiol Endocrinol Metab. 1991;260:E89–E94. doi: 10.1152/ajpendo.1991.260.1.E89. [DOI] [PubMed] [Google Scholar]
- 30.Larson DE, Ferraro RT, Robertson DS, Ravussin E. Energy metabolism in weight-stable postobese individuals. Am J Clin Nutr. 1995;62:735–739. doi: 10.1093/ajcn/62.4.735. [DOI] [PubMed] [Google Scholar]
- 31.Marra M, Scalfi L, Contaldo F, Pasanisi F. Fasting respiratory quotient as a predictor of long-term weight changes in non-obese women. Ann Nutr Metab. 2004;48:189–192. doi: 10.1159/000079556. [DOI] [PubMed] [Google Scholar]
- 32.Marra M, Scalfi L, Covino A, Esposito-del Puente Contaldo F. Fasting respiratory quotient as a predictor of weight changes in non-obese women. Int J Obes. 1998;22:601–603. doi: 10.1038/sj.ijo.0800612. [DOI] [PubMed] [Google Scholar]
- 33.Menshikova EV, Ritov VB, Ferrell RE, Azuma K, Goodpaster BH, Kelley DE. Characteristics of skeletal muscle mitochondrial biogenesis induced by moderate-intensity exercise and weight loss in obesity. J Appl Physiol. 2007;103:21–27. doi: 10.1152/japplphysiol.01228.2006. [DOI] [PubMed] [Google Scholar]
- 34.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–2000. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 35.Perdomo G, Commerford SR, Richard AMT, Adams SH, Corkey BE, O’Derty RM, Brown NF. Increased β-oxidation in muscle cells enhances insulin-stimulated glucose metabolism and protects against fatty acid-induced insulin resistance despite intramyocellular lipid accumulation. J Biol Chem. 2004;279:27177–27186. doi: 10.1074/jbc.M403566200. [DOI] [PubMed] [Google Scholar]
- 36.Petersen KF, Shubnan GI. Etiology of insulin resistance. Am J Med. 2006;199(Suppl):10S–16S. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pories WJ, Swanson MS, MacDonald KG, Long SB, Morris PG, Brown BM, Barakat HA, deRamon RA, Israel RG, Dolezal JM, Dohm GL. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg. 1995;222:339–352. doi: 10.1097/00000658-199509000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann NY Acad Sci. 2002;967:363–378. doi: 10.1111/j.1749-6632.2002.tb04292.x. [DOI] [PubMed] [Google Scholar]
- 39.Reaven GM. Banting Lecture 1988: role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 40.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 41.Schrauwen P, van Marken Lichtenbelt WD, Saris WH, Westerterp KR. Changes in fat oxidation in response to a high-fat diet. Am J Clin Nutr. 1997;66:276–282. doi: 10.1093/ajcn/66.2.276. [DOI] [PubMed] [Google Scholar]
- 42.Sebastian D, Herrero L, Serra D, Asins G, Hegardt FG. CPT 1 overexpression protects LE9 muscle cells from fatty acid-induced insulin resistance. Am J Physiol Endocrinol Metab. 2007;292:E677–E686. doi: 10.1152/ajpendo.00360.2006. [DOI] [PubMed] [Google Scholar]
- 43.Seidell JC, Muller DC, Sorkin JD, Andres R. Fasting respiratory exchange ratio and resting metabolic rate as predictors of weight gain: The Baltimore Longitudinal Study on Aging, bit. J Obes. 1992;16:667–674. [PubMed] [Google Scholar]
- 44.Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997;83:166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- 45.Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999;13:2051–2060. doi: 10.1096/fasebj.13.14.2051. [DOI] [PubMed] [Google Scholar]
- 46.Steinberg GR, Parolin ML, Heigenhauser GJ, Dyck DJ. Leptin increases FA oxidation in lean but not obese human skeletal muscle: evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab. 2002;283:E187–E192. doi: 10.1152/ajpendo.00542.2001. [DOI] [PubMed] [Google Scholar]
- 47.Tatarranni PA, Larson DE, Ravussin E. Body fat distribution and energy metabolism in obese men and women. J Am Coll Nutr. 1994;13:569–574. doi: 10.1080/07315724.1994.10718449. [DOI] [PubMed] [Google Scholar]
- 48.Thyfault JP, Kraus RM, Hickner RC, Howell AW, Wolfe RR, Dohm GL. Impaired plasma fatty acid oxidation in extremely obese women. Am J Physiol Endocrinol Metab. 2004;287:E1076–E1081. doi: 10.1152/ajpendo.00177.2004. [DOI] [PubMed] [Google Scholar]
- 49.Ukropcova B, McNeil M, Serada O, de Jonge L, Xie H, Bray G, Smith S. Dynamic changes in fat oxidation in human primary myocytes mirror metabolic characteristics of the donor. J Clin Invest. 2005;115:1934–1941. doi: 10.1172/JCI24332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weyer C, Snitker S, Rising R, Bogardus C, Ravussin E. Determinants of energy expenditure and fuel utilization in man: effects of body composition, age, sex, ethnicity, and glucose tolerance in 916 subjects. Int J Obes Relat Metab Disord. 1999;23:715–722. doi: 10.1038/sj.ijo.0800910. [DOI] [PubMed] [Google Scholar]
- 51.van der Vusse GJ, Reneman RS. Handbook of Physiology. Exercise: Regulation and Integration of Multiple Systems. Bethesda, MD: Am Physiol Soc; 1996. Lipid metabolism in muscle; pp. 952–994. sect. 12, chapt. 21. [Google Scholar]
- 52.World Health Organization (W.H.O.) Report of a WHO Consultation on Obesity. Washington, DC: World Health Organization; 1998. Obesity. Preventing and Managing the Global Epidemic. [PubMed] [Google Scholar]
- 53.Wortmann RL. Metabolic diseases of muscle. In: Wortmann RL, editor. Diseases of Skeletal Muscle. Philadelphia, PA: Lippincott, Williams, & Wilkins; 2000. pp. 157–187. [Google Scholar]
- 54.Zurlo F, Lillioja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF, Swinburn BA, Knowler WC, Bogardus C, Ravussin E. Low ratio of fat to carbohydrate oxidation as a predictor of weight gain: study of 24-h RQ. Am J Physiol Endocrinol Metab. 1990;259:E650–E657. doi: 10.1152/ajpendo.1990.259.5.E650. [DOI] [PubMed] [Google Scholar]
- 55.Zurlo F, Nemeth PM, Choksi RM, Sesodia S, Ravussin E. Whole-body energy metabolism and skeletal muscle biochemical characteristics. Metabolism. 1994;43:481–486. doi: 10.1016/0026-0495(94)90081-7. [DOI] [PubMed] [Google Scholar]