

Abstract
Purpose
To investigate herpes stromal keratitis (HSK) immunopathology by studying HSV-1–infected corneas that fail to develop HSK.
Methods
Plaque assay quantified HSV-1 in the tear film of infected mice. FACS analysis enumerated corneal leukocytic infiltrate and characterized infiltrate phenotypically after staining for activation and regulatory T cell (Treg) markers and for markers of antigen-presenting cell (APC) maturation. Treg cells were depleted in vivo using anti-CD25 mAb. Luminex analysis quantified the amount of cytokines and chemokines expressed in corneal tissue homogenate.
Results
Infected corneas without HSK exhibited a pronounced leukocytic infiltrate containing a significantly higher proportion and nearly identical absolute number of activated CD4+ T cells 15 days after infection when compared with those with HSK. Moreover, the frequency and absolute number of regulatory CD4+ T cells (Tregs) was lower in nondiseased corneas, and Treg depletion did not influence HSK incidence. The frequency of mature, immunogenic DCs and the ratio of mature DCs to CD4+ T cells were nearly identical in corneas with and without HSK. The authors observed a reduced population of neutrophils and reduced expression of neutrophil chemoattractants MIP-1β and keratinocyte chemoattractant and the neutrophil-attracting cytokine IL-6 in corneas without HSK.
Conclusions
These findings demonstrate that HSV-1–infected corneas can retain clarity in the presence of a substantial secondary leukocytic infiltrate, that activated CD4+ T cells, while necessary, are not sufficient for HSK development, that susceptibility to HSK is not determined by Tregs, and that clinical disease correlates with the accumulation of a critical mass of neutrophils through chemoattraction.
Herpes simplex virus type 1 (HSV-1)–induced stromal keratitis (HSK) is a blinding immunopathologic disease of the cornea characterized by recurrent bouts of destructive inflammation and progressive scarring in the corneal stroma.1 In mice, HSV-1 infection induces a transient neutrophilic primary infiltrate that dissipates 3 to 4 days postinfection (dpi). This primary polymorphonuclear (PMN) infiltrate contributes to control of replicating virus and coincides with the presence of a clinically apparent epithelial lesion.2 This primary neutrophilic infiltrate appears to occur in response to cytokines and chemokines produced by corneal parenchymal cells or cells of the innate immune system, or both, and is independent of T lymphocytes.2–5 A more chronic secondary leukocytic infiltrate is initiated after viral clearance from the cornea in normal mice but does not occur in T cell–deficient mice.6–8 The secondary infiltrate consists of neutrophils, CD4+ T cells, few CD8+ T cells, and antigen-presenting cells (APCs) consisting of CD11c+ dendritic cells (DCs) and F4/80+ macrophages.3,6,9,10 In most murine models of HSK, the secondary leukocytic infiltration of the cornea is orchestrated by CD4+ T cells.
Activation of naive CD4+ T cells in different microenvironments can result in differentiation along pathways leading to cells with distinct cytokine profiles. The involvement in HSK of CD4+ T cells expressing the Th1 cytokines IL-2 and IFN-γ has been established in mouse models.11,12 Extravasation of neutrophils into the infected cornea is facilitated by IFN-γ, apparently through the up-regulation of platelet endothelial cell adhesion molecule 1 on the local vascular endothelium,13 and is regulated indirectly by IL-2,13,14 presumably through the induction of chemotactic factors. CD4+ Th17 T cells have also been implicated in HSK because IL-17 induces corneal fibro-blasts to produce the neutrophil chemoattractant IL-8,15 and IL-6 regulates angiogenesis through the induction of vascular endothelial growth factor (VEGF) and neutrophilic infiltration through the induction of chemokines.15–19 In contrast, HSK severity appears to be ameliorated by CD4+ T cells expressing the Th2 cytokine IL-420 and by regulatory CD4+ T cells (Tregs), at least in part through the production of IL-10,21 which separately has been shown to mitigate HSK.4,22
Several chemokines have also been implicated in the secondary leukocytic infiltrate associated with HSK, including Mig (CXCL9), IP-10 (CXCL10), macrophage inflammatory protein (MIP)-1α (CCL3), MIP-1β (CCL4), MIP-2 (CXCL2), macrophage chemotactic protein (MCP)-1 (CCL2), MIP-3α (CCL20), and keratinocyte chemoattractant (KC).4,23–27 Many of these chemokines are produced by corneal parenchymal cells and by infiltrating inflammatory cells, and the relative contribution of these cellular sources to the chemokine milieu within the infected cornea likely changes as inflammation progresses. The combined effect of cytokine and chemokine production is an inflammatory infiltrate that, on achieving a critical mass, initiates neovascularization and destruction of the corneal architecture. However, in most humans who shed virus at the corneal surface and in some mice receiving a low-dose HSV-1 corneal infection, HSK fails to develop, suggesting that this cycle of leukocytic infiltration and activation is interrupted at some unknown point before the initiation of clinical disease.
Our previous study demonstrated that susceptibility to HSK was not associated with the magnitude of the HSV-specific CD4+ T-cell response generated in the draining lymph nodes or with the level of the delayed-type hypersensitivity response after HSV-1 corneal infection.28 However, HSK development was associated with a massive DC infiltration into the infected cornea between 7 and 14 dpi, which was abrogated by CD4+ T-cell depletion. These findings suggest that susceptibility to HSK is determined by the capacity of HSV-specific CD4+ T cells to infiltrate the cornea and to induce DC and neutrophil infiltration. We hypothesized that corneas that fail to develop HSK after HSV-1 infection would lack a critical mass of activated CD4+ T cells. However, this study demonstrates that HSV-1 infected corneas without HSK contained as many activated CD4+ T cells as those with HSK at the time of near maximal HSK severity. Corneas without HSK exhibited significantly fewer neutrophils and DCs and lower levels of known neutrophil and DC chemoattractants, suggesting that interference in the inflammatory process in these corneas occurs after CD4+ T-cell activation.
Materials and Methods
Animals
Female BALB/c wild type mice 6 to 8 weeks of age were purchased from The Jackson Laboratory (Bar Harbor, ME). All experimental animal procedures were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Corneal HSV-1 Infection
Corneal scarification was accomplished with a 30-gauge needle on mice that were under deep anesthesia induced by intraperitoneal injection of 2.0 mg ketamine hydrochloride and 0.04 mg xylazine (Phoenix Scientific, St. Joseph, MO) in 0.2 mL HBSS (BioWhittaker, Walkersville, MD). HSV-1 strain RE was grown in Vero cells, and intact virions were isolated on step gradients (Optiprep; Accurate Chemical & Scientific, Westbury, NY) according to the manufacturer’s instructions. Virus was applied directly to the eye at 1 × 103 plaque-forming units (PFUs) in 3 μMI (BioWhittaker).
HSK Scoring System
Mice were scored for herpes stromal keratitis (HSK) by slit lamp examination on alternate days between 7 and 15 days postinfection (dpi). A standard scale ranging from 1 to 4, based on corneal opacity, was used: 1+, mild corneal haze; 2+, moderate opacity; 3+, complete opacity; 4+, corneal perforation.
Quantification of Infectious Virus
Corneal surfaces were swabbed with sterile plastic applicators with cotton tips (Fisherbrand; Fisher Scientific, Pittsburgh, PA) at 2, 4, 6, 8, and 10 dpi, and swabs were placed in 0.5 mL sterile RPMI. Viral load was quantified using a standard plaque assay performed in duplicate. Samples were added in serial dilution to confluent Vero cells, incubated 1 hour at 37°C 5% CO2, and overlaid with 0.5% methylcellulose. Cultures were incubated for 72 hours at 37°C 5% CO2, fixed with 20% formaldehyde, and stained with crystal violet for 30 minutes each before rinsing with tap water.
Flow Cytometric Analysis
Corneas were excised at 15 dpi and incubated in PBS-EDTA at 37°C for 10 minutes. Stromas were separated from overlying epithelium and digested in 84 U collagenase type 1 (Sigma-Aldrich, St. Louis, MO) per cornea for 2 hours at 37°C and then were triturated to form a single-cell suspension. Suspensions were filtered through a 40-μm cell strainer cap (BD Labware, Bedford, MA) and washed. Suspensions were incubated with anti-mouse CD16/CD32 (Fcγ III/II receptor; clone 2.4G2; BD PharMingen, San Diego, CA), then stained with various leukocyte surface markers for 30 minutes on ice. The following antibodies were used: PerCP-conjugated anti-CD45 (30-F11), PE-conjugated anti-CD4 (RM4–5), anti-IA/IE (2G9), APC-Cy7-conjugated anti-CD8α (53–6.7), FITC-conjugated anti-CD69 (H1.2F3), anti-CD25 (7D4), anti-CD11c (HL3), APC-conjugated anti-F4/80, biotin-conjugated anti-CD80, pacific blue-conjugated anti-CD8α, streptavidin-PE (all BD PharMingen); APC-conjugated anti-Gr-1 (RB6–8C5; Caltag, Carlsbad, CA), and pacific blue-conjugated anti-CD40 using a labeling kit (Zenon Pacific Blue; Invitrogen, Carlsbad, CA). All isotype antibodies were obtained from BD PharMingen. Intracellular staining for regulatory T cells was performed according to standard protocol using Foxp3 staining kit (eBio-sciences, San Diego, CA). Briefly, after surface staining, cells were permeabilized using solution (Cytofix/Cytoperm; BD Biosciences, San Diego, CA) for 2 hours, then stained with APC-conjugated anti-Foxp3 (FJK16s; eBiosciences) for 30 minutes After staining, cells were fixed with 1% paraformaldehyde (PFA; Electron Microscopy Services, Fort Washington, PA) and analyzed on a flow cytometer (FACSAria with FACSDIVA data analysis software; BD Biosciences).
Regulatory T-Cell Depletion
Mice received by intraperitoneal injection 100 μg anti-CD25 mAb (PC61) in 500 μL PBS 3 days before infection. Control mice received 100 μg HLA-DR5 (HB151; American Type Culture Collection, Manassas, VA) in 500 μL PBS, 500 μL PBS alone, or no injection.
Multiplex Bead Array
Corneas with HSK were assayed individually, whereas two corneas without HSK were pooled per sample for assay. In both cases, the results are reported as the amount of protein per cornea. Corneas obtained at 15 dpi were quartered in sterile 1× PBS, then placed in 300 μS + complete protease inhibitor (Complex Mini Protease Inhibitor; Roche Applied Science, Indianapolis, IN). Samples were sonicated (Sonic Dismembrator, Model 100; Fisher Scientific, Pittsburgh, PA) four times for 15 seconds each, and the sonicator tip was rinsed with 75 μ L PBS + protease inhibitor, yielding a final volume of 600 μL/sample. Samples were microcentrifuged twice to remove cellular debris. Bio-Plex assay from Bio-Rad (Hercules, CA) was performed according to the manufacturer’s instructions, or samples were sent for luminex analysis by Millipore (St. Louis, MO). The following cytokines and chemokines were assayed: IL-6, IL-10, KC, MCP-1 (CCL2), and MIP-1β (CCL4).
Statistical Analyses
GraphPad Prism software was used for all statistical analyses. Where indicated, P values were calculated using the Student’s t-test when comparing two groups. P < 0.05 was considered significant. Results are presented as mean ± SEM.
Results
HSV-1 Titers in Corneal Tear Film Do Not Impact HSK Development
An infectious dose (1 × 103 PFU) of HSV-1 RE produced a 50% to 60% HSK incidence through 15 dpi. We hypothesized that rapid clearance of replicating HSV-1 from infected corneas might be an important factor in determining HSK susceptibility. To test this, we quantified tear film viral titers using viral plaque assay and retrospectively compared viral clearance from eyes that did or did not develop HSK (Fig. 1). No significant differences were observed in either viral burden or viral clearance kinetics between diseased and nondiseased corneas.
Figure 1.

Viral burden and clearance in corneal tear films were equivalent between HSV-infected corneas with and without HSK. Viral load and clearance from corneal tear films were assessed by standard plaque assay of eye swabs taken 2, 4, 6, 8, and 10 dpi. Data represent two independent experiments with at least 14 corneal samples per group.
Inflammatory Infiltrate in Nondiseased Corneas
In all corneas that developed HSK, corneal opacity and neo-vascularization were apparent by 15 dpi (data not shown). At 15 dpi, corneas that developed HSK (HSK score 2.0 ± 0.1) or did not develop HSK (HSK score 0) were excised, and the inflammatory infiltrate was compared by staining cells from dispersed corneas for various leukocyte markers followed by flow cytometric analysis (Fig. 2A). Surprisingly, corneas without HSK exhibited a substantial leukocytic infiltrate, though the magnitude of the infiltrate was reduced relative to corneas with HSK (Fig. 2B). Most infiltrating cells in corneas with or without HSK were PMNs, identified by their large size and high granularity, combined with high expression of Gr-1.
Figure 2.

Inflammatory cells infiltrate HSV-infected corneas without HSK but at reduced frequency compared to corneas with HSK. HSV-infected corneas with and without HSK at 15 dpi were disaggregated into single-cell suspensions and stained with anti-CD45, CD4, CD8α, CD11c, F4/80, and Gr-1 mAb. Cells were analyzed by flow cytometry. (A) Distinct live cell populations were identified based on FSC versus SSC, from which Gr-1bright (PMNs), CD11c+ DCs, F4/80+ macrophages, and CD4+ and CD8α + T cells could be gated. Isotype controls were used to aid gating (data not shown). (B) The total number of infiltrating cells per cornea is shown. Data represent the average of at least eight corneas per group from two or more independent experiments. **P < 0.01; ***P < 0.001.
Within nondiseased corneas, the total number of CD4+ T cells was reduced approximately threefold whereas the frequency of activated (CD69+) CD4+ T cells was increased by a similar margin (Fig. 3A), resulting in virtually identical numbers of activated CD4+ T cells in corneas with and without HSK (with HSK, 343 ± 7.6; without HSK, 340 ± 6.0; Fig. 3B). The frequency of activated CD8α+ T cells was also dramatically higher in corneas without HSK (Fig. 3A), resulting in a significantly higher absolute number of activated CD8α+ T cells in corneas without HSK (with HSK, 72 ± 2.4; without HSK, 216 ± 4.5; Fig. 3B). Thus, corneas with HSK exhibited a lower overall ratio of CD8α +/CD4+ T cells (with HSK, 0.16:1; without HSK, 0.64:1) and a lower ratio of activated CD8α+/activated CD4+ T cells (with HSK, 0.22:1; without HSK, 0.64:1; Table 1).
Figure 3.
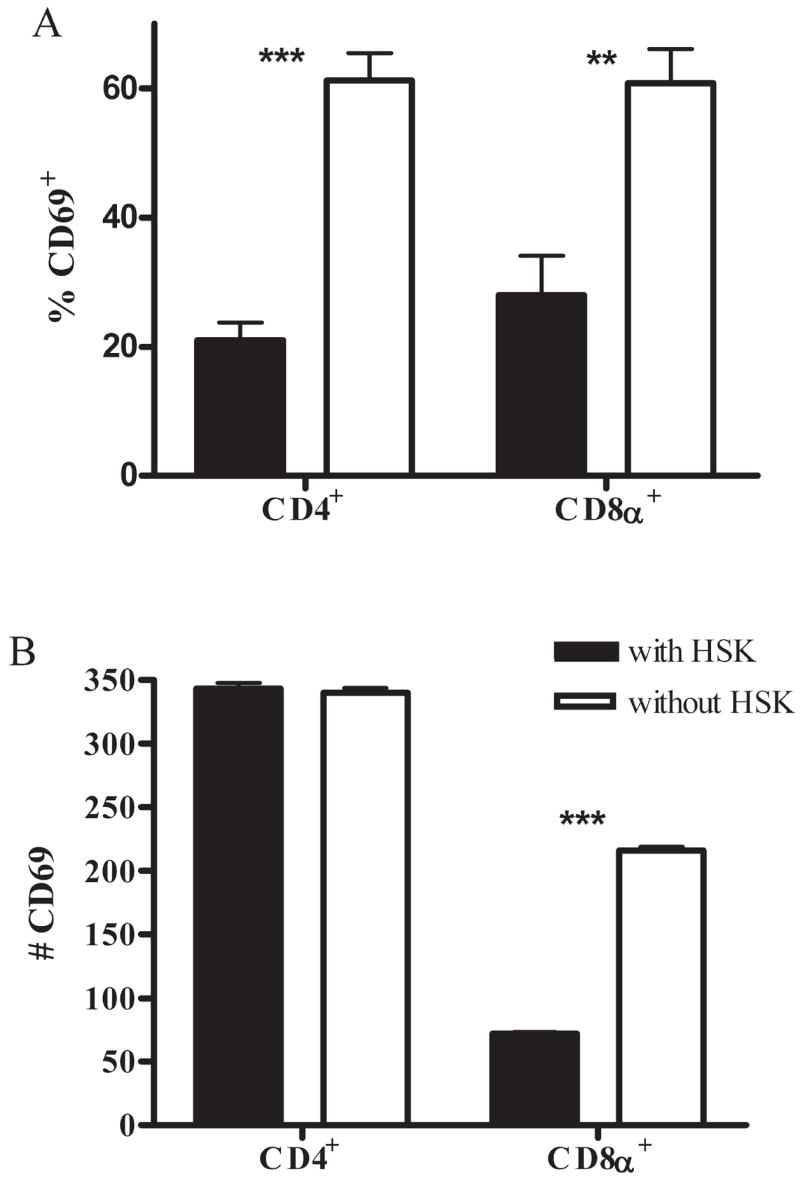
T cells in HSV-infected corneas without HSK are activated. Single-cell suspensions of HSV-infected corneas with and without HSK at 15 dpi were stained with anti-CD45, CD4, CD8α, and CD69 mAb. Flow cytometry was used to analyze the percentage (A) and number (B) of the total CD4+ and CD8α + T-cell populations that were CD69+. Data are representative of two independent experiments with at least seven mice per group. **P < 0.01; ***P < 0.001.
Table 1.
Ratios of T-Cell Subsets and APCs within HSV-Infected Corneas with and without HSK
| With HSK | Without HSK | |
|---|---|---|
| CD8α+/CD4+ | 0.16:1 | 0.64:1 |
| CD69+CD8α+/CD69+CD4+ | 0.22:1 | 0.65:1 |
| APC*/CD4+ | 2.13:1 | 0.87:1 |
| MHC class II+ CD11c+/CD4+ | 0.27:1 | 0.25:1 |
APCs equal the average number of CD11c+ plus F4/80+ cells.
Tregs in Infected Corneas
CD4+ T cells can be either pro-inflammatory effector T cells (Teff) or antiinflammatory Tregs; the two types of CD4+ T cells can be distinguished phenotypically, with Tregs expressing Foxp3 usually in conjunction with CD25. We hypothesized that the large number of activated CD4+ T cells in corneas without HSK would consist predominantly of Tregs, whereas those in corneas with HSK would consist primarily of Teff. To test this, cells from corneas with and without HSK were stained for Foxp3. Surprisingly, corneas with HSK contained a higher number of Tregs (Fig. 4) and a higher Treg/Teff ratio (1:2.75) than corneas without HSK (1:5.00). We also observed that extracts of corneas with HSK contained a greater amount of IL-10 than did corneas without HSK (Fig. 5), though values within diseased and nondiseased corneas were low.
Figure 4.

Few CD4+ T cells in HSV-infected nondiseased corneas are Foxp3+ Tregs. Single-cell suspensions of HSV-infected corneas with and without HSK at 15 dpi were stained with anti-CD45, CD4, and Foxp3 mAb. Flow cytometry was used to determine the total number of CD4+ T cells per cornea and to analyze the percentage of the total CD4+ T-cell population that was Foxp3+. Data are presented as the absolute number of CD4+Foxp3+ T cells per cornea and are representative of two independent experiments with at least seven mice per group. ***P < 0.001.
Figure 5.

Resistance to HSK is not associated with elevated levels of the inhibitory cytokine IL-10. HSV-infected corneas with and without HSK at 15 dpi were dissected and sonicated in PBS + protease inhibitor, yielding a final volume of one diseased cornea or two nondiseased corneas per 600 μL. Multiplex bead array for IL-10 expression was performed on this tissue extract. Data are representative of two independent experiments, with n values of six corneas with HSK and six samples (two corneas pooled per sample) of corneas without HSK. **P < 0.01.
To further explore a role for Tregs in HSK susceptibility, mice were treated systemically with anti-CD25 mAb 3 days before HSV-1 corneal infection, thus depleting Tregs rather than Teff. Additionally, nearly all CD4+CD25+ cells in infected corneas coexpressed Foxp3 (Fig. 6A), further ensuring specific depletion of Tregs and not Teff. Antibody treatment reduced the frequency of CD4+CD25+ cells by 95% and of CD4+Foxp3+ (both CD25+ and CD25+) Tregs in infected corneas by approximately 80% beyond 15 dpi (Fig. 6A), but it did not alter the incidence of HSK, further supporting the notion that susceptibility to HSK is not determined by Tregs. The frequency of CD4+ and CD8α + T cells in corneas with HSK was significantly increased by anti-CD25 treatment (Fig. 6B). However, infiltration of neutrophils was not significantly altered by Treg depletion (Fig. 6B), and HSK severity was similar in depleted and nondepleted mice (not shown).
Figure 6.

Depletion of CD4+CD25+ Tregs in HSV-infected corneas affects T-cell numbers in corneas but not HSK development. Mice were administered 100 μg anti-CD25 mAb (clone PC61) or anti-HLA-DR5 (control mAb) in 500 μL PBS by intraperitoneal injection 3 days before infection. (A) FACS dot plots comparing depleted corneal Treg infiltrate with a nondepleted control cornea. (B) At 15 dpi, single-cell suspensions of corneas that had developed HSK from both CD25-depleted and control mice were stained with anti-CD45, CD4, CD8α, Gr-1, and F4/80 mAb and were analyzed by flow cytometry to enumerate the total number of cells per cornea. Data are representative of two independent experiments, with n of 14 from depleted mice and 15 from control mice. *P < 0.05; ***P < 0.001.
Antigen-Presenting Cells in Infected Corneas
The number of APCs was reduced in corneas without HSK; with F4/80+, macrophages were reduced 8.90-fold and CD11c+ DCs were reduced 5.38-fold (Fig. 2). Likewise, the amount of MCP-1 (CCL2), an APC chemoattractant, was also reduced in corneas without HSK compared with corneas with HSK (Fig. 7). Moreover, the ratio of total APC (DCs + macrophages) to CD4+ T cells was significantly higher in corneas with HSK than in those without HSK (Table 1), consistent with the notion that a high overall APC/CD4+ ratio might predispose corneas to HSK development.
Figure 7.
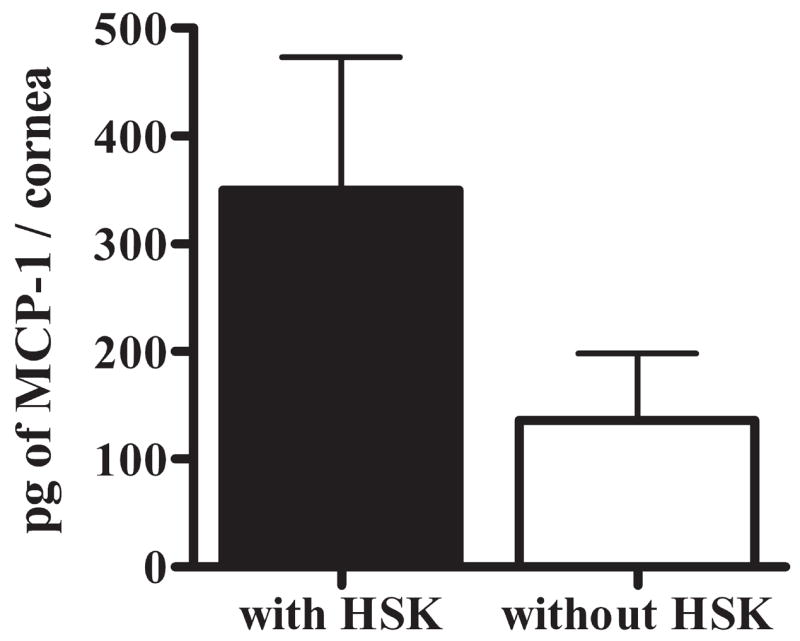
HSV-infected nondiseased corneas have reduced expression of the APC and lymphocyte chemoattractant MCP-1. HSV-infected corneas with and without HSK at 15 dpi were dissected and sonicated in preparation for multiplex bead array analysis for expression of the chemokine MCP-1 (CCL2) in corneal tissue extract. Data are representative of 2 independent experiments, with n values of six corneas with HSK and six samples (two pooled corneas per sample) of corneas without HSK.
Susceptibility to HSK could be influenced by qualitative differences in APCs because macrophages and DCs can be either immunogenic or tolerogenic, depending on their maturation phenotype.29,30 We compared the maturation phenotype of APCs in corneas with and without HSK. Higher frequencies of major histocompatibility complex (MHC) class II–positive macrophages and DCs were observed in corneas without HSK (Fig. 8), whereas the frequencies of macrophages and DCs that expressed the costimulatory molecules CD80 and CD40 were comparable in corneas with and without HSK. Moreover, the levels of expression of MHC class II, CD80, and CD40 were uniformly higher on DCs and macrophages in corneas without HSK. Therefore, most APCs in corneas that failed to develop HSK exhibited a mature immunogenic phenotype. Furthermore, the ratio of MHC class II–positive DCs to CD4+ T cells in corneas without HSK (0.25:1) was nearly identical to that in corneas with HSK (0.27:1; Table 1), consistent with the similar number of activated CD4+ T cells in corneas with and without HSK.
Figure 8.

CD11c+ dendritic cells and F4/80+ macrophages infiltrating HSV-infected nondiseased corneas are mature rather than tolerogenic. APC infiltrate was assessed at 15 dpi in HSV-infected corneas with and without HSK by flow cytometry after staining with anti-CD45, CD11c, F4/80, CD8α, MHC class II (IA/IE), CD80, and CD40 mAb for (A, C) the frequency of APCs per cornea expressing MHC class II and costimulatory molecules and (B, D) the amount of surface expression of MHC class II, CD80, and CD40, presented as the ratio of mean fluorescence intensity of corneas without HSK to corneas with HSK. (A, B) CD11c+ cells. (C, D) F4/80+ cells. Data represent the average of values from two independent experiments, with n of at least seven corneas per group. *P < 0.05.
Neutrophils in Infected Cornea
Neutrophils are considered the proximal mediators of corneal damage in HSK. Accordingly, the greatest reduction in the inflammatory infiltrate in corneas without HSK was observed in the Gr-1bright neutrophil population (9.77-fold reduction; Fig. 2). This dramatic reduction in neutrophil infiltration was accompanied by reduced levels of the neutrophil chemoattractants KC and MIP-1β (CCL4) and the neutrophil-attracting cytokine IL-6 in corneas that failed to develop HSK (Fig. 9).
Figure 9.
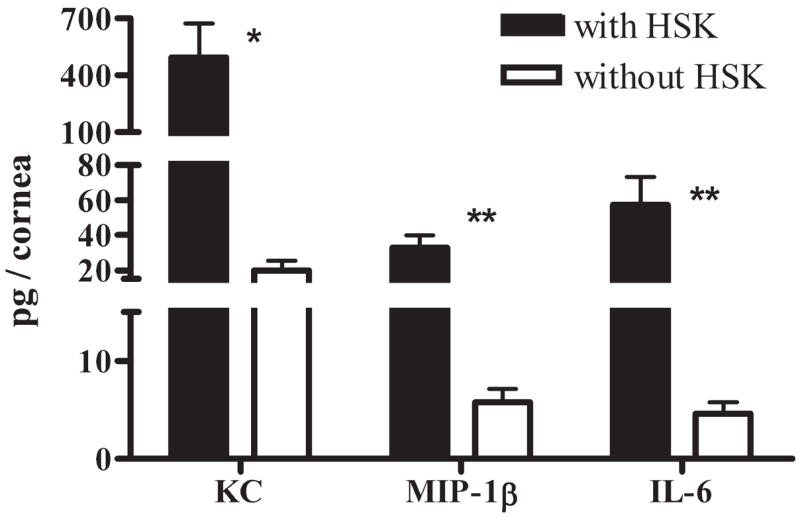
HSV-infected corneas without HSK have significantly reduced neutrophil chemoattractant expression. HSV-infected corneas with and without HSK at 15 dpi were dissected and sonicated for multiplex bead array analysis of corneal tissue extract. Results for corneas without HSK were halved to estimate the amount of protein per individual cornea. Representative data from one of two independent experiments are presented, with n values of six corneas with and six samples (two pooled corneas per sample) of corneas without HSK. *P < 0.05; **P < 0.01.
Discussion
HSK is a potentially blinding immunopathologic response to HSV-1 corneal infection. The key to developing effective prophylaxis is to define differences in the immune response that results in protection in some mice and immunopathology in others. Scientists have been dissuaded from such studies because the immune response in lymphoid organs declines before HSK onset so that at the peak of the immune response one cannot predict which mice will or will not develop HSK. Moreover, the HSV-specific T-cell response in the draining lymph nodes and the HSV-specific delayed-type hypersensitivity response in the skin after HSV-1 corneal infection are uniform among infected mice, despite models providing 50% HSK incidence.28 These studies suggest that HSK susceptibility is not determined at the inductive phase of the CD4+ T-cell response in the lymphoid organs but rather at the effector phase of the response within the infected cornea.
Our current studies demonstrate for the first time that corneas that fail to develop HSK do nonetheless develop an inflammatory infiltrate that is significant and quantifiable using current sensitive techniques. These findings establish the feasibility of studying differences in the inflammatory milieu in corneas that do or do not develop HSK. A key finding of this study is that the number of activated CD4+ T cells, as assessed by expression of the CD69 recent activation marker, is identical in corneas with and without HSK at the time of nearly maximal HSK severity, indicating that the mere presence of activated CD4+ T cells is not causal for disease and that the accumulation of activated CD4+ T cells is independent of accumulation of immunogenic APCs. Previous studies demonstrated a high Treg frequency in corneas with HSK and demonstrated that these cells attenuated the severity of HSK, at least in part through the production of IL-10,21 leading us to consider that the activated CD4+ T cells in corneas that failed to develop HSK might contain a higher frequency of Foxp3+ Tregs. However, our studies demonstrate that both the absolute number of Tregs and the ratio of Tregs/Teff are actually higher in corneas with HSK. Moreover, an 80% reduction of the Treg population in the cornea did not influence HSK incidence. Although the remaining 20% of Tregs could theoretically be capable of preventing HSK onset, an 80% reduction in Tregs did have an effect because depleted corneas that developed HSK had an increased number of CD4+ T cells, suggesting a role for Tregs in controlling CD4+ T-cell expansion or corneal infiltration. Thus, Tregs might regulate the severity of HSK in agreement with previous findings,21 but they do not appear to be a determining factor for susceptibility to HSK in our model.
We observed higher levels of IL-10 in corneas with HSK than in those without HSK. Although many cells produce IL-10, our finding would be consistent with the known capacity of Th1 cells (that mediate HSK) to produce IL-10 in an apparent attempt to dampen inflammatory tissue damage (for a review, see O’Garra and Vieira31). Although the low levels of IL-10 observed in corneas with HSK were apparently below the threshold required to prevent inflammatory damage to the cornea, the even lower level in corneas without HSK demonstrates that IL-10 is not a critical factor in determining susceptibility to HSK.
An interesting characteristic of HSK is the preponderance of CD4+ over CD8α+ T cells in the corneal infiltrate.32,33 Consistent with the possibility that a high CD4+/CD8α + T-cell ratio favors HSK development, we observed that corneas that failed to develop HSK exhibited significantly higher numbers of activated CD8α + T cells and a significantly lower CD4+/CD8α+ T-cell ratio. However, the functional significance of the small number of CD8+ T cells in nondiseased corneas remains to be established.
We next entertained the possibility that differences in corneal APCs determined HSK susceptibility. Indeed, the absolute number of macrophages and DCs and the ratio of DCs to CD4+ T cells were significantly higher in corneas with HSK. However, given that our previous study demonstrated that DC maturation within the infected cornea was important for HSK development,28 we characterized the corneal APCs for maturation marker expression. Surprisingly, the frequency of mature macrophages and DCs, as indicated by the expression of CD80 and CD40, was similar in corneas with and without HSK, and the level of expression of these molecules, as indicated by the mean fluorescence intensity, was actually higher on APCs from corneas without HSK. Moreover, the frequency of macrophages and DCs that expressed MHC class II and the level of MHC class II expression per cell were significantly higher in corneas without HSK, whereas the ratio of MHC class II–positive DCs to CD4+ T cells was nearly identical in corneas with and without HSK. We conclude from these observations that neither a lack of APC availability nor APC stimulatory capacity is a likely explanation for the failure of HSK development.
HSK corneal damage results from a second wave of infiltrating neutrophils that occurs after replicating virus is eliminated from the cornea.3,10 Neutrophils release matrix metalloproteinase (MMP), which breaks down the extracellular matrix of the corneal stroma and enhances neovascularization through the production of MMP and VEGF.34–38 The most obvious difference in the inflammatory infiltrate in corneas with and without HSK was the dramatically (approximately 10-fold) reduced neutrophil population in corneas without HSK. This reduced neutrophil infiltration was associated with a significant reduction in expression of the neutrophil chemoattractants MIP-1β and KC and the neutrophil-attracting cytokine IL-6. Neutrophils produce chemoattractants when exposed to IL-6, and neutrophils are necessary for HSV-1 clearance from cornea. Further, IL-6–deficient mice eliminate HSV-1 from the cornea with normal kinetics and fail to develop HSK.19 The combined results of these studies suggest that IL-6 is required for the second wave of neutrophil infiltration into the cornea that is associated with immunopathology but is not required for the first wave of neutrophils into the cornea that provides protection from replicating virus.
Importantly, our work highlights the study of HSV-1–infected, nondiseased corneas as a novel approach to elucidating HSK pathogenesis. Our findings support IL-6 as an important factor determining HSK susceptibility through the induction of neutrophil chemoattraction into the cornea and also point to a possible local inhibitory effect of CD8α + T cells within infected corneas on HSK progression.
Acknowledgments
Supported by National Eye Institute Grants R01 EY010359 (RLH) and P30-EY08099 (RLH); an unrestricted research grant (RLH) and a research grant (SJD) from Research to Prevent Blindness, Inc.; and a grant from the Eye and Ear Foundation of Pittsburgh (RLH).
Footnotes
Disclosure: S.J. Divito, None; R.L. Hendricks, None
References
- 1.Hendricks RL, Epstein RJ, Tumpey T. The effect of cellular immune tolerance to HSV-1 antigens on the immunopathology of HSV-1 keratitis. Invest Ophthalmol Vis Sci. 1989;30:105–115. [PubMed] [Google Scholar]
- 2.Tumpey TM, Chen SH, Oakes JE, Lausch RN. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol. 1996;70:898–904. doi: 10.1128/jvi.70.2.898-904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimeld C, Whiteland JL, Nicholls SM, Easty DL, Hill TJ. Immune cell infiltration in corneas of mice with recurrent herpes simplex virus disease. J Gen Virol. 1996;77(pt 5):977–985. doi: 10.1099/0022-1317-77-5-977. [DOI] [PubMed] [Google Scholar]
- 4.Tumpey TM, Cheng H, Yan XT, Oakes JE, Lausch RN. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J Leukoc Biol. 1998;63:486–492. doi: 10.1002/jlb.63.4.486. [DOI] [PubMed] [Google Scholar]
- 5.Yan XT, Tumpey TM, Kunkel SL, Oakes JE, Lausch RN. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest Ophthalmol Vis Sci. 1998;39:1854–1862. [PubMed] [Google Scholar]
- 6.Hendricks RL, Tumpey TM. Concurrent regeneration of T lymphocytes and susceptibility to HSV-1 corneal stromal disease. Curr Eye Res. 1991;10(suppl):47–53. doi: 10.3109/02713689109020357. [DOI] [PubMed] [Google Scholar]
- 7.Metcalf JF, Hamilton DS, Reichert RW. Herpetic keratitis in athy-mic (nude) mice. Infect Immun. 1979;26:1164–1171. doi: 10.1128/iai.26.3.1164-1171.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell RG, Nasisse MP, Larsen HS, Rouse BT. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest Ophthalmol Vis Sci. 1984;25:938–944. [PubMed] [Google Scholar]
- 9.Newell CK, Martin S, Sendele D, Mercadal CM, Rouse BT. Herpes simplex virus-induced stromal keratitis: role of T-lymphocyte subsets in immunopathology. J Virol. 1989;63:769–775. doi: 10.1128/jvi.63.2.769-775.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas J, Gangappa S, Kanangat S, Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol. 1997;158:1383–1391. [PubMed] [Google Scholar]
- 11.Hendricks RL, Tumpey TM, Finnegan A. IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J Immunol. 1992;149:3023–3028. [PubMed] [Google Scholar]
- 12.Stumpf TH, Shimeld C, Easty DL, Hill TJ. Cytokine production in a murine model of recurrent herpetic stromal keratitis. Invest Ophthalmol Vis Sci. 2001;42:372–378. [PubMed] [Google Scholar]
- 13.Tang Q, Hendricks RL. Interferon gamma regulates platelet endothelial cell adhesion molecule 1 expression and neutrophil infiltration into herpes simplex virus-infected mouse corneas. J Exp Med. 1996;184:1435–1447. doi: 10.1084/jem.184.4.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Q, Chen W, Hendricks RL. Proinflammatory functions of IL-2 in herpes simplex virus corneal infection. J Immunol. 1997;158:1275–1283. [PubMed] [Google Scholar]
- 15.Maertzdorf J, Osterhaus AD, Verjans GM. IL-17 expression in human herpetic stromal keratitis: modulatory effects on chemokine production by corneal fibroblasts. J Immunol. 2002;169:5897–5903. doi: 10.4049/jimmunol.169.10.5897. [DOI] [PubMed] [Google Scholar]
- 16.Banerjee K, Biswas PS, Kim B, Lee S, Rouse BT. CXCR2−/− mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL-6-induced neovascularization. J Immunol. 2004;172:1237–1245. doi: 10.4049/jimmunol.172.2.1237. [DOI] [PubMed] [Google Scholar]
- 17.Biswas PS, Banerjee K, Kinchington PR, Rouse BT. Involvement of IL-6 in the paracrine production of VEGF in ocular HSV-1 infection. Exp Eye Res. 2006;82:46–54. doi: 10.1016/j.exer.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem. 1996;271:736–741. doi: 10.1074/jbc.271.2.736. [DOI] [PubMed] [Google Scholar]
- 19.Fenton RR, Molesworth-Kenyon S, Oakes JE, Lausch RN. Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest Ophthalmol Vis Sci. 2002;43:737–743. [PubMed] [Google Scholar]
- 20.Heiligenhaus A, Li H, Schmitz A, Wasmuth S, Bauer D. Improvement of herpetic stromal keratitis with fumaric acid derivate is associated with systemic induction of T helper 2 cytokines. Clin Exp Immunol. 2005;142:180–187. doi: 10.1111/j.1365-2249.2005.02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172:4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 22.Tumpey TM, Elner VM, Chen SH, Oakes JE, Lausch RN. Interleu-kin-10 treatment can suppress stromal keratitis induced by herpes simplex virus type 1. J Immunol. 1994;153:2258–2265. [PubMed] [Google Scholar]
- 23.Thomas J, Kanangat S, Rouse BT. Herpes simplex virus replication-induced expression of chemokines and proinflammatory cytokines in the eye: implications in herpetic stromal keratitis. J Interferon Cytokine Res. 1998;18:681–690. doi: 10.1089/jir.1998.18.681. [DOI] [PubMed] [Google Scholar]
- 24.Shirane J, Nakayama T, Nagakubo D, et al. Corneal epithelial cells and stromal keratocytes efficiently produce CC chemokine-ligand 20 (CCL20) and attract cells expressing its receptor CCR6 in mouse herpetic stromal keratitis. Curr Eye Res. 2004;28:297–306. doi: 10.1076/ceyr.28.5.297.28682. [DOI] [PubMed] [Google Scholar]
- 25.Carr DJ, Chodosh J, Ash J, Lane TE. Effect of anti-CXCL10 monoclonal antibody on herpes simplex virus type 1 keratitis and retinal infection. J Virol. 2003;77:10037–10046. doi: 10.1128/JVI.77.18.10037-10046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wuest T, Austin BA, Uematsu S, Thapa M, Akira S, Carr DJ. Intact TRL 9 and type I interferon signaling pathways are required to augment HSV-1 induced corneal CXCL9 and CXCL10. J Neuroimmunol. 2006;179:46–52. doi: 10.1016/j.jneuroim.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wuest T, Farber J, Luster A, Carr DJ. CD4+ T cell migration into the cornea is reduced in CXCL9 deficient but not CXCL10 deficient mice following herpes simplex virus type 1 infection. Cell Immunol. 2006;243:83–89. doi: 10.1016/j.cellimm.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Hendricks RL. B7 costimulatory requirements of T cells at an inflammatory site. J Immunol. 1998;160:5045–5052. [PubMed] [Google Scholar]
- 29.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 30.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Garra A, Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–428. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- 32.Niemialtowski MG, Rouse BT. Predominance of Th1 cells in ocular tissues during herpetic stromal keratitis. J Immunol. 1992;149:3035–3039. [PubMed] [Google Scholar]
- 33.Niemialtowski MG, Godfrey VL, Rouse BT. Quantitative studies on CD4+ and CD8+ cytotoxic T lymphocyte responses against herpes simplex virus type 1 in normal and beta 2-m deficient mice. Immunobiology. 1994;190:183–194. doi: 10.1016/s0171-2985(11)80268-8. [DOI] [PubMed] [Google Scholar]
- 34.Kim B, Suvas S, Sarangi PP, Lee S, Reisfeld RA, Rouse BT. Vascular endothelial growth factor receptor 2-based DNA immunization delays development of herpetic stromal keratitis by antiangiogenic effects. J Immunol. 2006;177:4122–4131. doi: 10.4049/jimmunol.177.6.4122. [DOI] [PubMed] [Google Scholar]
- 35.Lee S, Zheng M, Kim B, Rouse BT. Role of matrix metalloprotein-ase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J Clin Invest. 2002;110:1105–1111. doi: 10.1172/JCI15755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Philipp W, Speicher L, Humpel C. Expression of vascular endothelial growth factor and its receptors in inflamed and vascularized human corneas. Invest Ophthalmol Vis Sci. 2000;41:2514–2522. [PubMed] [Google Scholar]
- 37.Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol. 2001;75:9828–9835. doi: 10.1128/JVI.75.20.9828-9835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng M, Schwarz MA, Lee S, Kumaraguru U, Rouse BT. Control of stromal keratitis by inhibition of neovascularization. Am J Pathol. 2001;159:1021–1029. doi: 10.1016/S0002-9440(10)61777-4. [DOI] [PMC free article] [PubMed] [Google Scholar]