

Abstract
The G59S missense mutation at the conserved microtubule-binding domain of p150glued, a major component of dynein/dynactin complex, has been linked to an autosomal dominant form of motor neuron disease (MND). To study how this mutation affects the function of the dynein/dynactin complex and contributes to motor neuron degeneration, we generated p150glued G59S knock-in mice. We found that the G59S mutation destabilizes p150glued and disrupts the function of dynein/dynactin complex, resulting in early embryonic lethality of homozygous knock-in mice. Heterozygous knock-in mice, which developed normally, displayed MND-like phenotypes after 10 months of age, including excessive accumulation of cytoskeletal and synaptic vesicle proteins at neuromuscular junctions, loss of spinal motor neurons, increase of reactive astrogliosis, and shortening of gait compared with wild-type littermates and age-matched p150glued heterozygous knock-out mice. Our findings indicate that the G59S mutation in p150glued abrogates the normal function of p150glued and accelerates motor neuron degeneration.
Keywords: dynactin, dynein, p150glued, motor neuron disease, mouse model, ALS
Introduction
Motor neurons rely heavily on microtubule-based transport of organelles, vesicles and molecules for their normal function and survival (Holzbaur, 2004). Intracellular cargos are transported either away from (anterograde) or toward (retrograde) the cell body by the kinesin and dynein motor protein complexes (Guzik and Goldstein, 2004). Dynactin, a macromolecular complex, has been proposed to facilitate dynein-mediated retrograde transport of vesicles and organelles along microtubules and provides a link between specific cargos, microtubules and cytoplasmic dynein (Schroer, 2004). The dynactin complex consists of at least 10 distinct components, including p45 (Arp1), p50 (dynamitin), and p150glued. Dynactin p150glued, encoded by the dynactin 1 (Dctn1) gene, is the largest subunit of the dynactin complex that binds directly to microtubules and the intermediate chain of dynein through its cytoskeleton-associated glycine-rich protein (CAP-Gly) and coiled-coil domains (Holzbaur and Vallee, 1994; Schroer, 2004). Recently, a single-base pair change in the Dctn1 gene (C957T) resulting in the substitution of serine for glycine at position 59 of p150glued has been associated with a slowly progressive, autosomal dominant form of lower motor neuron disease without sensory symptoms in a North American family (Puls et al., 2003). This G59S substitution located in the highly conserved CAP-Gly domain of p150glued potentially interferes with protein folding, decreases the binding of the mutant protein to microtubules, and leads to an alteration in dynein/dynactin-mediated transport (Puls et al., 2003; Levy et al., 2006). More missense mutations in Dctn1 have been identified since and are linked to amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), although how these mutations affect the structure and function of p150glued is unclear (Munch et al., 2004, 2005).
The evidence for a critical role for p150glued in vivo comes from the analysis of mutations in Drosophila with the homologous gene, glued. The heterozygous glued mutation leads to the disruption of axon morphology and fast axonal transport (Reddy et al., 1997; Martin et al., 1999), whereas null mutations are lethal early in development (Harte and Kankel, 1982). Furthermore, the integrity of the dynactin complex is essential for maintaining synapse stability at Drosophila neuromuscular junctions (NMJ) (Eaton et al., 2002). Overexpression of dynamitin disassembles the dynactin complex, disrupts axonal retrograde transport and induces accumulation of neurofilaments and synaptophysin at the cell periphery of motor neurons, leading to a late-onset progressive motor neuron disease in transgenic mice (LaMonte et al., 2002). Missense point mutations in cytoplasmic dynein heavy chain also result in motor neuron degeneration in heterozygous mutant mice (Hafezparast et al., 2003).
To study the pathogenic mechanism of the G59S mutation in vivo, we generated p150glued G59S knock-in mice and p150glued heterozygous knock-out mice. Whereas heterozygous p150glued knock-out mice appeared normal, heterozygous p150glued G59S knock-in mice developed a late-onset, slowly progressive motor neuron disease characterized by abnormal accumulation of neurofilaments and synaptic vesicle proteins at the NMJ, loss of motor neurons, and gait abnormalities. Our findings indicate that the G59S mutation likely exerts a dominant negative effect on the normal function of p150glued, leading to motor neuron degeneration in heterozygous p150glued knock-in mice.
Materials and Methods
Generation of p150glued knock-out and G59S knock-in mice
p150glued protein is encoded by the Dctn1 gene at mouse chromosome 6. DNA fragments containing Dctn1 were isolated from a mouse genomic library (Stratagene, La Jolla, CA). A 9.3 kb KpnI/SacII fragment carrying exons 2–8 of Dctn1 was subcloned into the pBluescript vector for later modifications. To construct the knock-in targeting vector, a 3.1 kb HindIII/SpeI fragment containing exons 2 and 3 of Dctn1 was modified via introduction of a new DdeI site in exon 2 by replacement of nucleotides GGGC with CTCA using QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA), resulting in the substitution of encoded amino acid Glycine with Serine (Fig. 1 A, Dctn1 /m). To construct the conditional knock-out construct, one copy of a Loxp site was inserted into the HindIII site in front of exon 2 and a 3.4 kb XbaI fragment, containing the neomycin-resistance gene flanked with two Loxp sites (lnl), was inserted into the SpeI site immediately after exon 3 (Fig. 1 A, Dctn1 /lnl). The targeting vector was linearized at a unique NotI site and transfected into 129/SvJ ES cells, which were later subjected to G418 selection for 7 d. The G418 resistant ES clones were picked and screened by Southern blot analysis for the correctly targeted clones. Two positive ES clones were expanded and injected into blastocysts. The resulting male chimera mice were bred with wild-type C57BL/6J female mice to obtain Dctn1 +/lnl mice. Dctn1 +/lnl mice were then crossed with Cre transgenic mice (EIIa-Cre) to obtain Dctn1 +/m and Dctn1 +/Δ animals in which both exons 2 and 3 were deleted (Fig. 1 A). Genomic DNA isolated from mouse livers were digested with HindIII and subjected to Southern blot analysis using a probe outside of the targeting vector. In addition to a 3.0 kb HindIII fragment for the wild-type (+) allele, 16.0 kb, 15.7 kb, and 5.7 kb HindIII fragments were detected for the G59S knock-in mutant (m) allele, the exons 2 and 3 deletion (Δ) allele, and the loxp and neomycin (lnl) allele, respectively (Fig. 1 B, arrow). All mutant Dctn1 mice are a hybrid of 129/SvJ and C57BL/6J strain backgrounds. Once confirmed by Southern blot, mouse genotypes were determined by PCR amplification of tail DNA (Dctn1-Ex3F: ACTTCCCCAGAGACTCCTGA and Dctn1-Ex4R: CAGTTTGCTGGTCTTTGCAG). The mice were housed in a 12-h light/dark cycle and fed regular diet ad libitum. All mouse work follows the guidelines approved by the Institutional Animal Care and Use Committees of the National Institute of Child Health and Human Development.
Figure 1.
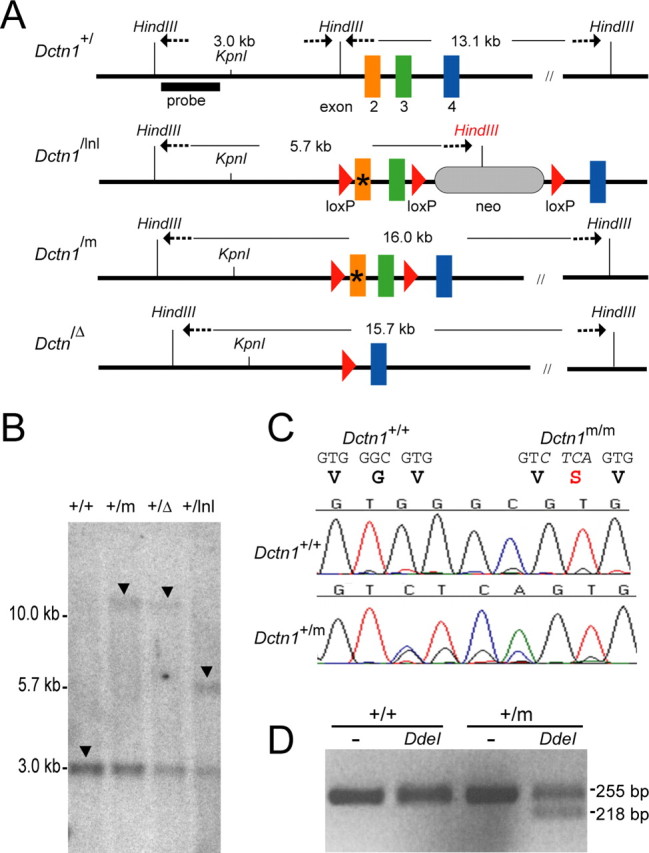
Generation of p150glued G59S knock-in mice. A, A schematic outline of mouse Dctn1 wild-type (+), floxed neomycin gene (lnl) insertion, G59S knock-in mutant (m), and exons 2 and 3 deletion (Δ) alleles. * represents the mutated region. B, Southern blot analysis of genomic DNA extracted from wild-type and heterozygous mutant mice confirmed the correct targeting of mutated Dctn1 alleles, which displayed 16.0, 15.7, 5.7, and 3.0 kb HindIII fragments (arrows) for the lnl, m, Δ, and + alleles, respectively. C, Chromatograms showed the partial sequence of exon2 amplified from RNA extracted from Dctn1 wild-type (+/+) and heterozygous knock-in (+/m) mice. D, Agarose gel electrophoresis of the RT-PCR products from wild-type (+/+) and Dctn1 +/m (+/m) after digestion with (+) or without (−) DdeI.
Transcription analysis of the mutant allele of p150glued G59S knock-in mice
Total RNA purified from the mouse brain by Trizol (Invitrogen) was used as the template for reverse-transcriptase and PCR (RT-PCR) amplification by a pair of Dctn1 specific primers residing in exon 2 (BamHI-Ex2F: cgcggatccTGTTGGAGCCACACTCTTTG) and 10 (EcoRI-Ex10R: ccggaattcTGTAGCGTTCCTTTGCCTCT), respectively. The PCR product was then either digested with BamH1 and EcoRI and subcloned into pBluescript vector for sequence analysis, or used as the template for a second round of PCR amplification using a reverse PCR primer located in exon 4 (Ex4R: CAGTTTGCTGGTCTTTGCAG). The nested PCR product was purified and subjected to DdeI digestion. The presence of the G59S allele of Dctn1 +/m was revealed by the appearance of a smaller DNA band after digestion with DdeI (Fig. 1 D).
Biochemical analysis of the p150glued G59S knock-in mice
Mouse brain or spinal cord was homogenized in TBS buffer (10 mm Tris-HCl pH 7.5, 150 mm NaCl, 5 mm EDTA) plus protease inhibitor cocktails (PI, Roche Bioscience, Palo Alto, CA). Aliquots of homogenates were extracted with 1% Triton X-100 or 2% SDS, respectively, and subjected to immunoblotting. Antibodies used in these studies included monoclonal anti-p150glued antibody (BD, Transduction Laboratories, Lexington, KY), polyclonal anti-p150glued antibody (Abcam, Cambridge, UK), dynein and p50 antibodies (Chemicon, Temecula, CA), β-tubulin antibody (Covance, Berkeley, CA), and β-actin antibody (Sigma, St Louis, MS).
Sucrose density gradient centrifugation
Brain lysates from wild-type and Dctn1 +/m mice were homogenized in 20 mm Tris-HCl, pH 7.4, and 1 mm EDTA with PI (Roche). Triton X-100 was added to a final concentration of 0.4%, and the homogenate was clarified by low-speed centrifugation. The resulting supernatant fraction was subjected to 5–20% linear sucrose density gradient centrifugation as described previously (Levy et al., 2006). The gradients were eluted in 1.0 ml fractions, which were resolved by SDS-PAGE and analyzed by Western blot.
Behavior analysis of the p150glued G59S knock-in mice
Rotarod test.
Mice were placed onto a rotating rod with auto acceleration from 0 rpm to 40 rpm in 4 min (San Diego Instruments, San Diego, CA). The length of time the mouse stayed on the rotating rod was recorded.
Grip strength measurement.
Mice were allowed to use their forepaws or hindpaws to pull or compress a triangular bar attached to a digital force gauge (Ametek, Largo, FL) set up to record the maximal pulling or compressing force. Six measurements were taken for each animal during each test.
Gait analysis.
As described previously (Wooley et al., 2005), the TreadScan Gait Analysis System (Clever Sys, Reston, VA) was used to assess the nature of the gait behaviors of the mice. Each mouse was placed into the chamber, onto the treadmill unit. With the speed set at 8 cm/s, each mouse was given 15 s of training time with the treadmill unit. The treadmill was then turned off and the mouse was given 1 min of rest. The treadmill was then turned on again at 8 cm/s and the movement of the mouse was recorded for 20 s at 100 frames per second. The TreadScan software grouped the frames into individual strides for each foot, usually producing between 35 and 50 strides. Stance time, swing time, brake time, propulsion time, stride time, stride length, percentage of stride, and percentage of stance were recorded. Each group of frames was then examined for erroneous data, such as multiple steps recorded as one stride, pauses in the movement resulting in sliding, or the nose and tail being mistaken for a foot. After all erroneous data were removed the rest of the data were exported to Microsoft Excel for calculation of the averages for each parameter of each mouse.
TUNEL assay.
Terminal deoxynucleotidyl transferase (TdT) –mediated deoxyuridine triphosphate (dUTP)-rhodamine nick end labeling (TUNEL) assay (Roche) was used to visualize cells undergoing programmed cell death as suggested by the manufacturer. Negative controls were treated similarly except for not being incubated with TdT enzyme. Slides were illuminated using a laser scanning confocal microscope (Zeiss LSM 510, Thornwood, NY).
Histology and immunohistochemical analysis.
Mice were perfused via cardiac infusion with 4% paraformaldehyde in cold PBS. To obtain frozen sections, tissues were removed and submerged in 30% sucrose for 24 h and sectioned at 40 μm thickness with a sliding microtome. For paraffin sections, tissues were embedded in paraffin and sectioned at 8 μm thickness by rotary microtome. Antibodies specific for GFAP and synaptophysin (Sigma), and SMI31 and SMI32 (Sternberger Monoclonal, Lutherville, MD) were used as suggested by the manufacturer followed by counterstaining with hematoxylin and eosin (HE). For immunohistochemical analysis, limb muscles from 10- to 24-month-old mice were dissected, fixed, and stained with a synaptophysin antibody (1:500) and SMI32 (1:3000), followed by Alexa Fluor 488-conjugated secondary antibody and Alexa Fluor 568-conjugated α-bungarotoxin (BTX). Z-serial images were collected with confocal microscope (Zeiss). The images presented represent single-projected images derived from overlaying each set of Z-images.
Motor neuron count
The L1 to L5 of lumbar spinal cord was sectioned at 40 μm thickness. Every 12 coronal sections was selected and stained with 0.1% Cresyl Violet (Nissl staining). The criteria for a motor neuron included a round, open, pale nucleus (not condensed and darkly stained), globular Nissl staining of the cytoplasm, and a diameter of ∼30–45 μm. More than 25 sections were counted for each animal.
Statistical analysis
Statistical analysis was performed using the StatView program (SAS Institute Inc, version 5.0). Data are presented as means ± SEM. Statistical significances were determined by comparing datasets of different groups using ANOVA or Log rank tests. Differences were considered significant with p < 0.05.
Results
Generation of p150glued G59S knock-in mice
To study the role of the p150glued G59S mutation in the development of motor neuron disease, we generated a series of Dctn1 mutant mice (Fig. 1 A). The targeted gene replacement of Dctn1 +/m, Dctn1 +/Δ, and Dctn1 +/lnl mice was verified by Southern blot in which 16.0, 15.7, 5.7, and 3.0 kb HindIII fragments were detected, corresponding to the G59S knock-in (m) allele, exon 2 and 3 deletion (Δ) allele, loxp and neomycin (lnl) insertion allele, and wild-type allele (+), respectively (Fig. 1 B, arrow). To examine the expression of Dctn1 +/m mutant allele, a pair of primers residing at exon 2 and 10 of Dctn1 was used for RT-PCR of total RNA extracted from Dctn1 +/m mouse brain. The replacement of GGGC with CTCA in the exon 2 of the mutant allele was confirmed by direct sequencing of purified RT-PCR product (Fig. 1 C, bottom). A second reverse primer located at exon 4 in combination with the same forward primer at exon 2 were used to amplify a 255 bp PCR product from both wild-type and Dctn1 +/m samples, which was then digested with DdeI. After DdeI digestion, a shorter 218 bp band was only detected in Dctn1 +/m samples, which further confirmed the expression of Dctn1 mutant allele (Fig. 1 D).
To compare the expression levels of both wild-type and mutant Dctn1 alleles in the Dctn1 +/m mouse brain, we subcloned the RT-PCR product of Dctn1 exons 2 through 10 into a plasmid vector and randomly picked 32 clones for sequencing. Among these 32 clones, 18 contained the mutated sequence, indicating an equal expression of both wild-type and mutant Dctn1 alleles in the Dctn1 +/m mouse brain. Except for the designed mutations at exon 2, no other sequence variation was found in Dctn1 mutant clones, suggesting that the genetic modification of Dctn1 does not alter the transcription of the mutant allele.
Embryonic lethality is observed in homozygous p150glued G59S knock-in mice
Previous studies suggest that the G59S mutation in p150glued compromises its binding affinity to microtubules and likely leads to dysfunction of the dynein/dynactin complex (Puls et al., 2003; Levy et al., 2006). To examine the functional consequence of the G59S mutation in p150glued, we intercrossed heterozygous Dctn1 +/m mice to obtain homozygous Dctn1 m/m mice. However, no Dctn1 m/m offspring were obtained from Dctn1 +/m crosses. The ratio of wild-type and Dctn1 +/m mice was 1:2, suggesting that the homozygous Dctn1 m/m mice are embryonically lethal. We then set up timed pregnant mating of heterozygous Dctn1 +/m mice and collected embryos at different gestation stages. At 9.5 d post coitus (dpc), the wild-type embryo had 21–29 pairs of somites, a well developed heart and CNS, and condensation of forelimb buds near the 8th–12th somite pairs (Fig. 2 A). The littermate Dctn1 m/m embryo, however, had severe gastrulation problems. The development of Dctn1 m/m embryos was likely arrested at approximately 8.0 dpc in which the neural tube was only partially formed and no typical somite could be identified (Fig. 2 B). The homozygous deletion of Dctn1 (Dctn1 Δ/Δ) also caused early embryonic lethality (Fig. 2 C,D). The development of Dctn1 Δ/Δ embryo was arrested at approximately 7.5 dpc (Fig. 2 D) compared with the wild-type littermate control at 8.5 dpc (Fig. 2 C). In contrast, no significant developmental alterations were observed in heterozygous Dctn1 +/m and Dctn1 +/Δ embryos compared with their wild-type littermate controls (data not shown).
Figure 2.
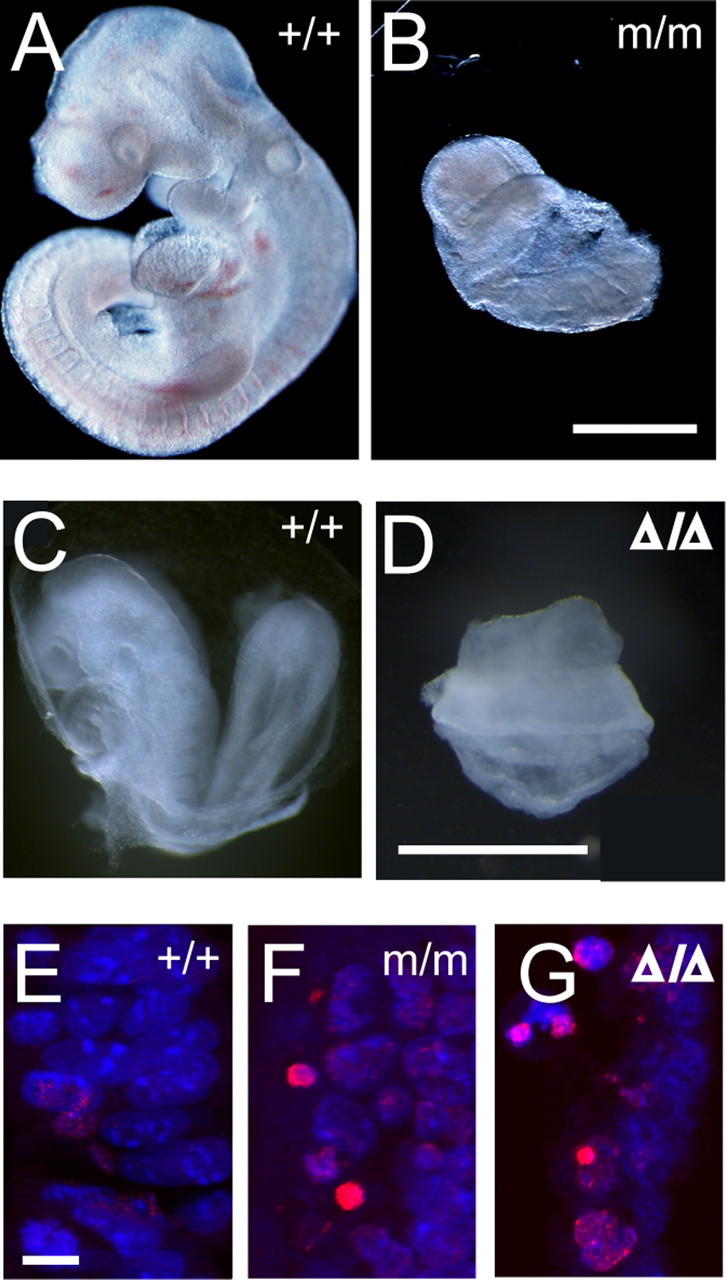
Homozygous Dctn1 knock-in mice were embryonic lethal. A, B, Wild-type (+/+) and Dctn1 m/m (m/m) embryos at 9.5 dpc. Scale bar, 500 μm. C, D, Wild-type (+/+) and Dctn1 Δ/Δ (Δ/Δ) were at 8.5 dpc. Scale bar, 600 μm. E–G, TUNEL staining (red) revealed increased programmed cell death in Dctn1 m/m (m/m; F) and Dctn1 Δ/Δ (Δ/Δ; G) embryos compared with wild-type controls (+/+; E). Nuclei were stained with ToPro-3 (blue). Scale bar, 10 μm.
Increased numbers of TUNEL positive cells were also observed in Dctn1 m/m (Fig. 2 F) and Dctn1 Δ/Δ (Fig. 2 G) embryos compared with wild-type (Fig. 2 E) and heterozygous littermates (data not shown), indicating an augmentation of apoptotic cell death in Dctn1-deficient embryos. The early embryonic lethality of Dctn1 m/m and Dctn1 Δ/Δ mice is consistent with the essential function of the dynein/dynactin complex in cell proliferation and suggests that the G59S mutation in p150glued severely disrupts the normal function of dynactin.
The G59S mutation destabilizes p150glued protein
Crystal structures of the CAP-Gly domain in the p150glued protein indicate that the Gly-59 residue is required for maintaining the folding of the three-layer β-sheet structure (Li et al., 2002). It may explain why over-expression of the p150glued G59S mutant protein induces the formation of aggregates in heterologous cell lines (Levy et al., 2006). We examined the expression of p150glued protein in Dctn1 +/+, Dctn1 +/m, Dctn1 +/Δ, and Dctn1 +/lnl mouse brains using antibodies that specifically recognize the C-terminal (Fig. 3 A, top) or N-terminal (Fig. 3 A, middle) of p150glued. The p150glued C-terminal antibody (C-p150) recognized a doublet of 150 and 135 kDa bands in mouse brain as previously described (Tokito et al., 1996). The 135 kDa band is encoded by an alternative splicing variant of Dctn1 gene that lacks the first 5 coding exons (Tokito et al., 1996). The p150glued N-terminal antibody (N-p150) raised against the microtubule binding domain of p150glued, reacts only with wild-type full-length p150glued but not with the p150glued G59S mutation or N-terminal truncated p135 variant (Levy et al., 2006). We found that the level of p150glued was reduced by 50% in adult Dctn1 +/m, Dctn1 +/Δ, and Dctn1 +/lnlmouse brains and spinal cords (Fig. 3 A,B). Moreover, p150glued protein was not detectable in Dctn1 m/m mouse embryos collected from 7 to 9 dpc (Fig. 3 C,D). These results suggest that p150glued G59S mutant protein is either quickly degraded or forms detergent insoluble aggregates.
Figure 3.

The G59S mutation destabilized p150glued protein. A, Western blot analyses revealed the expression of p150glued/p135 (top; detected by the p150glued C-terminal antibody, C-p150) and p150glued alone (middle; detected by p150glued N-terminal antibody, N-p150) in brains and spinal cords of wild-type (+/+), Dctn1 +/m (+/m), Dctn1 +/Δ (+/Δ), and Dctn1 +/lnl (+/lnl) mice. The expression of β-tubulin (bottom) was used as a loading control. B, Bar graph shows reduced accumulation of p150glued in Dctn1 +/m and Dctn1 +/Δ brains compared with wild-type controls, whereas the expression of alternative spliced p135 was slightly increased in these animals. Data are means ± SEM. C, Genotyping of wild-type (+/+), Dctn1 +/m (+/m), and Dctn1 m/m (m/m) embryos by PCR amplification of genomic DNA prepared from yolk sacs. D, Western blot analyses using p150glued C-terminal antibody revealed the absence of p150glued protein from Dctn1 m/m (m/m) embryos (top). The expression of β-actin (bottom) was used as a loading control.
The Dctn1 p135 isoform is particularly enriched in neural tissues (Tokito et al., 1996), which may explain why it is undetectable in 8.0 dpc mouse embryos (Fig. 3 D). The level of p135 was comparable among wild-type, Dctn1 +/m and Dctn1 +/Δ mouse brains (Fig. 3 A,B). Although the exact function of p135 remains elusive, it may regulate the function of dynactin complex by competing with p150glued for binding with dynein and other dynactin subunits (Melloni et al., 1995).
Dynein/dynactin complex remains intact in Dctn1 +/m mice
The vertebrate dynactin complex has been reported to migrate as a ∼19S protein heteromultimer resolved by 5–20% sucrose gradient sedimentation (Levy et al., 2006). To test the integrity of the dynactin complex in Dctn1 +/m mice, we performed sedimentation analysis on a 5–20% sucrose gradient using brain (Fig. 4 A,B) and spinal cord (data not shown) extracts from wild-type and Dctn1 +/m mice. The distribution of the dynactin complex, including p150glued, p135, and p50, covered a relatively broader region from fraction 4–7 compared with that of dynein complex, but all proteins in the complex peaked near the 19S fraction (fraction 6) (Fig. 4 A,B). We found that the migration patterns of p150glued, p135, p50, and dynein intermediate chain from Dctn1 +/m brains were very similar to those from wild-type controls (Fig. 4 A,B), indicating that the G59S mutation in p150glued causes no obvious alteration in the composition of the dynactin and dynein complexes in Dctn1 +/m mice.
Figure 4.

Dynein/dynactin complex remains intact in Dctn1 +/m mice. Density gradient centrifugation showed that p150glued and its alternative splicing variant, p135, predominantly migrated at ∼19S in a 5–20% sucrose gradient similar to dynein and p50 in wild-type (+/+) (A) and Dctn1 +/m (+/m) (B) samples prepared from brains. The expression of β-tubulin was used here as a loading control.
Accumulation of neurofilament and synaptophysin at neuromuscular junctions of Dctn1 +/m mice
Retrograde transport of neurofilaments (NF) along axons has been observed in vivo and in vitro cell culture models (Glass and Griffin, 1991; Watson et al., 1993). The dynein/dynactin complex associates with NF and is responsible for retrograde transport of NF (Shah et al., 2000). Knock-down of dynein with siRNA in vitro significantly diminishes the occurrence of retrograde NF movement without affecting anterograde NF movement, resulting in an accumulation of NF at the axon terminals (He et al., 2005). Because of the significant reduction of p150glued in Dctn1 +/m and Dctn1 +/Δ mice, we examined whether the retrograde transport of NF was affected in motor neurons of these mice. The endplates of neuromuscular junctions (NMJ) of gastrocnemius muscle, in which axons of spinal motor neurons terminate, were visualized by α-bungarotoxin (BTX) staining that specifically labels acetylcholine receptors (AChRs) at the postsynaptic sites. SMI32, a mouse monoclonal antibody reacting with a nonphosphorylated epitope in neurofilament H (NF-H), was used to detect the expression of NF at axon terminals. SMI32 immunoreactivity was restricted to thick axons that terminated at entry points of NMJ of wild-type mice at 10 months of age (Fig. 5 A, top), whereas a significant accumulation of SMI32 immunoreactivity was observed at the NMJ of littermate Dctn1 +/m mice that overlapped with BTX staining (Fig. 5 A, middle). The abnormal accumulation of NF at NMJ of Dctn1 +/m mice indicates that the G59S mutation in p150glued further compromises the normal function of dynactin, because the loss of one allele of Dctn1 gene in 13-month-old Dctn1 +/Δ mice did not cause a similar accumulation of NF (Fig. 5 A, bottom).
Figure 5.

Accumulation of neurofilament and synaptophysin at the NMJ of Dctn1 +/m mice. A, Motor neurons axons revealed by SMI32 staining terminated at endplates visualized by BTX staining. Increased accumulation of NF was observed at NMJ of gastrocnemius muscle sections from Dctn1 +/m (+/m 10m) mice at 10 months of age compared with wild-type (+/+ 10 m) littermate controls and Dctn1 +/Δ (+/Δ 13 m) mice at 13 months of age. B, Similar to NF, elevated accumulation of synaptophysin (Syn) was observed at NMJ of gastrocnemius muscle sections from Dctn1 +/m (+/m 10 m) mice at 10 months of age compared with wild-type (+/+ 10 m) littermate controls and Dcrn1 +/Δ (+/Δ 13 m) mice at 13 months of age. Muscle section thickness, 10 μm. Scale bars, 20 μm.
In addition to NF, synaptophysin, an integral membrane protein associated with small synaptic vesicles is also retrogradely transported by the dynein and dynactin complex (Li et al., 2000). Similar to NF, a significant accumulation of synaptophysin immunoreactivity was observed at the NMJ of Dctn1 +/m mice compared with wild-type littermate controls (Fig. 5 B, top and middle) and 13-month-old Dctn1 +/Δ mice (Fig. 5 B, bottom).
Motor neuron degeneration of Dctn1 +/m mice
To investigate whether the G59S mutation in p150glued leads to motor neuron degeneration, we counted the numbers of motor neurons in lumbar spinal cords from 5 pairs of 4 and 16-month-old Dctn1 +/m mice and their wild-type littermates (Fig. 6 A–E). The criteria for a motor neuron included a round, open, pale nucleus (not condensed and darkly stained), globular Nissl staining of the cytoplasm, and a diameter of ∼30–45 μm (Fig. 6 C,D). More than 25 coronal sections evenly sampled from L1 to L5 of the lumbar spinal cord were counted for each animal. There was no significant difference in numbers of spinal motor neurons between 4-month-old Dctn1 +/m mice and their wild-type littermates (+/+: 15.18 ± 0.31, n = 147 vs +/m: 15.87 ± 0.26, n = 150, p = 0.09). However, as shown in Figure 6 E, the number of motor neurons per section in Dctn1 +/m mice was reduced significantly (12.98 ± 0.22, n = 141, p < 0.001) compared with wild-type littermate controls (15.10 ± 0.57, n = 149). The number of motor neurons in age-matched Dctn1 +/Δ mice (n = 3) was also slightly decreased but not statistically significant compared with wild-type controls (14.26 ± 0.68, n = 90, p = 0.7).
Figure 6.
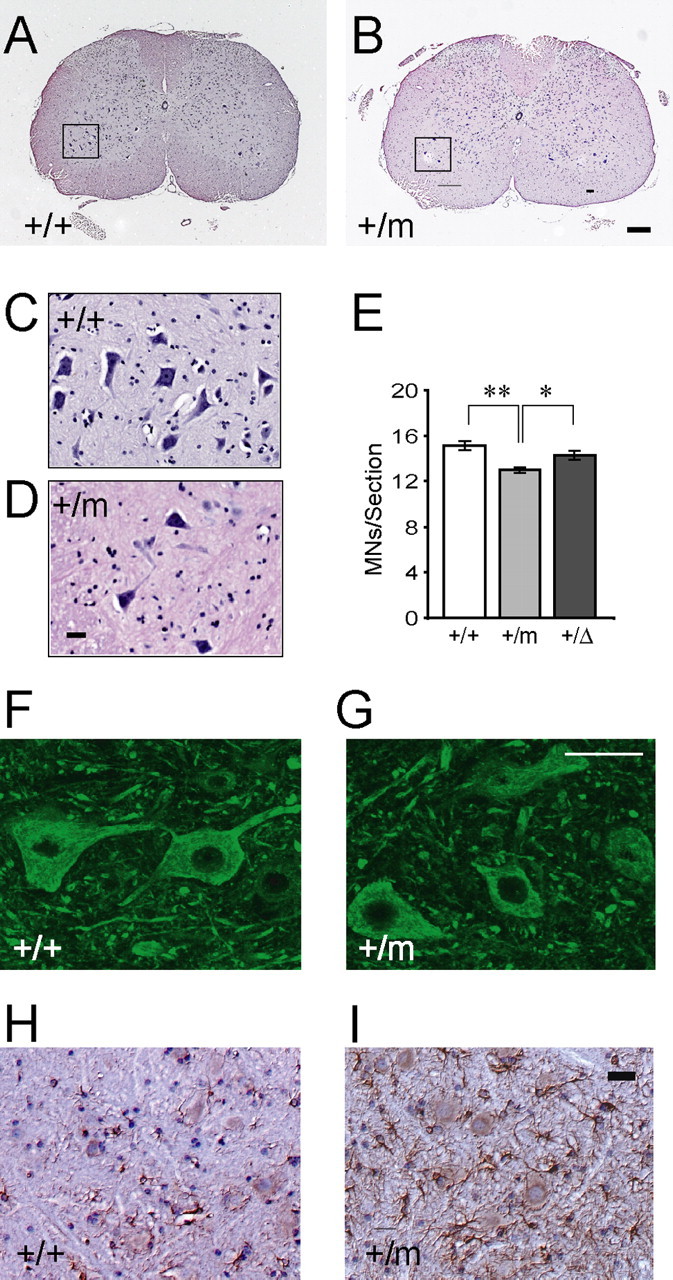
Motor neuron degeneration in Dctn1 +/m mice (A–D) HE staining of coronal sections of lumbar spinal cords revealed motor neurons in wild-type (A, C) and Dctn1 +/m (B, D) mice at 16 months of age. The insets (C, D) showed motor neurons under higher magnification of the boxed area (A, B). Scale bars: main, 100 μm; insets, 50 μm. E, Box graph of numbers of motor neurons per lumbar spinal cord section of wild-type (+/+), Dctn1 +/m (+/m), and Dctn1 +/Δ (+/Δ) mice. *p < 0.01 and **p < 0.001, respectively. F, G, Representative images of p150glued staining with the antibody against the C-terminal of p150glued in the wild-type (+/+) and Dctn1 +/m (+/m) lumbar spinal cords. Scale bar, 50 μm. H, I, Representative images of GFAP staining in the wild-type (+/+) and Dctn1 +/m (+/m) lumbar spinal cords. All the sections were counterstained with HE. Scale bar, 20 μm.
To examine whether mutant p150glued forms intracellular aggregation, we stained the spinal motor neurons of Dctn1 +/m mice with an antibody against the C-terminal of p150glued (Fig. 6 G). The staining pattern of p150glued in spinal motor neurons was very similar between wild-type (Fig. 6 F) and p150glued G59S mutant mice. No obvious aggregation of p150glued was detected within the soma of spinal motor neurons in p150glued G59S mutant mice (Fig. 6 G). To identify any other neuropathological abnormalities associated with motor neuron degeneration, we examined spinal cord sections of 16-month old Dctn1 +/m mice with a series of neuropathological markers. More glial fibrillary acidic protein (GFAP)-positive cells were observed in the spinal cord of Dctn1 +/m mice compared with wild-type controls (Fig. 6 H,I). But, no significant alteration in ubiquitin or phosphorylated neurofilament staining was found in Dctn1 +/m motor neurons (data not shown).
Motor behavior defects of Dctn1 +/m mice
To evaluate whether the observed motor neuron degeneration leads to movement disorders in Dctn1 +/m mice, a cohort of 21 male mice (9 for wild-type and 12 for Dctn1 +/m) was examined with a battery of motor behavioral tests. Wild-type and Dctn1 +/m mice showed no significant differences in performance in either the accelerating rotarod or grip strength at 10 and 16 months of age (data not shown). Because older patients carrying the G59S mutation in p150glued had steppage gait (Puls et al., 2005), and SOD1 G93A mice, a well established mouse model for ALS, developed significantly shorter stride length (Gurney et al., 1994; Puttaparthi et al., 2002; Puls et al., 2005), we examined the gait of Dctn1 +/m mice using the Treadscan Gait Analysis system. We quantified the stride length of wild-type and Dctn1 +/m mice at 4 and 16 months of age. The stride length of Dctn1 +/m mice at 4 months of age was comparable between wild-type and mutant mice (+/+: 58.39 ± 0.71 mm vs +/m: 57.33 ± 1.01 mm, n = 5 each, p = 0.39), but it was significantly shorter at 16 months of age (47.98 ± 0.85 mm, n = 12, p = 0.03) compared with that of wild-type controls (50.75 ± 0.86 mm, n = 9).
The G59S Mutation in Dctn1 does not affect the progression of motor neuron degeneration in SOD1G93A transgenic mice
Similar to the G59S mutation in p150glued, two missense mutations in cytoplasmic dynein heavy chain also result in progressive motor neuron degeneration in heterozygous mutant mice (Hafezparast et al., 2003). Interestingly, these dynein mutations prolong the survival of SOD1G93A transgenic mice (Kieran et al., 2005; Teuchert et al., 2006), raising the question as to whether the G59S mutation in p150glued causes a similar effect. To address this question, we cross-bred SOD1G93A mice, the same strain of mice used in Kieran's and Teuchert's studies (Kieran et al., 2005; Teuchert et al., 2006), with Dctn1 +/m mice and examined the onset of paralysis of SOD1G93A and Dctn1 +/m double transgenic mice. We found that the onset of paralysis was comparable between SOD1G93A single transgenic mice with SOD1 G93A and Dctn1 +/m double mutant mice (log rank test, p = 0.49) (Fig. 7), suggesting that deficiency in Dctn1 gene does not significantly affect the pathogenesis of SOD1G93A transgenic mice.
Figure 7.

The G59S mutation in p150glued does not affect the survival of SOD1G93A transgenic mice. Kaplan–Meier plot of cumulative probability of survival of SOD1G93A/Dctn1 +/+ (n = 10) and SOD1G93A/Dctn1 +/m mice (n = 15).
Discussion
The growth and maintenance of the axon, as well as the movement of cargo between the cell body and the distal tip of the axon, rely on the mechanism of axonal transport (Guzik and Goldstein, 2004). The dynein/dynactin complex plays an essential role in retrograde axonal transport (Allan, 1996; Karki and Holzbaur, 1999; Schroer, 2004). Mutations in cytoplasmic dynein heavy chain and over-expression of dynactin p50 subunit have been shown to affect axonal transport and induce progressive motor neuron degeneration (LaMonte et al., 2002; Hafezparast et al., 2003). The present study demonstrates for the first time that a mouse model carrying a MND-linked G59S substitution in the dynactin p150glued subunit develops many symptoms related to ALS and MND, such as motor neuron degeneration, reactive astrogliosis, and abnormal gait. Because Dctn1 +/m mice contain one copy of wild-type allele and one copy of G59S mutant allele, it faithfully replicates the genetic mutation in humans and may serve as a useful animal model for studying the pathogenic mechanism of MND and testing potential therapeutics.
We provided evidence to further demonstrate that p150glued is required for the cellular functions of cytoplasmic dynein (King and Schroer, 2000). Dctn1 m/m and Dctn1 Δ/Δ mice died before 8.5 dpc, similar to cytoplasmic dynein heavy chain knock-out mice (Harada et al., 1998). The G59S mutation in p150glued does not affect the integrity of the dynein/dynactin complex, except for inducing self-aggregation when over-expressed in cell lines (Levy et al., 2006). In sucrose density gradient centrifugation assays, we did not observe any abnormal distribution of dynein, p50, and p150glued extracted from brain or spinal cord of Dctn1 +/m mice, nor could we detect any aggregated forms of p150glued from Dctn1 +/m mouse brains. In fact, we observed an ∼50% reduction of p150glued in Dctn1 +/m mouse brains and failed to detect any p150glued from Dctn1 m/m mouse embryos, suggesting that the G59S mutation in p150glued leads to a rapid degradation of mutant p150glued. Protein structure analysis indicates that the Gly59 and the adjacent Phe88 residues play an important role in the folding of CAP-Gly domain (Li et al., 2002). The substitution of Gly59 with the polar Ser59 residue may hinder its interaction with the side chain of Phe88 and lead to protein misfolding and degradation (Puls et al., 2003). It is not unusual that missense mutations and short in-frame deletions or insertions cause protein misfolding and eventually degradation in genetic diseases (Bross et al., 1999). Misfolded proteins may aggregate if the cellular degradation pathways are compromised, which may explain why p150glued G59S mutant protein aggregates when over-expressed in heterologous cell lines (Levy et al., 2006). We could not detect any p150glued aggregates in the brain or spinal cord of Dctn1 +/m mice, indicating that the mutant protein is primarily targeted to the degradation pathway in vivo. However, we cannot completely rule out the presence of the p150glued G59S mutant protein or its aggregates inside cells.
It is still unclear how the G59S mutation in p150glued affects the activity of dynein. A recent study suggests that the CAP-Gly domain of p150glued enables stable binding of dynein at the plus end of microtubules; whereas the basic domain of p150glued facilitates dynein processivity along a microtubule (Culver-Hanlon et al., 2006). The G59S mutation seems to weaken the binding of p150glued with microtubules (Levy et al., 2006), which may alter the movement of dynein along a microtubule. The speed of retrograde transport in Dctn1 +/m motor neurons remains to be determined. The increased accumulation of NF and synaptophysin at the tips of motor neuron axons of Dctn1 +/m mice may result from either a reduction of the speed in retrograde transport or a partial loss of the capacity of dynein/dynactin-mediated retrograde transport, as shown previously in dynein knock-down neurons (He et al., 2005).
The most distinct clinical phenotype of patients carrying the G59S mutation in p150glued is early bilateral vocal fold paralysis that affects the abductor and abductor laryngeal muscles (Puls et al., 2005). Muscle weakness and atrophy in the face, hands, and distal legs were observed later, reflecting motor neuron degeneration in the ventral horn of the spinal cord and hypoglossal nucleus of the medulla. P50 and dynein aggregates were also observed in a subset of motor neurons from an autopsy study on one patient. Consistent with these clinical observations, we detected a significant loss of motor neurons and increased astrogliosis at the lumbar spinal cord of Dctn1 +/m mice. Because age-matched Dctn1 +/Δ mice did not develop any of these neuropathological abnormalities as observed in Dctn1 +/m mice, our data indicate that a 50% reduction of p150glued alone is not sufficient to cause motor neuron degeneration at the age having been examined. Neither does the increase of p135 account for the motor behavioral and neuropathological deficits observed in Dctn1 +/m mice, because a comparable increase of p135 was also found in Dctn1 +/Δ mice. The G59S mutation may exert additional stress to motor neurons through further disruption of the function of dynein/dynactin complex. Alternatively, these results may be also consistent with a toxic-gain-of-function mechanism because the NMJ phenotypes and motor neuron loss were worse in the heterozygous knock-in mice than in the knock-out animals. However, because very little or no p150glued G59S protein was detected in the mutant mouse it is hard to argue the mutant protein may cause any significant effect in cells. Nonetheless, to clearly address this issue, p150glued G59S transgenic mice (developed by Dr. Philip Wong at the Johns Hopkins University) will breed into p150glued heterozygous knock-out background. If the G59S mutation is a dominant-negative mutation, an acceleration of disease progression will be expected from these mice because of a loss of 50% endogenous wild-type protein.
Axonal transport deficits have also been reported in SOD1G93A transgenic mice (Warita et al., 1999), which could be rescued by mutations in dynein, resulting in longer survival of SOD1G93A transgenic mice (Kieran et al., 2005; Teuchert et al., 2006). The detailed molecular interaction between these two mutant proteins is unclear. There is evidence that the aggregates of mutant SOD1 interact with cytoplasmic dynein and alter its subcellular localization in motor neurons (Ligon et al., 2005), which may inhibit dynein activity and lead to motor neuron degeneration. Whether mutations in dynein affect its interaction with SOD1G93A remains unknown. Alternatively, slower retrograde axonal transport as observed in dynein mutant neurons (Kieran et al., 2005) may neutralize the impairment of fast anterograde axonal transport in motor neurons of SOD1G93A transgenic mice and prolong the survival of these mutant mice. Because the G59S mutation in p150glued, which caused a loss of at least 50% of dynein/dynactin activity, did not affect the onset of paralysis or death of SOD1G93A transgenic mice, our data indicate that partial loss of dynein/dynactin-mediated retrograde axonal transport is not sufficient to delay the motor neuron degeneration of SOD1G93A mice. The mechanism by which the p150glued G59S mutation exerts a pathogenic effect is likely different from that of dynein mutations. Alternatively, one can argue that the effects of heterozygous p150glued G59S mutation might be too subtle to affect the severe and rapid disease progression in SOD1G93A mice.
In summary, our study indicates that the G59S mutation in p150glued may play a dominant negative role in the normal function of dynactin, which leads to motor neuron degeneration in the heterozygous mutant mice. The 150glued G59S knock-in mice may serve as a useful tool for studying the pathogenic mechanisms of ALS and MND.
Footnotes
This work was supported by the intramural research program of National Institute on Aging/National Institutes of Health. We thank the Transgenic Mouse Core Facility of the Johns Hopkins University for blastocyte injection, Drs. Philip Wong and Fiona Laird (Johns Hopkins University School of Medicine, Baltimore, MD) for sharing their unpublished data, and the National Institutes of Health Fellows Editorial Board for editing this manuscript.
The authors declare no competing financial interests.
References
- Allan V. Motor proteins: a dynamic duo. Curr Biol. 1996;6:630–633. doi: 10.1016/s0960-9822(09)00434-5. [DOI] [PubMed] [Google Scholar]
- Bross P, Corydon TJ, Andresen BS, Jorgensen MM, Bolund L, Gregersen N. Protein misfolding and degradation in genetic diseases. Hum Mutat. 1999;14:186–198. doi: 10.1002/(SICI)1098-1004(1999)14:3<186::AID-HUMU2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Culver-Hanlon TL, Lex SA, Stephens AD, Quintyne NJ, King SJ. A microtubule-binding domain in dynactin increases dynein processivity by skating along microtubules. Nat Cell Biol. 2006;8:264–270. doi: 10.1038/ncb1370. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Glass JD, Griffin JW. Neurofilament redistribution in transected nerves: evidence for bidirectional transport of neurofilaments. J Neurosci. 1991;11:3146–3154. doi: 10.1523/JNEUROSCI.11-10-03146.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Guzik BW, Goldstein LS. Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr Opin Cell Biol. 2004;16:443–450. doi: 10.1016/j.ceb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141:51–59. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte PJ, Kankel DR. Genetic analysis of mutations at the Glued locus and interacting loci in Drosophila melanogaster . Genetics. 1982;101:477–501. doi: 10.1093/genetics/101.3-4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbaur EL. Motor neurons rely on motor proteins. Trends Cell Biol. 2004;14:233–240. doi: 10.1016/j.tcb.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL, Vallee RB. DYNEINS: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol. 1999;11:45–53. doi: 10.1016/s0955-0674(99)80006-4. [DOI] [PubMed] [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur EL. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Pfister KK, Brady ST, Dahlstrom A. Cytoplasmic dynein conversion at a crush injury in rat peripheral axons. J Neurosci Res. 2000;61:151–161. doi: 10.1002/1097-4547(20000715)61:2<151::AID-JNR6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Li S, Finley J, Liu ZJ, Qiu SH, Chen H, Luan CH, Carson M, Tsao J, Johnson D, Lin G, Zhao J, Thomas W, Nagy LA, Sha B, DeLucas LJ, Wang BC, Luo M. Crystal structure of the cytoskeleton-associated protein glycine-rich (CAP-Gly) domain. J Biol Chem. 2002;277:48596–48601. doi: 10.1074/jbc.M208512200. [DOI] [PubMed] [Google Scholar]
- Ligon LA, LaMonte BH, Wallace KE, Weber N, Kalb RG, Holzbaur EL. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. NeuroReport. 2005;16:533–536. doi: 10.1097/00001756-200504250-00002. [DOI] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melloni RH, Jr, Tokito MK, Holzbaur EL. Expression of the p150Glued component of the dynactin complex in developing and adult rat brain. J Comp Neurol. 1995;357:15–24. doi: 10.1002/cne.903570103. [DOI] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- Munch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G, Ludolph AC. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58:777–780. doi: 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttaparthi K, Gitomer WL, Krishnan U, Son M, Rajendran B, Elliott JL. Disease progression in a transgenic model of familial amyotrophic lateral sclerosis is dependent on both neuronal and non-neuronal zinc binding proteins. J Neurosci. 2002;22:8790–8796. doi: 10.1523/JNEUROSCI.22-20-08790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S, Jin P, Trimarchi J, Caruccio P, Phillis R, Murphey RK. Mutant molecular motors disrupt neural circuits in Drosophila . J Neurobiol. 1997;33:711–723. doi: 10.1002/(sici)1097-4695(19971120)33:6<711::aid-neu1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- Shah JV, Flanagan LA, Janmey PA, Leterrier JF. Bidirectional translocation of neurofilaments along microtubules mediated in part by dynein/dynactin. Mol Biol Cell. 2000;11:3495–3508. doi: 10.1091/mbc.11.10.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuchert M, Fischer D, Schwalenstoecker B, Habisch HJ, Bockers TM, Ludolph AC. A dynein mutation attenuates motor neuron degeneration in SOD1(G93A) mice. Exp Neurol. 2006;198:271–274. doi: 10.1016/j.expneurol.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Tokito MK, Howland DS, Lee VM, Holzbaur EL. Functionally distinct isoforms of dynactin are expressed in human neurons. Mol Biol Cell. 1996;7:1167–1180. doi: 10.1091/mbc.7.8.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–131. doi: 10.1016/s0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- Watson DF, Glass JD, Griffin JW. Redistribution of cytoskeletal proteins in mammalian axons disconnected from their cell bodies. J Neurosci. 1993;13:4354–4360. doi: 10.1523/JNEUROSCI.13-10-04354.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley CM, Sher RB, Kale A, Frankel WN, Cox GA, Seburn KL. Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve. 2005;32:43–50. doi: 10.1002/mus.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]