

Abstract
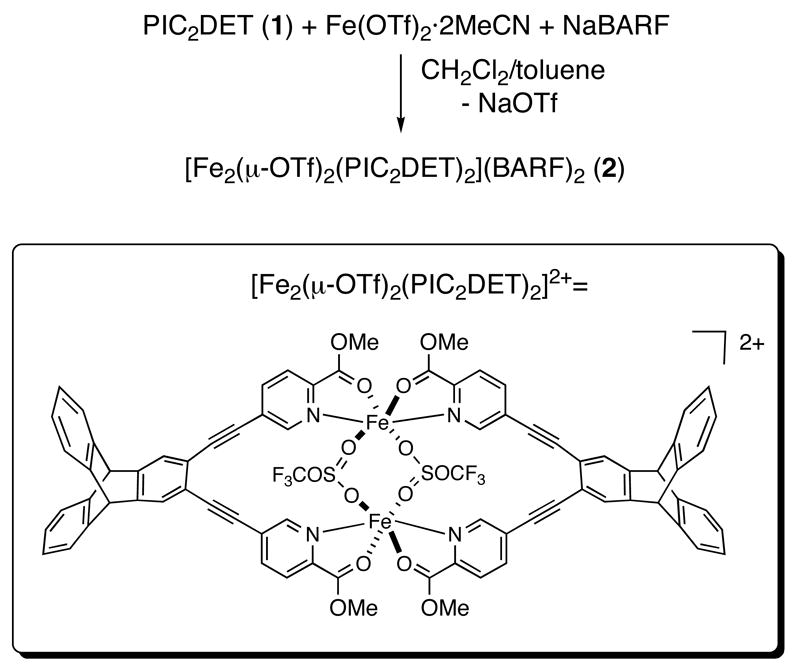
The synthesis and characterization of the diiron(II) complex [Fe2(μ-OTf)2-(PIC2DET)2](BARF)2 (2), where PIC2DET is a 2,3-diethynyltriptycene-linked dipicolinic methyl ester ligand, are described. The dication in 2, contains, [Fe2(μ-OTf)2(PIC2DET)2]2+ two symmetry-equivalent iron atoms with octahedral coordination geometries. Each metal ion has a N2O4 atom donor set that includes four atoms from two picolinic ester N,O chelate rings, as well as two oxygen atoms from the bridging trifluoromethanesulfonate groups. The Fe2(μ-OTf)2 core of 2 is stabilized by two PIC2DET ligands that bind the two metal ions in a head-to-head fashion, leading to an Fe···Fe distance of 5.173(1)Å. Molar conductivity data for 2 are consistent with Fe2(μ-OTf)2(PIC2DET)2]2+ retaining its identity in acetone solutions, where it behaves as a 2:1 electrolyte. 1H NMR spectroscopic, solution (d6-acetone) and solid-state magnetic susceptibility data all indicate that the iron atoms of 2 are high-spin (S = 2). A fit of the magnetic data (2 – 300K) to a spin-only isotropic exchange Hamiltonian H = −2JS1·S2 are consistent with weak antiferromagnetic coupling between the two iron atoms with J ~ −0.99(2) cm−1 and g = 2.10(1).
Keywords: diiron complexes, dinucleating ligands, picolinic esters
1. Introduction
The design of organic molecules that are capable of forming metal coordination complexes with controlled nuclearity is an active area of research. Ligands that are based on the bis(2-pyridyl)diethynylbenzene scaffold (Figure 1), where two 2-pyridyl groups are linked through a 1,2-diethynylarene spacer, furnish a variety of metal complexes. The coordination modes of these ligands are well understood, and mononuclear1,2 and dinuclear3–5 metal complexes derived from these molecules have been synthesized and structurally characterized. In addition to applications in supramolecular chemistry, these ligands have found use in catalysis6 and are cytotoxic toward a number of human cancer cell lines, which may be attributed to their ability to bind metal ions in a controlled fashion.7
Figure 1.
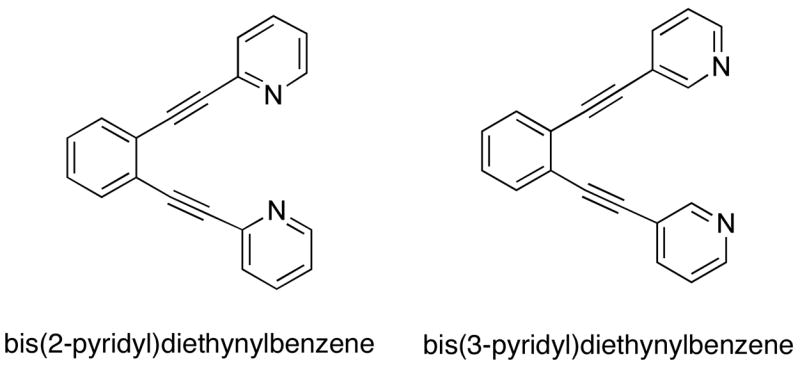
Bis(pyridyl)diethynylbenzene ligands
By contrast, much less is known about the coordination modes of ligands based upon the bis(3-pyridyl)diethynylbenzene scaffold (Figure 1). To date, only a few examples of metal complexes derived from these ligands have been characterized. One report described how mononuclear copper complexes could be constructed from the 3-pyridyl-derived ligands, even though their larger N···N distance should make coordination of both nitrogen atoms to a single metal ion more difficult than for 2-pyridyl-based ligands.8 To adopt such a chelating coordination mode, substantial bending of the pendant alkyne arms was postulated to occur, which would shorten the N···N distance and bring the two heterocycles closer to one another. Subsequently, three dinuclear complexes with similar coordination geometries were crystallographically characterized. These include two examples derived from the N,O-chelating diquinolinic ester ligand Et2BCQEBEt (Figure 2), the diiron(II) complex [Fe2(μ-O2CArTol)3-(Et2BCQEBEt)](OTf), which has a Fe···Fe distance of 3.58 Å, and the dicopper(I) complex [Cu2(μ-I)2(Et2BCQEBEt)] with a Cu···Cu distance of 2.58 Å.9 The most recent example is a dinuclear iron-sodium complex [FeNa(μ-O2CTrp)3(PIC2DET)] derived from the dipicolinic ester ligand PIC2DET (Figure 2), with an Fe···Na distance of 3.18 Å.10 In all three cases the heteroaryl rings of the bis(3-pyridyl)diethynylbenzene ligand adopt nearly coplanar conformations to position two metal ions a relatively short distance (~ 3 Å) apart from one another.
Figure 2.
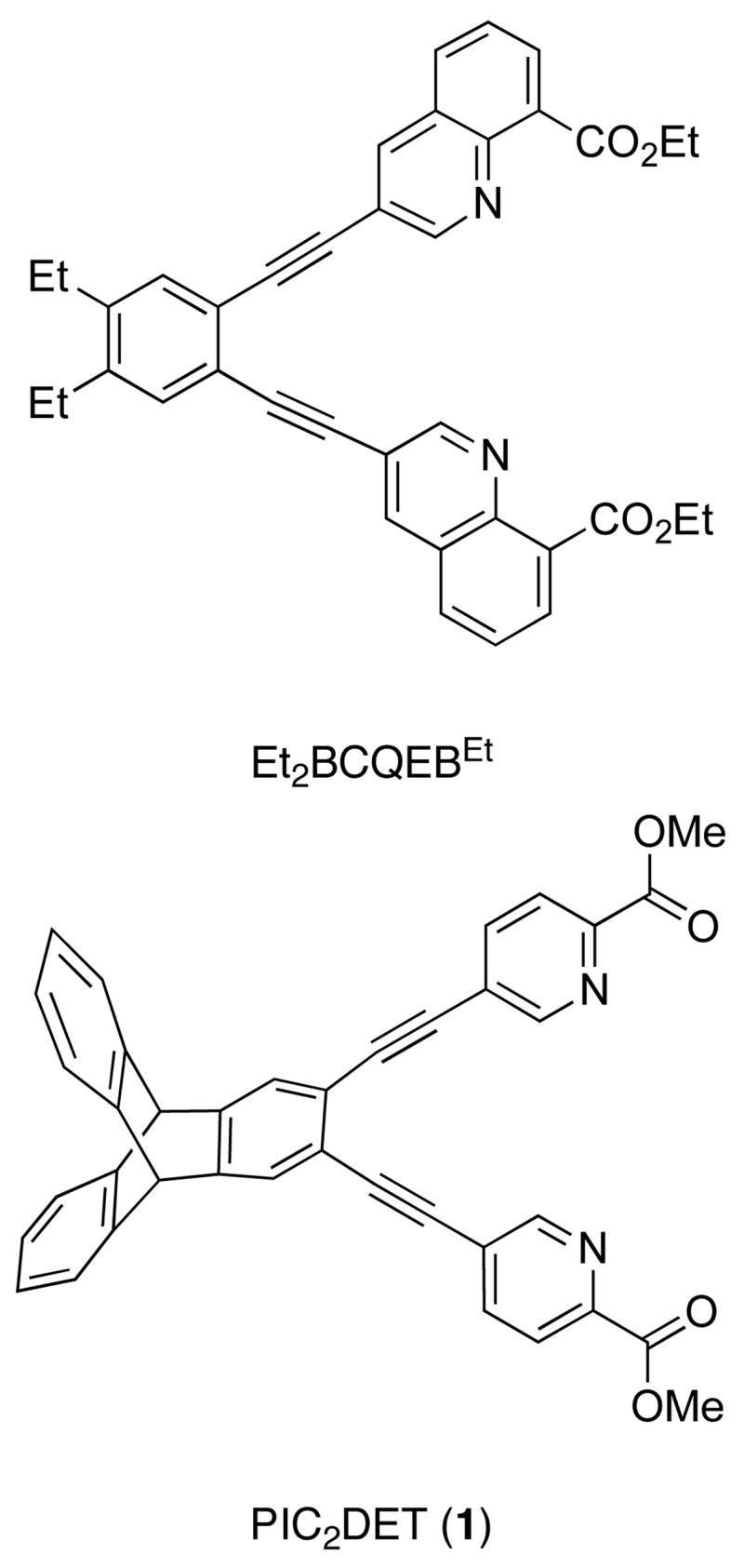
Ligands based on the bis(3-pyridyl)diethynylbenzene scaffold
In the present report, we describe the synthesis and characterization of the diiron(II) complex [Fe2(μ-OTf)2(PIC2DET)2](BARF)2 (2) which reveals a new coordination mode for ligands derived from the bis(3-pyridyl)diethynylbenzene scaffold. We describe how the ligand PIC2DET can support a dinuclear structure with a substantially larger metal-metal distance, simply by rotating its two pyridine rings away from the plane of the arene linker and by binding two ligands to two metal ions in a head-to-head fashion. In addition to the structure of 2, we also report molar conductivity data, 1H NMR spectra, and solution and solid-phase magnetic susceptibility data for the compound, which is a unique example of a diiron(II) complex.
2. Experimental
General Considerations
All reagents were obtained from commercial sources and used as received unless otherwise noted. Dichloromethane (CH2Cl2), toluene, and pentane were saturated with nitrogen and purified by passage though activated alumina columns under argon.11 The synthesis and isolation of 2 was performed in an MBraun drybox under a nitrogen atmosphere. Synthesis of the dipicolinic ester ligand PIC2DET (1) was carried out as described elsewhere.12
[Fe2(μ-OTf)2(PIC2DET)2](BARF)2 (2)
A mixture of PIC2DET (1) (50 mg, 87 μmol), NaBARF (78 mg, 87 μmol), and CH2Cl2 (5 mL) was treated with Fe(OTf)2·2MeCN (38 mg, 87 μmol), resulting in an orange heterogeneous mixture. The reaction was maintained for 1 h with stirring, treated with toluene (2 mL), and cooled to −25 °C. After 1 h, the reaction was filtered to remove a colorless solid. Vapor diffusion of pentane into the orange filtrate gave air-stable, orange-yellow dichroic blocks of the complex 2·4CH2Cl2·C7H8 (115 mg, 31.0 μmol, 71%) that were isolated after 2 days by filtration. X-ray crystallographic data for the orange-yellow blocks are provided below (Tables 1 and 2). 1H NMR (500 MHz, d6-acetone) δ 60.33, 12.96, 8.63, 7.81, 7.77, 7.69, 7.24, 6.21, 3.03. IR (KBr, cm−1) 3429 (w), 3068 (w), 2965 (w), 2221 (m), 1670 (s), 1610 (m), 1589 (m), 1567 (m), 1462 (s), 1448 (s), 1323 (s), 1357 (s), 1323 (s), 1279 (s), 1224 (s), 1128 (s), 1040 (m), 1022 (s), 945 (m), 888 (m), 867 (w), 839 (m), 796 (w), 745 (m), 713 (m), 696 (m), 682 (s), 670 (s), 630 (s), 621 (m), 581 (w), 518 (w), 479 (w), 449 (w). Elemental analysis was performed on a sample that had been dried in vacuo at 100 °C. Anal. Calcd. for C142H72B2F54Fe2N4O14S2: C, 51.97; H, 2.21; N, 1.71. Found: C, 51.40; H, 2.13; N, 1.97.
Table 1.
Summary of X-ray Crystallographic Data for 2·4CH2Cl2·C7H8.
| 2·4CH2Cl2·C7H8 | |
|---|---|
| Formula | C153 H88 B2Cl8 F54Fe2N4 O14S2 |
| Formula Weight | 3713.31 |
| Space Group | P21/n |
| a, Å | 11.884(2) |
| b, Å | 17.517(4) |
| c, Å | 37.374(8) |
| β, deg | 96.62(3) |
| V, Å3 | 7729.0(3) |
| Z | 2 |
| ρcalc, g/cm3 | 1.596 |
| T, °C | 173(2) K |
| μ(Mo Kα), mm−1 | 0.482 |
| θ limits, deg | 1.75 – 27.00 |
| total no. of data | 65573 |
| no. of unique data | 16853 |
| no. of params | 1405 |
| GOF on F2 | 1.039 |
| R1 (%)a | 6.70 |
| wR2 (%)b | 17.52 |
| max, min peaks, e/Å3 | 0.916, −0.903 |
Table 2.
Selected Bond Lengths (Å) and Angles (deg) for 2·4CH2Cl2·C7H8.a
| Bond length | Bond angle | ||
|---|---|---|---|
| Fe1···Fe1A | 5.173(1) | O2–Fe1–N1 | 76.08(9) |
| Fe1–N1 | 2.155(2) | O2–Fe1–N2 | 92.23(9) |
| Fe1–N2 | 2.157(2) | O2–Fe1–O7 | 164.56(8) |
| Fe1–O2 | 2.167(2) | N2–Fe1–O4 | 76.20(8) |
| Fe1–O4 | 2.154(2) | N1–Fe1–N2 | 159.45(9) |
| Fe1–O5 | 2.096(2) | O5–Fe1–O7 | 89.24(8) |
| Fe1–O7 | 2.131(2) | S1–O5–Fe1 | 133.7(1) |
| O5–S1 | 1.454(2) | O7A–S1–O5 | 112.4(1) |
| O7–S1A | 1.451(2) | ||
Numbers in parentheses are estimated standard deviations of the last significant figure. Atoms are labeled as indicated in Figures 3 and 4.
X-ray Crystallographic Studies
Intensity data were collected on a Bruker (formerly Siemens) SMART APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (γ = 0.71073 Å), controlled by a Pentium-based PC running the SMART software package. Single crystals were mounted on the tips of glass fibers, coated with paratone-N oil, and cooled to 173 K under a stream of N2 maintained by a KRYO-FLEX low-temperature apparatus. Data collection and reduction protocols were described previously.13 The structure was solved by direct methods and refined on F2 by using the SHELXTL-97 software incorporated in the SHELXTL software package.12 Empirical absorption corrections were applied by using the SADABS program,14 and the structures were checked for higher symmetry with the PLATON software.15 All non-hydrogen atoms were located and their positions refined with anisotropic thermal parameters by least-squares cycles and Fourier syntheses. All hydrogen atoms were assigned to idealized positions and given thermal parameters equivalent to either 1.5 (methyl hydrogen atoms) or 1.2 (all other hydrogen atoms) times the thermal parameter of the carbon atom to which they were attached. The trifluoromethyl groups of the bridging triflate moiety and the BARF counteranions were positionally disordered due to rotation about the C–S bond. These CF3 groups adopted two or three alternative conformations. For CF3 groups with two conformations, the fluorine atoms were refined at 50% occupancy and for three conformations, 33% occupancy. One CH2Cl2 molecule (C1S) was refined at full occupancy. Another area of residual electron density was refined as containing either two non-overlapping CH2Cl2 molecules (C2S and C3S) that were refined at 50% occupancy, or a single toluene molecule refined at 50% occupancy. Hydrogen atoms were not assigned to the three solvent molecules with 50% occupancy but their contributions were included in computing the overall formula and crystal density. The molecular structure of the dication [Fe2(μ-OTf)2(PIC2DET)2]2+, and several views highlighting the coordination geometry, are depicted in Figs. 3–4.
Figure 3.
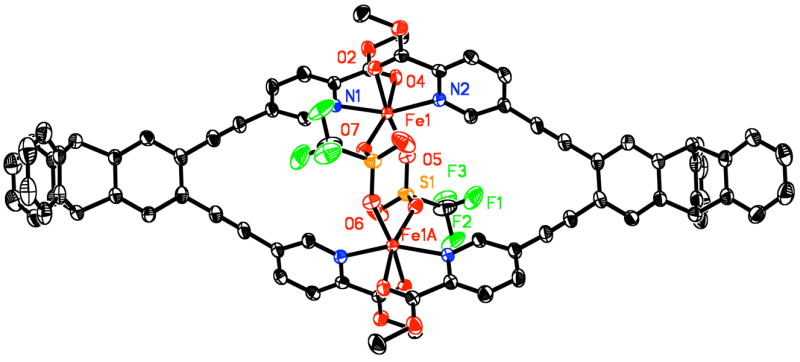
ORTEP diagram of [Fe2(μ-OTf)2(PIC2DET)2]2+ showing 50% probability thermal ellipsoids for all non-hydrogen atoms.
Figure 4.
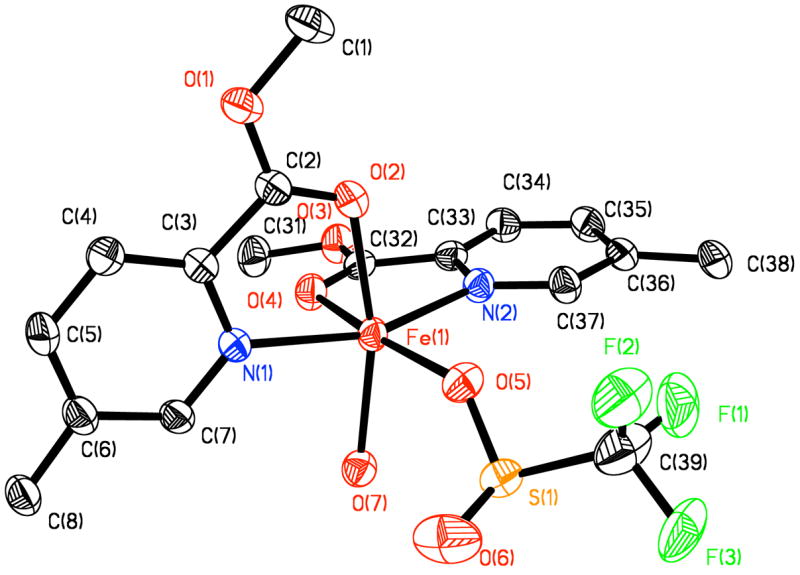
One half of the [Fe2(μ-OTf)2(PIC2DET)2]2+ core showing 50% probability thermal ellipsoids for all non-hydrogen atoms. The diethynyltriptycene groups of PIC2DET are abbreviated for clarity.
Physical Measurements
IR spectra were recorded on a Thermo Nicolet Avatar 360 spectrometer with OMNIC software. Conductivity measurements were performed with a Fisher Scientific portable conductivity meter (Model 09-326) under ambient atmosphere. 1H NMR spectra were recorded on a Varian 500 MHz spectrometer in the Massachusetts Institute of Technology Department of Chemistry Instrument Facility.
Magnetic Susceptibility Study
Magnetic susceptibility measurements were performed on a powdered sample of 2 (44.5 mg) that was dried in vacuo at 100 °C to remove lattice solvent. Data were collected between 2 and 300 K with an applied magnetic field of 1.0 T using a Quantum Design MPMS SQUID magnetometer housed in the Massachusetts Institute of Technology Center for Material Science and Engineering. The powdered sample was suspended inside a plastic straw holder for the measurements. The magnetic moment of the holder was determined to be less than 0.01 % of the total sample moment over the same range of temperatures, so its contribution to the overall susceptibility was not significant. A diamagnetic correction of −4388 × 10−6 emu mol−1 was calculated from Pascal’s constants.16 The molar susceptibility data were fit to the expression given in eq 1,17 which was derived from the spin-only isotropic exchange Hamiltonian H =−2JS1·S2, where S1 = S2 = 2 and x = J/kT.
| (1) |
3. Results
1. Synthesis
The diiron(II) complex [Fe2(μ-OTf)2(PIC2DET)2](BARF)2 (2) (Scheme 1), where BARF is tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, was initially identified by X-ray crystallography as a minor byproduct from the reaction of [Fe2(μ-O2CTrp)3(PIC2DET)](OTf)10 and NaBARF. With knowledge of its composition, a higher-yielding, rational synthetic route to the complex was devised. Reaction of PIC2DET (1) with one equiv of Fe(OTf)2·2MeCN and one equiv of NaBARF in dichloromethane led to the formation of a cloudy orange solution. Addition of toluene to the reaction mixture followed by cooling to −25 °C resulted in precipitation of a colorless solid (NaOTf) that was removed by filtration. Vapor diffusion of pentane into the orange filtrate gave orange-yellow dichroic blocks of the complex 2·4CH2Cl2·C7H8 that were isolated in 71% yield. Compound 2 appears to be stable in the presence of dioxygen and shows no color change consistent with oxidative degradation, even after being stored under ambient atmosphere for several weeks.
Scheme 1.
2. Description of the Structure
X-ray analysis of the orange-yellow dichroic blocks revealed that the diiron(II) complex 2 crystallizes in the monoclinic space group P21/n, with a = 11.884(2) Å, b = 17.517(4) Å, c = 37.374(8) Å, β = 96.62(3)°, V = 7729.0(3) Å3, and Z = 2 (Table 1). The two halves of [Fe2(μ-OTf)2(PIC2DET)2]2+ are related by an inversion center and the Fe···Fe distance in the dication is 5.173(1) Å (Figure 3). The asymmetric unit contains one half of the [Fe2(μ-OTf)2(PIC2DET)2]2+ dication and one BARF counteranion (Figure 4). Each iron atom has a distorted octahedral coordination environment with an N2O4 donor atom set consisting of two cis oxygen atoms and two trans nitrogen atoms of the N,O-chelating picolinic ester groups, as well as two cis-positioned oxygen atoms from the bridging triflates. Deviation from ideal octahedral geometry is due primarily to the picolinic esters, which form two five-membered chelate rings with bite angles of approximately 76.2°. The Fe1–N1 and Fe1–N2 bond distances are identical within experimental error, 2.155(2) and 2.157(2) Å, respectively. Bond lengths to the carbonyl oxygen atoms are also similar, with an Fe1–O2 distance of 2.167(2) Å and Fe1–O4, 2.154(2) Å. Distances between the iron atom and the two triflate oxygen atoms are more disparate, with Fe1–O5 and Fe1–O7 lengths of 2.096(2) and 2.131(2) Å, respectively. These data are significant because 2 is one of only a handful of crystallographically characterized picolinic esters bound to a metal ion.18
3. Molar Conductivity
To gain insight into the behavior of the diiron complex 2 in solution, molar conductivity data were collected in acetone at 24 °C. The ΛM values measured vary from 194 to 373 S·cm2·mol−1 for concentrations of 2.0 to 0.15 mM (Table 2). Although the range for these ΛM values is broad, these numbers match those of other 2:1 electrolytes in acetone in the 10−3 to 10−4 M concentration range19 and are characteristic of ion pairing.20 Furthermore, the slope derived from an Onsager plot of the data is consistent with the properties a 2:1 electrolyte.21 Thus, the conductivity results suggest that the structure of the [Fe2(μ-OTf)2(PIC2DET)2]2+ dication revealed by the crystal structure is retained in solution.
4. Magnetic Studies
1H NMR spectroscopic data were acquired for 2 dissolved in d6-acetone. The spectrum contains several paramagnetically shifted signals and is illustrated in Figure 5. The resonance are well resolved and range from 60 to 3 ppm, consistent with high-spin (S = 2) iron(II) centers. Moreover, the small number of resonances in the spectrum is consistent with the 2 having a symmetrical structure in solution. A solution-phase magnetic susceptibility measurement performed in d6-acetone at 293 K by Evan’s method22 was also consistent with the iron ions of 2 remaining high-spin in solution. The analysis returned a value of 5.2 μB per iron center, only slightly higher than the spin-only value of 4.90 μB computed for an S = 2 species.23
Figure 5.
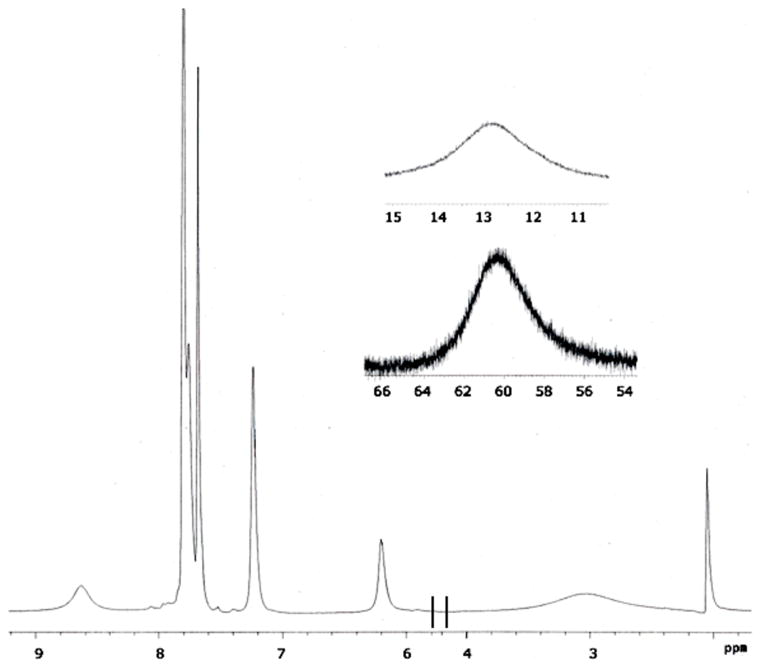
1H NMR spectrum of 2 (500 MHz) at 293 K as a d6-acetone solution with insets showing resonances at 60 and 13 ppm.
Variable-temperature magnetic susceptibility data for 2 was collected to gain insight about the compounds behavior in the solid state. Again, the data are consistent with 2 possessing two non-interacting high-spin (d6, S = 2) ferrous ions. Illustrated in Figure 6 is a plot of χM versus T and μeff versus T for a solid sample of 2. The effective magnetic moment for 2 is 7.1 μB at 296 K and remains fairly constant upon cooling to 50 K (7.3 μB), then drops dramatically to 3.6 μB upon cooling to 2 K. This high μB value at 2 K indicates that excited magnetic states are populated even at low temperatures.
Figure 6.
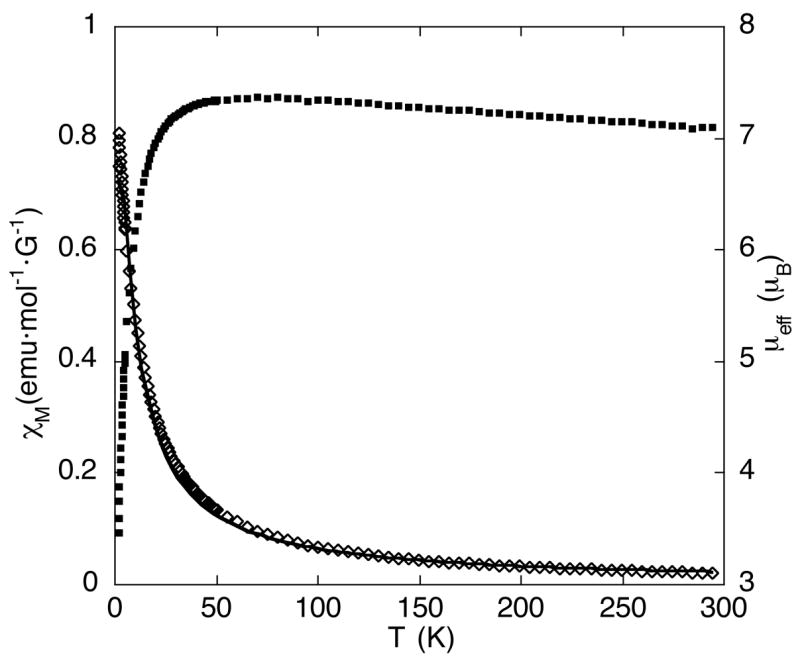
Plot of the molar susceptibility (χM) and effective magnetic moment (μeff) versus temperature for 2. The solid line corresponds to the best fit described in the text.
The solid state temperature-dependent magnetic data displayed in Figure 6 were fit to an expression derived from the spin-only isotropic exchange Hamiltonian H = −2JS1·S2, where S1 = S2 = 2. The best fit was obtained for J = −0.99(2) cm−1 and g = 2.10(1). It should be noted that this analysis does not take into account the effect of zero-field splitting, which can be of the same order of magnitude as the exchange coupling for diiron(II) complexes.24 Although the fit is therefore an oversimplification, the weak antiferromagnetic coupling interaction determined by this analysis is consistent with the absence of an efficient exchange pathway between the two iron(II) ion, which are separated by > 5Å.
4. Discussion
Unlike picolinic acids, which display a variety of coordination modes, the coordination chemistry of picolinic esters has not been extensively explored.18 The syntheses of several picolinic ester containing transition metal complexes with the general formula MCl2L2, where M = Co(II), Fe(II) or Mn(II) and L = ethyl picolinate, have been reported and magnetic susceptibility measurements for this series of compounds are consistent with a weak ligand field and high-spin electronic states.25 At the time, no crystal structures were available to confirm the stereochemistry of the complexes, which could exist in any of five distinct geometric isomers (A–E) shown in Figure 7, not counting enantiomers. Recently, the crystal structure of a high-spin mononuclear Co(II) complex CoCl2L2 was reported, where L is isopropyl picolinate.18 The stereochemistry of this complex features trans-positioned nitrogen donor atoms and cis oxygen donors (Figure 7, stereoisomer C), analogous to the of 2. The structural data provided here for compound 2 agree with previous observations that picolinic esters form high spin metal complexes and our study provides the first structural parameters for an iron(II) compound of this type. Moreover, the results may indicate a preference for placing the nitrogen donor atoms in trans positions for such octahedral dipicolinic ester complexes, and the ability of ligands like 1 to form complexes with this stereochemistry by dimerizing in head-to-head fashion may stabilize the dinuclear forms of complexes of ligand 1.
Figure 7.
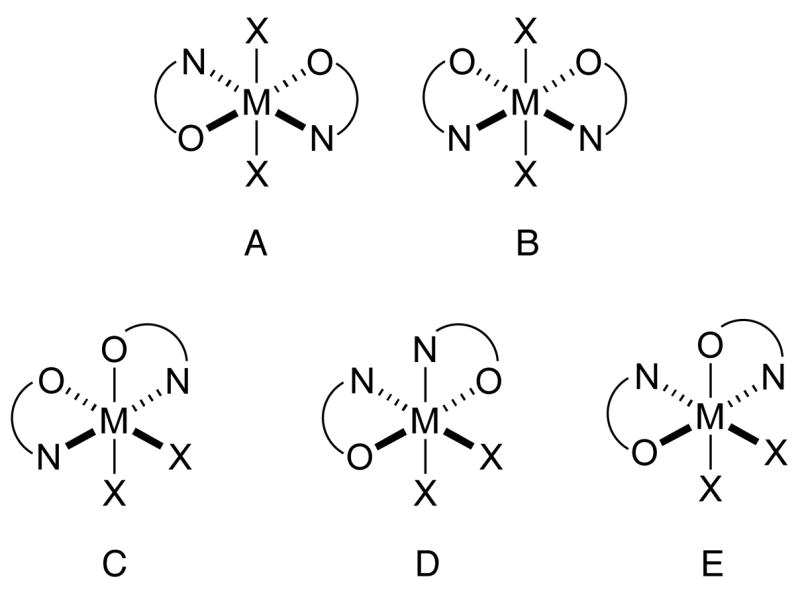
Possible geometric isomers for a metal complex of the general formula MX2L2, where L is a N-O chelating ligand.
Compound 2 is also the first example of a diiron(II) complex that is bridged by two triflate groups, and the magnetic exchange coupling between the two metal ions is correspondingly weak. Fitting the temperature-dependent magnetic data for 2 reveaked a weak magnetic exchange interaction between the two iron(II) ions. The magnitude of the coupling constant (~ −1 cm−1) is similar to that of several other antiferromagnetically coupled diiron(II) complexes that contain bridging groups and are separated by a distance of > 4 Å, such as the diiron(II) tetracarboxylate complex [Fe2(μ-O2CArTol)2(O2CArTol)2-(pyridine)2], where O2CArTol is 2,6-di(p-tolyl)benzoate, which is bridged by two carboxylates at an Fe···Fe distance of 4.22 Å and has a similar antiferromagnetic exchange coupling constant of −0.90 cm−1.26
Several results are consistent with the dication of 2 retaining its structure in d6-acetone. First, the 1H NMR spectrum in that solvent contains only nine resonances, indicating that in solution 2 adopts a symmetrical structure in which the two sides of the PIC2DET ligand are magnetically equivalent, as would be expected for the dication of 2. Secondly, molar conductivity data indicate that 2 is a 2:1 electrolyte in solution. Finally, the solid-state susceptibility measurement agrees approximately with the value measured in d6-acetone solution, which lends further support to the hypothesis that 2 retains its structure in solution.
Implications for the Bis(3-pyridyl)diethynylbenzene Scaffold
The structure of 2 illustrates a new type of dinucleating binding mode for ligands derived from the bis(3-pyridyl)diethynylbenzene scaffold. Previous work revealed that these ligands are able to position two metal ions a short distance apart (~ 3 Å) from one another. To illustrate, space-filling representations of the dimetallic bis(3-pyridyl)diethynylbenzene cores of the three coordination complexes known to date are depicted in Figure 8 (structures a–c). In each of these complexes, the pyridyl rings of the ligand adopt a conformation that places them nearly coplanar with the benzene ring of the linker. Also shown in Figure 8 is a representation of the core of 2 (structure d). By inspection of this model, it is immediately apparent that the M···M distance is larger for d than for the other three complexes. To achieve this larger distance, the pyridyl rings of 1 rotate approximately 40° out of the plane of the benzene linker. Furthermore, this ligand is able to adopt such a conformation without placing strain on the two alkyne bonds of the ligand, as evidenced by the four torsion angles about the triple bond in the alkyne arms, which are all > 175 °. In fact, the torsion angles in all four of these structures vary only slightly from linearity, from 173.6 to 178.8°. The situation is much different if one were to prepare mononuclear complexes with ligands derived from the bis(3-pyridyl-diethynylbenzene scaffold. To obtain such structures by coordinating both nitrogen atoms of the ligand to a single metal ion, substantial bending of the pendant alkyne groups would be required, to a much greater extent than shown with models a–d. Therefore, coordination modes similar to a–d are likely to be preferred in the structures.
Figure 8.
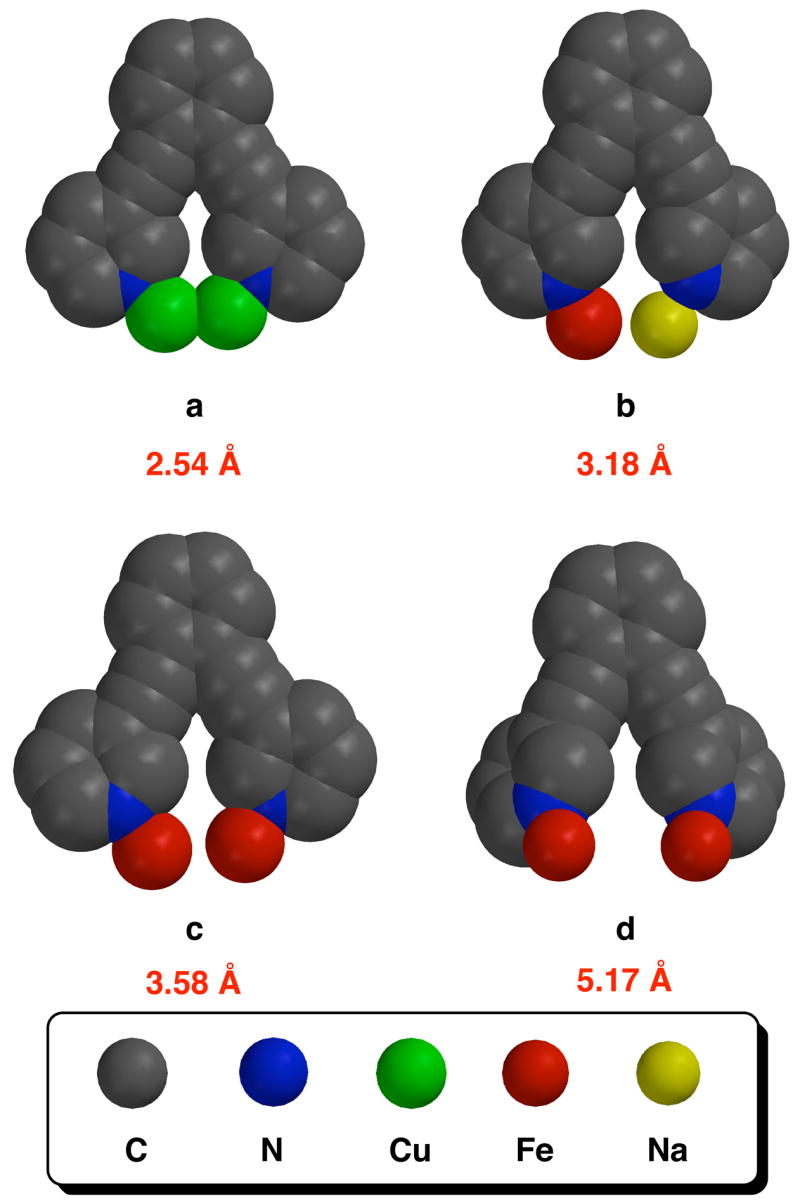
Space-filling models of the dimetallic bis(3-pyridyl)diethynylbenzene cores of four coordination complexes generated from crystallographic coordinates: (a) [Cu2(μ-I)2(Et2BCQEBEt)];9 (b) [FeNa(μ-O2CTrp)3(PIC2DET)];10 (c) [Fe2(μ-O2CArTol)3-(Et2BCQEBEt)](OTf);9 (d) [Fe2(μ-OTf)2(PIC2DET)2](BARF)2 (2). Approximate metal-metal distances are listed below each model.
Ligands derived from the bis(3-pyridyl)diethynylbenzene scaffold were devised to construct structural models of dimetallic enzyme active sites having short metal-metal distances (~ 3 Å). These ligands are especially useful in mimicking enzyme cores with two syn N-donor ligating groups, such as the active sites of the hydroxylase components of bacterial multicomponent monooxygenases,27 which contain two syn histidine donors, because the N-donors are held rigidly in a syn orientation by the diethynylbenzene backbone. This topography is realized in structures a–c, which also have the desired 1:2 ligand-to-metal stoichiometries. The complex [Fe2(μ-O2CArTol)3(Et2BCQEBEt)](OTf) is arguably the closest structural model of the active site of these hydroxylases synthesized to date. The structure of 2 is much different, since two PIC2DET ligands bind to two iron(II) atoms in a head-to-head fashion, resulting in a ligand-to-metal stoichiometry of 1:1. In forming this structure, two octahedral metal centers are produced with the stereochemistry shown in Figure 7 without placing strain on the alkyne groups. In terms of modeling the coordination mode of the enzyme active sites, this result is undesired. To circumvent such behavior in future work, pyridine groups with more than two ligating functionalities may be required block additional binding sites on the metal atoms and to prevent the head-to-head dimerization process encountered in this investigation.
Conclusions
The synthesis and structure of the diiron(II) complex [Fe2(μ-OTf)2(PIC2DET)2] (BARF)2 (2) are described. Solid and solution phase magnetic susceptibility measurements indicate that 2 contains a pair of high-spin iron(II) centers that are weakly antiferromagnetically coupled. Molar conductivity and 1H NMR spectroscopic data are consistent with the dication of 2 retaining its identity in solution. The structure reveals a new mode of coordination for bis(3-pyridyl)diethynylarene-based ligands that supports the largest M···M distance observed to date in such molecules. Future syntheses of dinuclear complexes with ligands like 1 having metal-metal distances on the order of 3 Å may require three or more ligating groups per metal atom in order to occupy additional coordination sites and thus prevent the kind of head-to-head dimerization encountered here.
Table 2.
Molar Conductivities of 2 in Acetone at 24 °C.
| [2] in acetone (mM) | ΛM(S·cm2·mol−1) |
|---|---|
| 2.0 | 194 |
| 1.5 | 219 |
| 1.0 | 240 |
| 0.5 | 278 |
| 0.25 | 328 |
| 0.15 | 373 |
Acknowledgments
This work was supported by Grant GM32134 from the National Institute of General Medicine Sciences. J.J.K. was a National Institute of Health postdoctoral fellow (F32 GM069236-01). We thank Dr. Shaoyan Chu for assistance with SQUID instrumentation and Dr. Datong Song for help with X-ray crystallography.
Footnotes
Supporting Information Available: X-ray crystallographic data for 2 including tables, ORTEP diagrams, and a CIF file. This material is available free of charge via the Internet at http://pubs.acs.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bosch E, Barnes CL. Inorg Chem. 2001;40:3097. doi: 10.1021/ic010058u. [DOI] [PubMed] [Google Scholar]
- 2.Shotwell S, Windscheif PM, Smith MD, Bunz UHF. Org Lett. 2004;6:4151. doi: 10.1021/ol048609i. [DOI] [PubMed] [Google Scholar]
- 3.Kawano T, Kuwana J, Du CX, Ueda I. Inorg Chem. 2002;41:4078. doi: 10.1021/ic025574e. [DOI] [PubMed] [Google Scholar]
- 4.Bosch E, Schultheiss N, Rath N, Bond M. Cryst Growth Des. 2003;3:263. [Google Scholar]
- 5.Kawano T, Kuwana J, Ueda I. Bull Chem Soc Jpn. 2003;76:789. [Google Scholar]
- 6.Kawano T, Kuwana J, Shinomaru T, Du CX, Ueda I. Chem Lett. 2001:1230. [Google Scholar]
- 7.Lin CF, Lo YH, Hsieh MC, Chen YH, Wang JJ, Wu MJ. Biorg Med Chem. 2005;13:3565. doi: 10.1016/j.bmc.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 8.Rawat DS, Benites PJ, Incarvito CD, Rheingold AL, Zaleski JM. Inorg Chem. 2001;40:1846. doi: 10.1021/ic010014l. [DOI] [PubMed] [Google Scholar]
- 9.Kuzelka J, Farrell JR, Lippard SJ. Inorg Chem. 2003;42:8652. doi: 10.1021/ic034928e. [DOI] [PubMed] [Google Scholar]
- 10.Kodanko JJ, Xu D, Song D, Lippard SJ. J Am Chem Soc. 2005;127:16004. doi: 10.1021/ja054943d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Orgaometallics. 1996;15:1518. [Google Scholar]
- 12.Kodanko JJ, Morys AJ, Lippard SJ. Org Lett. 2005;7:4585. doi: 10.1021/ol051700h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuzelka J, Mukhopadhyay S, Spingler B, Lippard SJ. Inorg Chem. 2004;43:1751. doi: 10.1021/ic035009r. [DOI] [PubMed] [Google Scholar]
- 14.Sheldrick GM. SADABS: Area-Detector Absorption Correction University of Gottingen. [Google Scholar]
- 15.Spek AL. PLATON. A Multipurpose Crystallographic Tool Utrecht University; [Google Scholar]
- 16.Carlin RL. Magnetochemistry. Springer-Verlag; Berlin: 1986. [Google Scholar]
- 17.Kahn O. Molecular Magnetism. VCH; New York, NY: 1993. [Google Scholar]
- 18.March R, Clegg W, Coxall RA, Cucurull-Sanchez L, Lezama L, Rojo T, Gonzalez-Duarte P. Inorg Chim Acta. 2003;353:129. [Google Scholar]
- 19.Geary WJ. Coord Chem Rev. 1971;7:81. [Google Scholar]
- 20.Hammack WS, Conti AJ, Hendrickson DN, Drickamer HG. J Am Chem Soc. 1989;111:1738. [Google Scholar]
- 21.Feltham RD, Hayter RG. J ChemSoc. 1964:4587. [Google Scholar]
- 22.Sur SK. J Magn Reson. 1989;82:169. [Google Scholar]
- 23.Drago RS, Drago RS. Physical Methods for Chemists. 2. Saunders College Pub; Ft. Worth: 1992. [Google Scholar]
- 24.Boca R. Coord Chem Rev. 2004;248:757. [Google Scholar]
- 25.Hay RW, Clark CR. Transition Met Chem. 1979;4:28. [Google Scholar]
- 26.Lee D, Lippard SJ. Inorg Chem. 2002;41:2704. doi: 10.1021/ic020186y. [DOI] [PubMed] [Google Scholar]
- 27.Sazinsky MH, Lippard SJ. Acc Chem Res. 2006;39:558. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]