

Abstract
In this letter, we report metal-enhanced e-type fluorescence. Eosin in close proximity to silver island films (SiFs) shows enhanced e-type fluorescence with an approximately two-fold higher intensity observed from SiFs, as compared to a control sample. Our findings suggest two complementary mechanisms for the enhancement: surface plasmons can radiate e-type delayed fluorescence efficiently and enhanced absorption also facilitates enhanced emission from both S1 and T1 states. This observation is helpful in our understanding not only for studying the interactions between plasmons and fluorophores but also for our laboratories continued efforts to develop a unified plasmon-lumophore description.
For many years, our laboratories have been studying the near-field interactions of fluorophores with metal nanoparticles (such as Ag, Au, Cu, Al, and alloys), a phenomena named metal-enhanced fluorescence (MEF) by Geddes in 2002.1 According to our current mechanistic interpretation of MEF, nonradiative energy transfer occurs from excited distal fluorophores to the surface plasmon electrons on noncontinuous films, in essence, an induced mirror dipole. The surface plasmons, in turn, radiate the photophysical characteristics of the coupling fluorophores. We have also reported metal-enhanced phosphorescence (MEP) at low temperature, whereby two mechanisms are thought to account for the enhanced phosphorescence signatures: (i) nonradiative energy transfer is thought to occur from excited distal triplet-state luminophores to surface plasmons in noncontinuous silver films, which, in turn, radiate fluorophore/lumophore phosphorescence emission highly efficiently with a reduced phosphorescence decay time, and (ii) an overall increase in the net system absorption also partially contributes to the enhanced singlet and triplet emission. These observations have also led us to postulate the first unified plasmon-fluorophore description.2
Nearly all previous MEF studies were exclusively focused on prompt fluorescence emission,3 where excited singlet state populations rapidly decayed to ground states. Subsequently, we have now questioned whether surface plasmons can, in fact, radiate and therefore amplify e-type (eosin-type) fluorescence, as well as promote enhanced emission through an enhanced net system absorption. e-type fluorescence is well known to arise from thermal activation of the S1 state from the T1 state (reverse-intersystem crossing), and hence its decay time (excited state lifetime) is under-pinned by the decay of the T1 state, which typically has a long decay time in the millisecond range. Because of its long decay time, e-type delayed fluorescence has been widely used to investigate the rotational diffusion time of biological macromolecules in membranes on micro-millisecond time scales.4 However, for these types of studies, the quantum yield of e-type fluorescence emission generally hinders its widespread use in membrane and protein biophysics. In this letter, we subsequently address this constraint and report our observations of eosin in close proximity to silver island films (SiFs).
In this study, silver island films were prepared as we have previously published.3 300 μl of eosin (1.0×10−6M) immobilized in 4% (w/w) polyvinyl alcohol (PVA) was sandwiched between both the glass slides and the silver island film coated slides, respectively. The top inset of Fig. 1 shows the experimental sample geometry. Fluorescence spectra were collected on a Varian Cary Eclipse fluorometer at an angle of 45° to the surface. Excitation was incident to the bottom of the glass surface, 473 nm, with a 20 nm slit. The e-type fluorescence and phosphorescence spectra were collected in the Varian fluorometer in phosphorescence mode, with argon gas purging to remove O2.
FIG. 1.
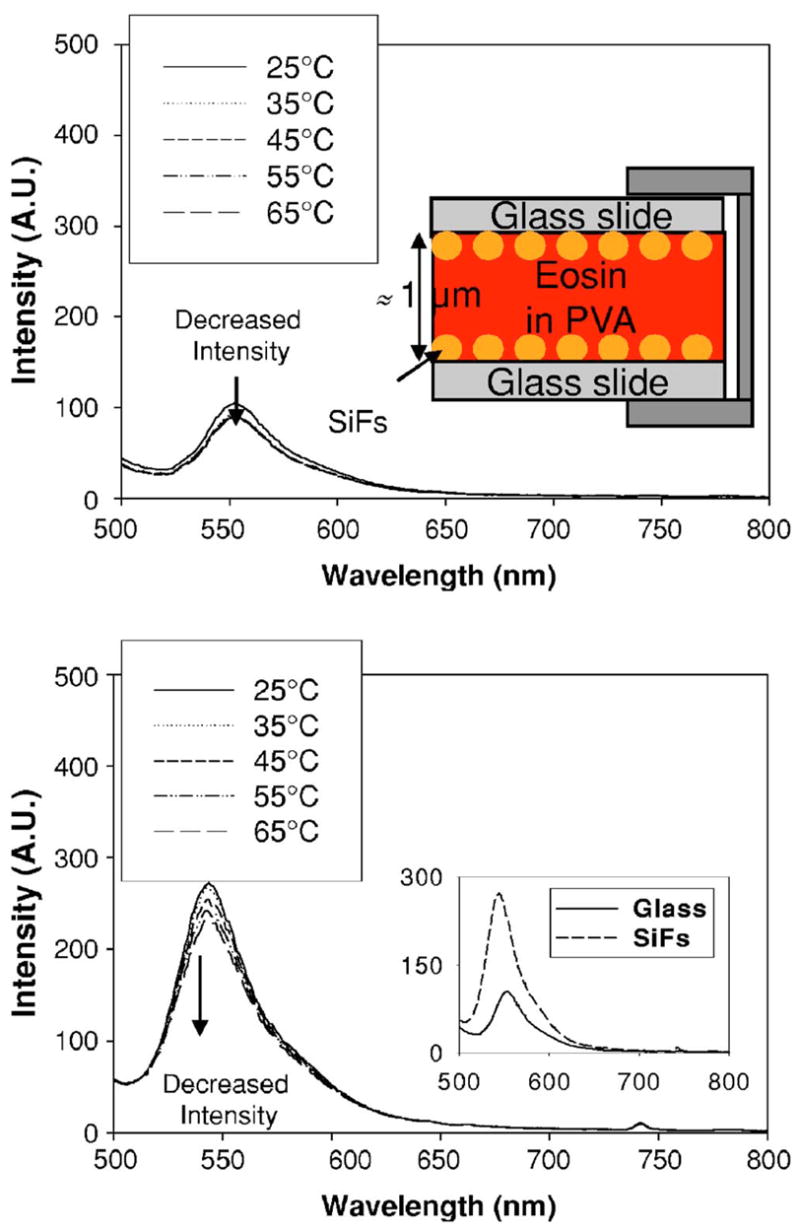
(Color online) Fluorescence emission spectra, λex=473 nm, of eosin immobilized in PVA, sandwiched between unsilvered slides at different temperatures (top), the experimental sample geometry (top inset), and between silvered slides at different temperatures (bottom) and also at 25 °C (bottom insert). SiFs—silver island films and PVA—polyvinyl alcohol solution.
Figure 1 shows the fluorescence emission spectra of eosin from glass and SiFs at different temperatures. The fluorescence was quenched with an increased temperature due to a simple oxygen quenching collision rate effect.5 It was also observed that the enhanced fluorescence intensity was approximately threefold brighter from the silver as compared to glass (Fig. 1 bottom inset). These findings of metal-enhanced S1 fluorescence of eosin are consistent with our previous reported findings for fluorophores sandwiched between silver nanostructures.6 Figure 2 shows e-type fluorescence and phosphorescence spectra after argon purging in a cuvette at different temperatures. We can observe not only e-type delayed fluorescence at ≈550 nm, which is spectrally identical to prompt S1 fluorescence spectra, as shown in Fig. 1, but also the phosphorescence emission peak is evident at ≈690 nm. The delayed emission band readily disappears if the solution is not deoxygenated. The e-type fluorescence peak readily increases with increased temperature, while the phosphorescence peak subsequently decreases. On SiFs, we have observed different extents of e-type fluorescence and phosphorescence from both glass slides and SiFs at different temperatures, with argon purging. The inset of Fig. 2 shows an ≈2.5-fold brighter e-type fluorescence as compared to phosphorescence, ≈1.9-fold brighter on SiFs.
FIG. 2.
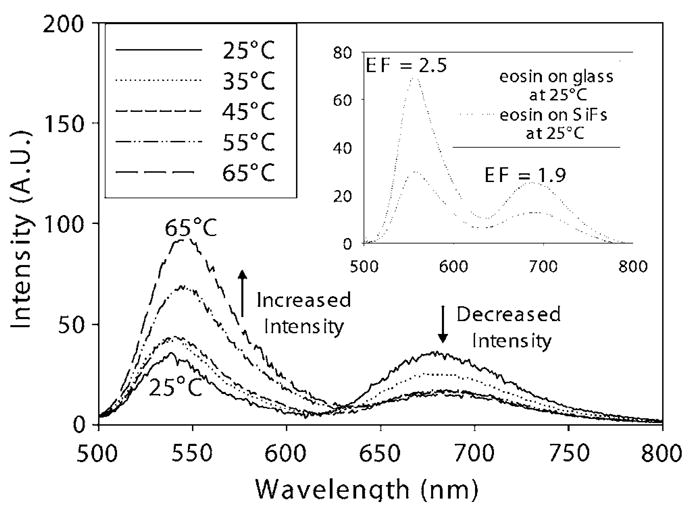
e-type fluorescence and phosphorescence emission spectra, ex =473 nm, of eosin in a cuvette at different temperatures. Eosin immobilized in PVA, sandwiched between two silvered and unsilvered slides at 25 °C (inset). EF—enhancement factor.
Metal-enhanced fluorescence and e-type fluorescence can also be seen visibly, as shown in the real-color photographs of Fig. 3. The eosin fluorescence emission intensity on glass is a light greenish; on SiFs, it is brighter than on glass due to the MEF effect. After heating the sample for 2 min, the eosin fluorescence emission on glass is much brighter than the preheating emission. This indicates more e-type fluorescence with increased temperature. The eosin emission is even more pronounced on SiFs after heating, indicating that e-type fluorescence emission was enhanced by surface plasmons (Fig. 3), as well as an increased net absorption effect, discussed below.
FIG. 3.

(Color online) Color photographs of eosin emission from glass and SiFs before and after 2 min heating. λex=473 nm. SiFs—silver island films. The real-color photographs were taken through an emission filter (488 nm razor edge).
Figure 4, top shows an eosin absorbance spectrum on glass and SiFs. We can see that when eosin is in close proximity to SiFs, the absorption band at 530 nm is increased approximately threetimes. It is well known that when a lumophore is placed close to metal, there is often a very strong net absorption effect, a consequence of the localized electromagnetic field around the metallic particles. In essence, conducting metallic particles can modify the free space absorption condition in ways that increase the incident electric field felt by a lumophore,7 i.e., enhanced plasmon absorption effect.8
FIG. 4.
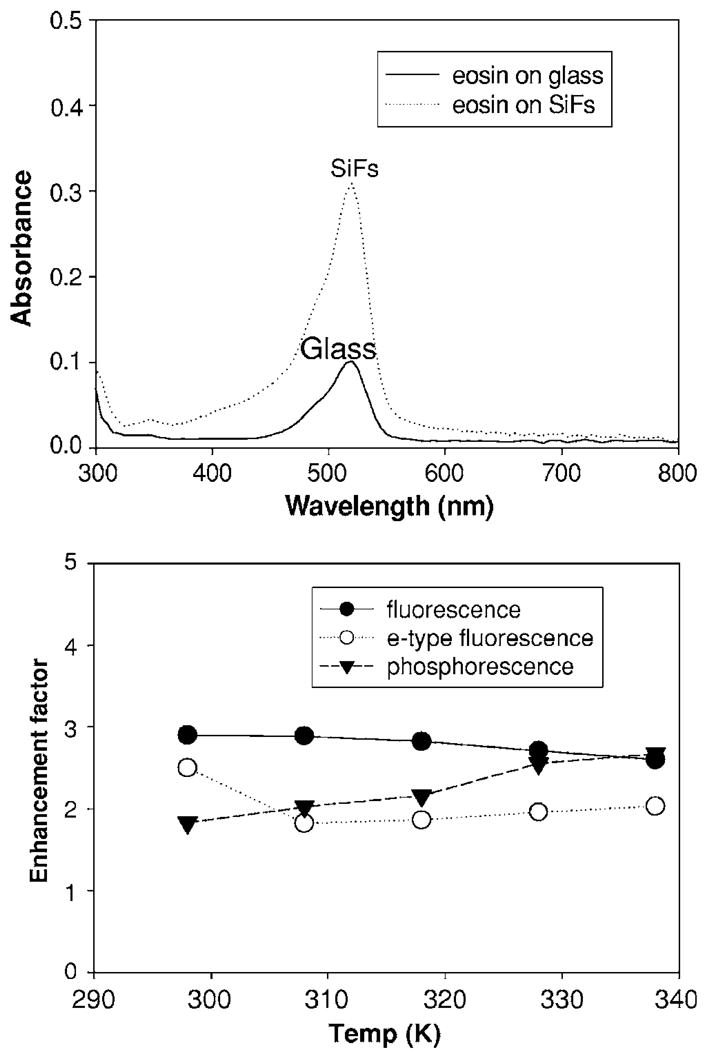
Absorption spectra of eosin immobilized in PVA, sandwiched between two silvered and unsilvered slides, respectively (top). Enhancement factor of fluorescence, e-type fluorescence, and phosphorescence at different temperatures (bottom). The enhancement factor for each type of emission was calculated by dividing the emission maximum intensity on silver by that measured on glass (control sample). In all cases, the spectra were identical when normalized.
Figure 4, bottom summarizes the enhancement factors of fluorescence, e-type fluorescence, and phosphorescence on SiFs, as compared to a glass control sample at different temperatures. The fluorescence enhancement factor decreased slightly with increased temperature, as expected. In a previous paper, we reported no changes in plasmon resonance at low temperatures, where at cooler temperatures, a reduced “dampening” or reduced “quenching” of the fluorophore facilitates MEF through changes in the nonfree electron component of the effective dielectric function of the metal.9 In this regard, it is therefore easy to correlate the trends in enhanced fluorescence with temperature. However, the phosphorescence enhancement factor is slightly increased with increasing temperature. In our system, e-type fluorescence and phosphorescence are competitive processes both resulting in a depopulation of the triplet state.10 Subsequently, we speculate that with increased temperatures, more T1 to S1 thermal activations afford for more recycling of states back to the triplet state, which, in turn, results in further enhanced phosphorescence. In this regard, reverse-intersystem crossing followed by intersystem crossing again back to the triplet state has been reported for eosin in the absence of metal,10 giving us some confidence in our interpretations.
We have also measured the e-type fluorescence lifetime in close proximity to SiFs. Since the populations of the singlet and triplet states are in thermal equilibrium, the lifetime of the e-type fluorescence is similar to the phosphorescence lifetime (millisecond) range.11 Because phosphorescence directly results from the triplet state, it has a shorter lifetime than e-type fluorescence, which still has to further decay from the S1 state. The lifetimes were calculated from the decay data (not shown) using single exponential decay curve analysis. We observed a reduced e-type fluorescence lifetime (τon SiFs ≈ 0.6 ms) for fluorophores near to silver as compared to glass (τon glass ≈ 2.0 ms). We also observed a reduced phosphorescence lifetime (τon SiFs ≈ 0.4 ms) for fluorophores near to silver as compared to glass (τon glass ≈ 1.0 ms), which we have similarity observed previously for Rose Bengal at 77 K,12 as expected; the MEP lifetime is shorter than the metal-enhanced e-type fluorescence lifetime, i.e., 0.4 ms vs. 0.6 ms, respectively. These findings are consistent with our previously reported findings and trends for nanosecond decay time fluorophores with S1 emission sandwiched between SiFs.13
In conclusion, we report metal-enhanced e-type fluorescence, which is thought due to both enhanced absorption and emission. Eosin in close proximity to SiFs can undergo enhanced e-type fluorescence, with up to a 2.5-fold increase observed as compared to an identical control sample containing no silver. This observation is helpful in the development of long-lived, high quantum yield and photostable probes for biophysical applications. In this regard, our laboratory has recently developed MEF solution-based probes which harness the significant benefits of MEF but in a solution-based nanoparticle architecture.14 It is therefore very likely that these nanoball architectures could also incorporate e-type fluorescent probes for downstream biophysical applications.
Acknowledgments
Support from the NIH, the National Institute of Neurological Disorders & Stroke, under Grant No. NS055187 is acknowledged.
References
- 1.Geddes CD, Lakowicz JR. J Fluoresc. 2002;12:121. doi: 10.1023/A:1021341305229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aslan K, Previte MJR, Zhang YX, Geddes CD. Biophys J. 2007;371A:371A. [Google Scholar]
- 3.Aslan K, Lakowicz JR, Geddes CD. Curr Opin Chem Biol. 2005;9:538. doi: 10.1016/j.cbpa.2005.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brand L, Witholt B. Methods Enzymol. 1967;11:776. [Google Scholar]
- 5.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Academic; New York: 1999. [Google Scholar]
- 6.Zhang Y, Aslan K, Previte MJ, Geddes CD. Chem Phys Lett. 2006;432:528. doi: 10.1016/j.cplett.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hao E, Schatz GC. J Chem Phys. 2004;120:357. doi: 10.1063/1.1629280. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J, Huang l, Wang Y, Zhu J. Physica B. 2005;362:103. [Google Scholar]
- 9.Zhang Y, Aslan K, Previte MJ, Geddes CD. J Fluoresc. 2007 doi: 10.1007/s10895-007-0196-y. [DOI] [PubMed] [Google Scholar]
- 10.Edidin M. Annu Rev Biophys Bioeng. 1974;3:179. doi: 10.1146/annurev.bb.03.060174.001143. [DOI] [PubMed] [Google Scholar]
- 11.Garland PB, Moore CH. Biochem J. 1979;183:561. doi: 10.1042/bj1830561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Aslan K, Previte MJR, Malyn SN, Geddes CD. J Phys Chem B. 2006;110:25108. doi: 10.1021/jp065261v. [DOI] [PubMed] [Google Scholar]
- 13.Aslan K, Leonenko Z, Lakowicz JR, Geddes CD. J Fluoresc. 2005;15:643. doi: 10.1007/s10895-005-2970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aslan K, Wu M, Lakowicz JR, Geddes CD. J Am Chem Soc. 2007;129:1524. doi: 10.1021/ja0680820. [DOI] [PMC free article] [PubMed] [Google Scholar]