

Abstract
5′-O-[N-(Salicyl)sulfamoyl]adenosine (Sal-AMS, 1) is a potent inhibitor of the bifunctional enzyme salicyl-AMP ligase in Mycobacterium tuberculosis. This inhibitor acts by disrupting the biosynthesis of the mycobactin siderophores that are essential for the process of iron acquisition. To aid with in vitro metabolism and in vivo pharmacokinetic studies of Sal-AMS, a stable deuterium-labelled Sal-AMS analog (Sal-AMS-d4) was synthesized. This deuterium-labelled analog was used as an internal standard to conduct in vitro plasma and microsomal stability studies. Sal-AMS was found to be stable for 24 h in human plasma and 1 h in human liver microsomes at 37°C.
Keywords: 5′-O-[N-(Salicyl)sulfamoyl]adenosine, adenylation inhibitor, siderophore, tuberculosis, mass spectrometry
Introduction
Mycobacterium tuberculosis, the etiological agent of tuberculosis (TB), is the leading cause of bacterial infectious disease mortality.1 Of late, an increase in the number of multidrug resistant and extensively drug resistant TB cases has been a cause of great concern since these are resistant to both first- and second-line TB drugs. Hence, the development of new antitubercular agents exhibiting novel mechanisms of action is urgently needed.
Iron is required for almost all known bacterial pathogens, and this micronutrient is highly restricted in a human host where the concentration is too low to support bacterial colonization and growth.2 Bacteria and other microorganisms have developed unique solutions to this problem of iron sequestration. They synthesize and secrete small molecule iron chelators termed siderophores that extract iron from host proteins and the resulting siderophore-iron complex is then imported by dedicated bacterial receptors. M. tuberculosis is dependent upon two series of structurally related peptidic siderophores, collectively referred to as the mycobactins for iron acquisition.3,4 5′-O-[N-(Salicyl)sulfamoyl]adenosine (Sal-AMS, 1, Figure 1) has recently been described as a compound that inhibits mycobactin synthesis by blocking the second biosynthetic step catalyzed by the bifunctional enzyme MbtA, a salicyl-AMP ligase encoded by the gene Rv2384. This bisubstrate nucleoside inhibitor exhibited nanomolar enzyme inhibition as well as potent inhibition of whole-cell M. tuberculosis growth rivaling the first-line agent isoniazid.5,6
Figure 1.

Structures of Sal-AMS (1) and its corresponding deuterium-labelled analog Sal-AMS-d4 (2).
Herein we report the synthesis of a stable isotopically labelled standard of Sal-AMS (Figure 1) to support in vitro metabolism and in vivo pharmacokinetic studies. Installation of the label on the salicyl moiety was deemed useful to identify potential metabolites of the salicyl group. Additionally, tandem MS/MS indicated the most abundant fragment contained salicylate.
Results and discussion
Sal-AMS was subjected to tandem MS/MS to identify the optimal site to incorporate a label (Figure 2). Application of a medium collision energy to the precursor ion of Sal-AMS [M-H]- at 465.1 afforded daughter ions at m/z 345.1 and 198.1 corresponding to the two expected molecular fragmentation patterns. The base peak at 198.1 is consistent with the N-(sulfonyl)salicylamide moiety derived from cleavage of the bond between O-5′ and S, whereas the ion at 345.1 corresponds to 5′-O-(sulfamoyl)adenosine resulting from cleavage of the labile N-acyl bond. Based upon the above-observed fragmentation pattern in MS/MS, potential for metabolism of the salicylate moiety, and ability to readily incorporate four deuterium atoms, Sal-AMS-d4 2 was prepared as described below.
Figure 2.
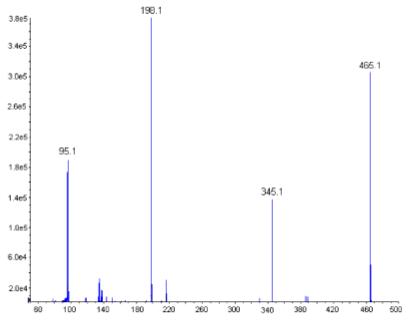
Fragmentation pattern of Sal-AMS. This figure is available in color online at www.interscience.wiley.com/journal/jlcr.
The synthesis of the desired deuterium-labelled analog, began with the silylation of adenosine 3 to tris-TBS derivative 4 followed by regioselective deprotection of 5′-O-TBS employing 50% aq TFA to afford 5 as described.5 Sulfamoylation of 5 in dimethylacetamide provided 6.7 3,4,5,6-Tetradeuterosalicylic acid was activated with CDI then treated with excess 6 and DBU to yield 7 as the triethylammonium salt. Deprotection of 7 with TBAF followed by ion exchange mediated by Dowex-50WX8 cation-exchange resin furnished Sal-AMS-d4 2 as the sodium salt. Compound 2 was fully characterized by HRMS, 1H NMR and 13C NMR. The isotopic purity of the final deuterium-labelled analog was observed to be 98.2%.
A simple, rapid and accurate liquid chromatographic-TOF mass spectrometric method for quantification of Sal-AMS (1) in human plasma and liver microsomes with Sal-AMS-d4 (2) as an internal standard was developed and validated. Sal-AMS was monitored by TOF-MS at 465.083 and Sal-AMS-d4 was monitored at 469.108. Alternatively, one could employ MS/MS (tandem mass spectrometry) detection with the transitions of 465.1 > 198.1 and 469.1 > 202.1, respectively.
Sal-AMS (1) was incubated at 37°C with human plasma at a concentration of 5 μM and aliquots were collected at 0, 0.25, 0.5, 1, 2, 3, 4, 6, 12 and 24 h. The internal standard 2 (100 μL, 5 μM in ammonium formate buffer pH 4.3) was added and the plasma was extracted on a solid phase cartridge (Phenomenex Strata™X 30 mg/1 ml) that was pre-conditioned with 1.0 mL methanol, followed by 2.0 mL of ammonium formate buffer. The extraction cartridge was washed with 2.0 mL ammonium formate buffer. Sal-AMS and Sal-AMS-d4 were eluted with 1 mL of 80% acetonitrile in water. The liquid was completely removed by purging under nitrogen and redissolved in 200 μL of 20% acetonitrile in acetate buffer. The liquid was centrifuged at 12 000 rpm for 10 min and 15 μL of the supernatant was injected into the LC/MS system. Chromatographic separation was achieved on a Polaris C18 column and a mobile phase consisting of acetonitrile/water (90:10 v/v) and 10 mM ammonium formate buffer (pH 4.3) at a flow rate of 0.3 mL/min. The effluents were detected at m/z 465.1 and 469.1 for 1 and 2, respectively. This method is fast, as the run-cycle time was <15 min run to run and the described method was linear over the Sal-AMS concentration range of 0.1-10 μM corresponding to 0.046-4.66 μg/mL (r>0.9986, Figure 4). The stability of Sal-AMS in these processed samples was evaluated by comparing the change in the concentration of Sal-AMS over a 24 h time period (Figure 3). Analytes were considered stable if the deviation of mean test responses were within 15% of freshly prepared samples. This study indicates that Sal-AMS in plasma is stable for at least 24 h at 37°C.
Figure 4.
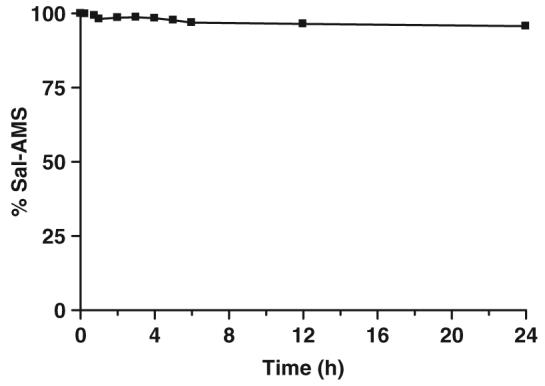
Stability of Sal-AMS in human plasma over 24 h.
Figure 3.
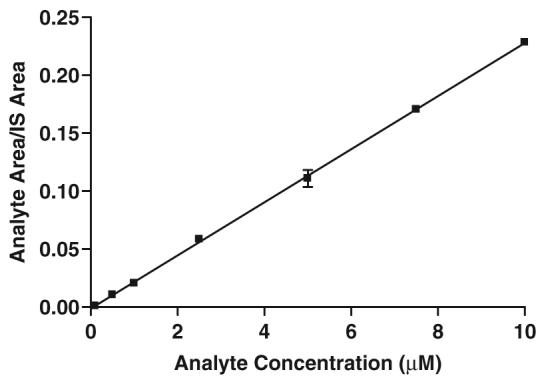
Standard calibration curve.
Additionally, Sal-AMS was incubated at 37°C with pooled human liver microsomes (0.5 mg/ml) at a concentration of 2 μM Sal-AMS in 0.1 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH. Metabolism rate of Sal-AMS was measured in duplicates in a final volume of 200 μL. After a 3 min pre-incubation of substrate and microsomes at 37°C, the reaction was started with the addition of 1 mM NADPH. Sal-AMS metabolism was monitored at 5, 10, 20, 30 and 60 min and the reactions were stopped with 200 μL ice-cold methanol containing 2 μM internal standard 2. A control reaction was also run (water replacing NADPH) to monitor stability of the compound in a microsomal incubation without enzyme cofactors. After the incubation samples were quenched, they were centrifuged at 13 000 × g at 4°C for 5 min to precipitate the microsomal protein. The supernatant was then passed through a 0.2 μm nylon spin filter to prepare for analysis on LC-MS. The stability of Sal-AMS in these processed samples was evaluated by comparing the change in the concentration of Sal-AMS by the LC-MS method described above. No Sal-AMS degradation was observed with a 3% variation of Sal-AMS present at each time point and control. The stability study indicates that Sal-AMS is not degraded by NADPH-dependent oxidative metabolism in human liver microsomes.
Experimental
General
Sal-AMS (1) was synthesized as described.6 Salicylic acid-d4 was purchased from Cambridge Isotope Laboratories. HPLC grade water and methanol were purchased from Fisher Scientific. High-purity ammonium formate and formic acid were purchased from Fluka. Drug-free (blank) buffered human plasma was obtained from Fairview Hospital, Minneapolis, MN, and stored at -20°C prior to use. Pooled human liver microsomes were obtained from BD Biosciences (Franklin Lakes, NJ). Chromatographic separation was carried out on an Agilent 1100 HPLC with a Polaris C18 column 3 μm, 50 × 2.0 mm (Varian). A gradient mobile phase consisting of acetonitrile/water (90:10 v/v) and 10 mM ammonium formate (pH 4.3) was delivered with a flow rate of 0.3 mL/min. The gradient program is given in Table 1. The total run time for each sample analysis was 15 min. The sample injection volume was 15 μL. Mass spectra were obtained on an Agilent (Model G1969A) LC time-of-flight (TOF) mass spectrometer equipped with an electrospray ionization source and a TOF detector in negative ion mode. The spray voltage and capillary temperature were 4000 V and 350°C, respectively. Sal-AMS and the deuterated internal standard eluted at 9.3 min (Figure 5).
Table 1.
LC-MS gradient program
| Time (min) | % Solvent Aa | % Solvent Bb |
|---|---|---|
| 0 | 98.0 | 2.0 |
| 3.5 | 93.5 | 6.5 |
| 3.6 | 80.0 | 20.0 |
| 8.0 | 80.0 | 20.0 |
| 8.5 | 40.0 | 60.0 |
| 12.0 | 40.0 | 60.0 |
| 12.5 | 98.0 | 2.0 |
| 15.0 | 98.0 | 2.0 |
Solvent A: 10 10mM mM ammonium formate (pH 4.3).
Solvent B: MeCN/H H2O (90:10 v/v).
Figure 5.
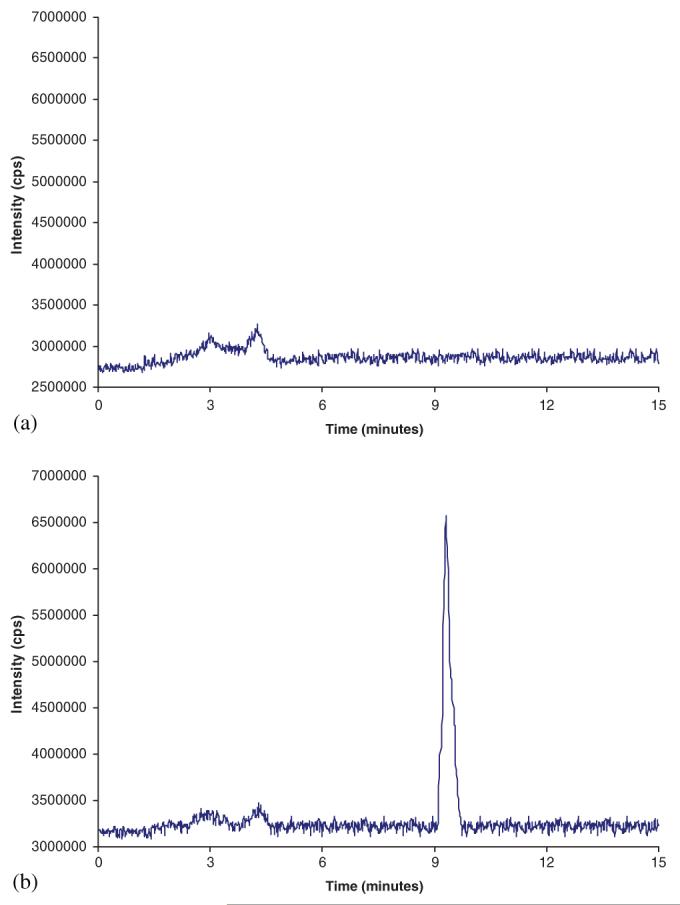
(a) Chromatogram of blank plasma and (b) chromatogram of plasma spiked with 2.5 μM Sal-AMS. This figure is available in color online at www.interscience.wiley.com/journal/jlcr.
Synthesis of Sal-AMS-d4
2′,3′-O-Bis(tert-butyldimethylsilyl)-5′-O-(sulfamoyl)adenosine (6)
To a cooled solution of 2′,3′-O-bis(tert-butyldimethylsilyl)adenosine (4.0 g, 8.0 mmol, 1.0 equiv) in dimethylacetamide (20 mL) was added sulfamoyl chloride (1.88 g, 16 mmol, 2.0 equiv) and the reaction mixture was stirred at rt for 3 h. The resulting mixture was partitioned between H2O (100 mL) and EtOAc (100 mL) and the aqueous layer extracted with EtOAc (2 × 100 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. Purification by flash chromatography (95:5 CH2Cl2/MeOH) afforded the title compound (3.3 g, 73%) as a white solid: 1H NMR, 13C NMR and HRMS agreed with literature values.5
5′-O-N-[(2-Hydroxy-3,4,5,6-tetradeutero)benzoyl]sulfamoyladenosine sodium salt (2)
To a solution of 3,4,5,6-tetradeuterosalicylic acid (60 mg, 0.42 mmol, 1.0 equiv) in MeCN (5 mL) was added CDI (85.7 mg, 0.50 mmol, 1.2 equiv) and the reaction stirred at 60°C for 2 h. The solution was cooled to rt, then 6 (339.2 mg, 0.59 mmol, 1.4 equiv) and DBU (128.5 μL, 0.92 mmol, 2.2 equiv) were added and the reaction stirred an additional 2 h at rt. The reaction mixture was concentrated in vacuo. Purification by silica gel flash chromatography (80:20:0.5 EtOAc/MeOH/Et3N) afforded 2′,3′-O-bis(tert-butyldimethylsilyl)-5′-O-N-[(2-hydroxy-3,4,5,6-tetradeutero)benzoyl]sulfamoyladenosine triethylammonium salt.
To the above product in THF (5 mL) was added TBAF (1.0 M in THF, 1.0 mL, 1.05 mmol, 2.5 equiv) and the reaction stirred 30 min at rt. Purification by flash chromatography (80:20:0.5 EtOAc/MeOH/Et3N) afforded the title compound as the triethylammonium salt (2 · Et3N). Conversion to the sodium salt was achieved by passage of an aqueous solution of 2 · Et3N (0.5 mL) through a short column (20 × 50 mm) of Dowex 50WX8-100 cation-exchange resin in the sodium form and the compound containing fractions were pooled and lyophilized to afford the title compound (150 mg, 72% overall yield) as a white flocculent solid: m.p.>260°C (dec); 1H NMR (600 MHz, CD3OD) δ 4.31-4.38 (m, 2H), 4.40-4.42 (m, 2H), 4.71 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.16 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 68.6, 71.5, 75.2, 83.7, 88.3, 119.3, 119.6, 140.2, 150.0, 152.9, 156.4, 161.1, 174.3 (C3-C6 of the salicyl are not observed due to C-D coupling); HRMS (ESI-) calcd for C17H13D4N6O8S [M-H]- 469.1090, found 469.1085 (error 1.1 ppm).
Calibration curves
The primary stock solutions of Sal-AMS (10 mM) and the internal standard d4-Sal-AMS (5 μM) were prepared in water. Working standard solutions were prepared from the primary standard solutions by further dilution with water. These working standard solutions were used to prepare a calibration curve. A seven-point standard calibration curve (0.1-10 μM) for Sal-AMS was prepared by spiking blank plasma with an appropriate amount of Sal-AMS.
Data analysis
Data were analyzed on Agilent’s AnalystQS software. For quantification, the peak area ratios of the target ions of the drug with those of the internal standard were compared with weighted (1/c) least-squares calibration curves in which the peak area ratios of the calibration standards were plotted versus their concentrations. The net difference in concentration from time 0 to 24 h is ≤4.3%, demonstrating that the Sal-AMS is stable for at least 24 h at 37°C when incubated with human plasma.
Conclusion
A deuterium-labelled internal standard for Sal-AMS, Sal-AMS-d4, was successfully synthesized and characterized. A simple, sensitive and rapid LC-MS method has been developed for the determination of Sal-AMS in human plasma and liver microsomes. This method will be useful for monitoring Sal-AMS plasma levels, particularly in pharmacokinetic studies. Sal-AMS is stable for at least 24 h at 37°C when incubated with human plasma and 1 h at 37°C with human liver microsomes. The stability of Sal-AMS in human plasma is noteworthy since the acylsulfamate linkage has the potential to undergo enzymatic hydrolysis. Additionally, the lack of metabolism of Sal-AMS was notable as this compound contains numerous metabolically liable functional groups.
Scheme 1.

Reaction Conditions: (a) TBSCI, Imidazole, DMF, 100%; (b) TFA, THF/H2O, 0° C, 70%; (c) Sulfamoyl chloride, DMA, 73%; (d) 3,4,5,6-tetradeuterosalicylic acid, CDI, DBU, MeCN; (e) TBAF, THF; (f) Dowex 50WX-Na+, 72% (3 steps).
Acknowledgement
This research was supported by a grant from NIH (R01AI070219) and funding from the Center for Drug Design in the Academic Health Center of the University of Minnesota to C.C.A. We thank Jessica Keller for performing microsomal stability studies.
References
- [1].World Health Organization Fact sheet on tuberculosis. 2005 http://www.who.int/mediacentre/factsheets/fs104/en/print.html.
- [2].Ratledge C, Dover LG. Annu. Rev. Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. DOI: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- [3].Vergne AF, Walz AJ, Miller MJ. Natl. Prod. Rep. 2000;17:99–116. doi: 10.1039/a809397k. DOI: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- [4].Snow GA. Bacteriol. Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Nat. Chem. Biol. 2005;1:29–32. doi: 10.1038/nchembio706. DOI: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- [6].Somu RV, Boshoff H, Qiao CH, Bennett EM, Barry CE, Aldrich CC. J. Med. Chem. 2006;49:31–34. doi: 10.1021/jm051060o. DOI: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- [7].Okada M, Iwashita S, Koizumi N. Tetrahedron Lett. 2000;41:7047–7051. DOI: 10.1016/S0040-4039(00)01130-8. [Google Scholar]