

Abstract
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma of childhood. This paper is focuses on imaging for diagnosis, staging, and follow-up of noncraniofacial RMS.
Keywords: Rhabdomyosarcoma, Imaging, Children
Introduction
Rhabdomyosarcoma (RMS) is the most common paediatric soft-tissue sarcoma and constitutes 3–5% of all malignancies in childhood [1]. In children, the age-standardized annual incidence rate for RMS is between 4 and 7 per million [2]. This review focuses on the imaging of all RMS occurring outside the head and neck region, which comprise 40% of RMS; around 15% are genitourinary (GU) nonbladder prostate tumours (GU-NBP, i.e. paratesticular, vaginal and uterine tumours), 10% are bladder prostate tumours (BP), 15% occur in the limbs, and 20% occur in other sites (i.e. thoracic or abdominal tumours).
Clinical symptoms vary widely, based on the location of the primary tumour, but in general presenting symptoms often are indolent, with nonspecific or minimal symptoms, at the start mimicking innocent general paediatric diseases. Often the duration or progression of symptoms alerts the physician to the presence of a malignant tumour. Site-specific symptoms in GU-NBP tumours are a paratesticular mass in boys that may be painful or not, while girls may present with a grape-like (botryoid) vaginal extrusion of mucosanguineous tissue or micturition problems. BP tumours may present with urinary retention, haematuria, constipation or an abdominal mass. Limb tumours often present with a painless swelling and/or with enlarged regional lymph nodes, while presentation of tumours in other locations may vary from biliary obstruction in tumours of the biliary tract, to painless masses, all depending on their primary site.
Patients with metastatic disease present with more general symptoms of fatigue, weight loss, and low blood counts. As soon as a RMS is suspected an extensive work-up must be performed to get a histological diagnosis and correct staging of the tumour. Imaging of the primary site with US, MRI or CT is required in all patients at the start of the diagnostic work-up [3]. Once the diagnosis has been confirmed histologically, the most frequently involved metastatic sites will be investigated; CT of the lungs, imaging of regional lymph nodes, and a technetium bone scan are recommended for every patient. Furthermore, bone marrow aspirates and trephines should be performed in all patients, while examination of the cerebral spinal fluid is only required in patients with paraspinal and parameningeal locations. As soon as the work-up has been completed, the patient can be stratified to receive treatment according to the child’s risk group, based on the six significant prognostic factors for localized RMS that came out of a retrospective European analysis (European Paediatric Soft-tissue Sarcoma Study Group, EpSSG); histology, postsurgical status according to the Intergroup Rhabdomyosarcoma Study Group (IRSG), tumour site, node involvement, tumour size, and patient age (Table 1).
Table 1.
EpSSG prognostic factors [28]
| Favourable | Unfavourable | |
|---|---|---|
| Histology | Embryonal | Alveolar |
| IRSG status | Higher grades more unfavourable | |
| Tumour site | Head and neck non-parameningeal | All other sites |
| Orbital | ||
| Genitourinary – nonbladder/prostate | ||
| Node involvement | N0 | N1 |
| Tumour size (cm) | ≤5 | >5 |
| Age (years) | <10 | ≥10 |
Pathology
RMS is a fast-growing, primitive, high-grade, malignant mesenchymal tumour. Depending on their degree of differentiation, the tumour cells manifest features that more or less can be found in the cells of skeletal muscle. These features, essential for the diagnosis, are the presence of myofibrils and cross striations (on light and electron microscopy) and/or positive immunohistochemical staining for markers of muscle differentiation such as desmin and myoD1. For detailed descriptions of the histopathological aspects of RMS, reference should be made to Weiss and Goldblum [4] and Fletcher et al. [5].
Based on morphology, RMS is traditionally subdivided into embryonal, alveolar and pleomorphic. Pleomorphic RMS, in contrast to embryonal and alveolar RMS, almost exclusively occurs in adults (median age sixth decade), and is therefore not discussed further. Embryonal RMS is the most common type (60–70% of all RMS). The cells show a close resemblance to various stages in the embryogenesis of normal skeletal muscle (Fig. 1). Subtypes are botryoid RMS and spindle-cell RMS (Fig. 2). When arising in the submucosa, embryonal RMS may present as a fast-growing exophytic, polypoid mass. This macroscopic variant is called botryoid RMS (grape-like) and, due to its growth pattern (primary exophytic and not invasive), has a better prognosis.
Fig. 1.
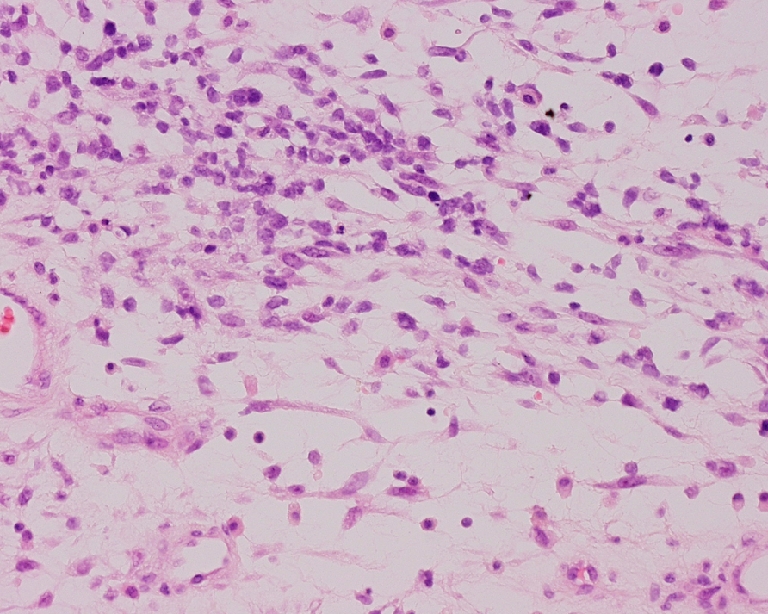
Embryonal RMS: small cells and primitive spindle-shaped cells resembling the first stages of developing normal skeletal muscle (H&E, original magnification 10×20)
Fig. 2.
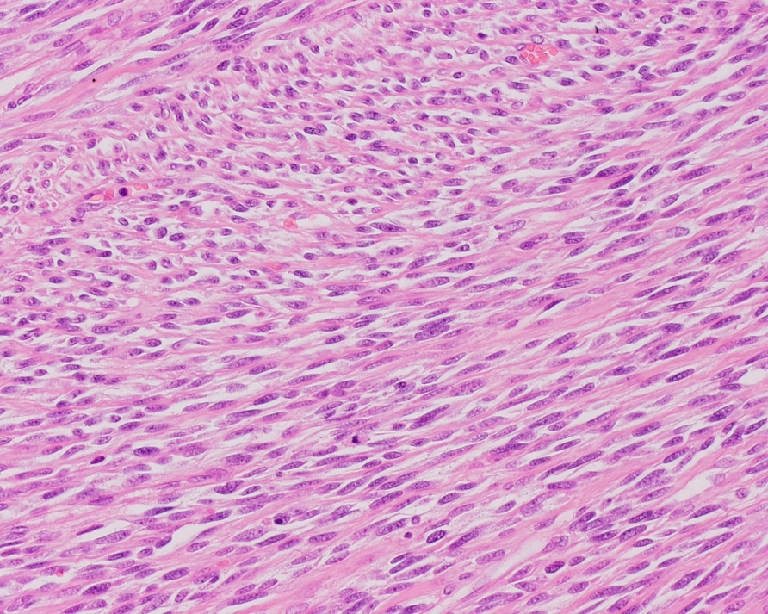
Embryonal RMS, spindle-cell type: closely packed spindle cells arranged in perpendicular crossing fascicles giving a leiomyosarcoma-like appearance (H&E, original magnification 10×20)
Spindle-cell RMS comprising about 4% of all RMS, seems to behave less aggressively and is most often encountered in a paratesticular location (about 30% of all paratesticular RMS) and the head and neck [6, 7]. Histologically spindle cell RMS is characterized by parallel orientation of spindle cells with an eosinophilic, fibrillary cytoplasm and an elongated, hyperchromatic nucleus. The cells are arranged in broad fascicles (fibro-/leiomyosarcoma-like) or in short, interdigitating fascicles (storiform) and whirls with, in contrast to the broad fasciculated variant (leiomyomatous type), an abundant, partly hyalinized collagenous matrix.
Alveolar RMS is composed of ill-defined, dense aggregates of poorly differentiated round or oval tumour cells that frequently show loss of cohesion (Fig. 3). This loss of cohesion and the presence of thin fibrous septa result in an alveolar pattern. In cases where the tumour cells do not show loss of cohesion, the term ‘solid alveolar RMS’ is used. Alveolar RMS represents about 20% of all RMS and has two specific translocations with specific fusion transcripts that can be detected by RT-PCR. The majority (about 55%) show a t(2;13)(q35;q14) translocation with the corresponding fusion transcript PAX3-FKHR [8]. In about 22% a t(1;13)(p36;q14) translocation is found with fusion transcript PAX7-FKHR. In rare cases, RMS with a more or less alveolar pattern lacks these translocations.
Fig. 3.
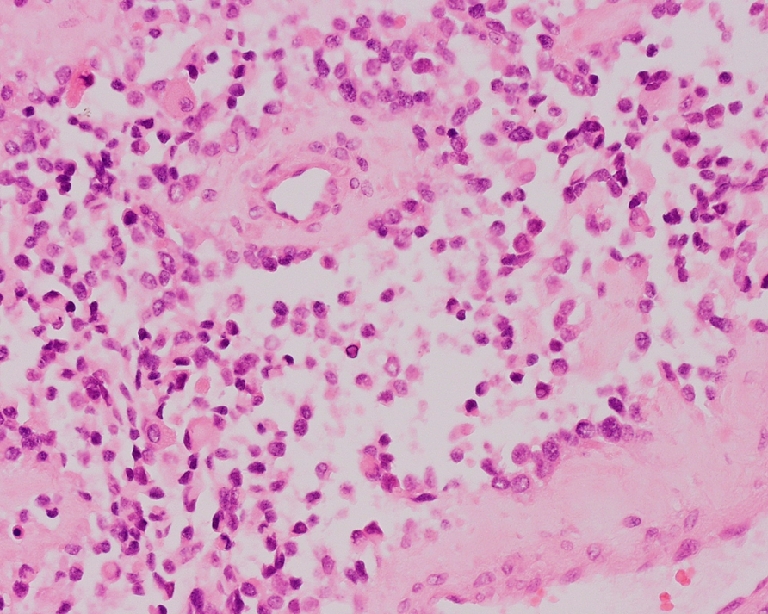
Alveolar RMS: cluster of primitive cells with loss of cellular cohesion and bordered by dense fibrous septa, resulting in an alveolar pattern (H&E, original magnification 10×20)
Both embryonal and alveolar RMS may show rhabdoid tumour-like features and anaplasia (focal or diffuse) [9, 10]. More recently, under the heading sclerosing RMS, a variant with hyalin sclerosis has been described (Fig. 4) [11]. It is unclear whether this is a distinct subtype.
Fig. 4.
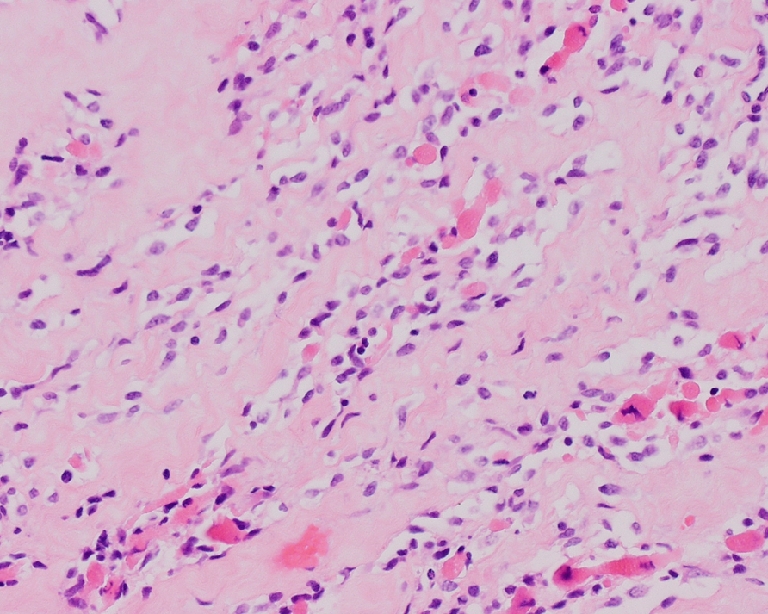
Sclerosing RMS: small cells, primitive spindle-shaped cells and eosinophilic rhabdomyoblasts in a background of hyaline sclerosis (H&E, original magnification 10×20)
In 1995, the IRSG and the International Society of Paediatric Oncology (SIOP) suggested a prognostically more significant classification (Table 2) [12].
Table 2.
RMS: IRSG and SIOP classification, extended with new variants [12]
| Group | Prognosis | Subtype |
|---|---|---|
| I | Better prognosis | Embryonal RMS, botryoid type |
| Embryonal RMS, spindle-cell type | ||
| II | Intermediate prognosis | Embryonal RMS (remaining) |
| III | Worse prognosis | Alveolar RMS |
| IV | Unclear prognosis | RMS with rhabdoid features |
| Embryonal RMS with diffuse anaplasia | ||
| Sclerosing RMS |
Imaging findings
Radiography
As RMS is a soft-tissue tumour, conventional radiology plays an insignificant role in its diagnosis. Localized bony erosion adjacent to the primary site is a recognized complication; this area may be hot on a 99mTc-MDP bone scan in the absence of metastatic disease in the skeleton. In contrast to the initial diagnostic work-up, where for the assessment of pulmonary metastases CT is mandatory, AP and lateral chest radiographs are used in the follow-up period.
Ultrasonography
US is often the first imaging modality used in children with soft-tissue masses because it is readily available, has high resolution, and can easily assess extent and vascularity of a mass. One should not forget that most soft-tissue lesions are benign, can readily be diagnosed with US, and do not need further diagnostic work-up or even treatment.
On US, RMS in general shows as a well-defined, slightly hypoechoic inhomogeneous mass that can show significantly increased flow (Fig. 5). In the particular case of paratesticular RMS, US is the imaging modality of choice, although CT of the abdomen is also necessary to evaluate for retroperitoneal lymphadenopathy. In all other RMS locations additional imaging using CT or MRI is mandatory.
Fig. 5.

A 6-year-old boy with a mass in the left scrotum. US image shows an ill-defined heterogeneous mass surrounding the testis (open arrow). The mass shows increased flow (solid arrow). Histopathology: embryonal RMS
US is also of use in image-guided biopsies. Recently Sebire and Roebuck [13] systematically reviewed the pathological diagnosis of paediatric tumours from image-guided needle-core biopsies. They concluded that image-guided biopsy material was sufficient to come to a diagnosis in 94% (95%; CI 92–96%) of patients. Complications needing treatment, mostly haemorrhage requiring transfusion, were reported in only 1% of patients. For image-guided biopsies the material obtained should be transported fresh to the pathology department. Fixation should not be performed as this precludes further cytogenetic studies.
CT
In order to assess pulmonary metastases from RMS, CT of the chest is a mandatory examination. However, assessment of pulmonary involvement can sometimes be difficult. Although criteria such as number and size of lesions, morphology (noncalcified, round and well-defined) and location (inferior lobes, subpleural spaces, branching vessels) have been applied in adult patients, none has shown 100% specificity. According to the EpSSG guidelines for evaluating chest CT the following criteria with respect to the diagnosis of pulmonary metastases should be applied: one pulmonary or one pleural nodule over 10 mm in diameter, two or more well-defined nodules of 5–10 mm or five or more lesions >5 mm [3]. This comes with the assumption that there is no other medical explanation for these lesions. When there is a high suspicion that a small lesion is metastatic, and appears to be the only site of metastatic disease, biopsy may be performed. In the EpSSG protocol, lung biopsy is not recommended.
With the introduction of multidetector CT (MDCT) the advantage of MRI of being a multiplanar imaging modality has been overtaken, and by virtue of its underlying physics, CT is superior to MRI in detecting osseous changes. The drawback of CT in children is the use of ionizing radiation. Since the seminal papers of Paterson et al. [14] and Brenner et al. [15], we should be aware of the theoretical risk of CT-induced cancer fatalities and take this into consideration especially in the work-up of children with cancer, as they have already proven their tendency to present with a malignancy early in life [14, 15].
MRI
With its superior ability to depict soft-tissue changes, MRI is the primary imaging modality in RMS [16]. Although imaging protocols should be tailored for each individual patient, they should at least consist of axial T1-W and T2-W images (for anatomic detail and assessment of neurovascular structures), T1-W images perpendicular to the axial plane, and imaging after gadolinium administration. It is important that at least two series should be identical, one before and one after contrast agent administration, to be able to discern enhancement. Contrast-enhanced series are mandatory and ideally be performed with fat saturation.
The imaging characteristics of RMS are relatively nonspecific. Like most soft-tissue tumours they have intermediate signal intensity on T1-W images (Fig. 6). On T2-W images they tend to be of intermediate-to-high signal intensity. If the tumour contains a high number of septa it may have a lobular shape. RMS in general show strong enhancement on postcontrast imaging (Fig. 6). In very rare instances the tumour may show a predominantly cystic appearance (Fig. 7). Dynamic series are useful in order to assess tumour vascularity, and to differentiate between postchemotherapy/surgery residual disease and fibrosis.
Fig. 6.

A 13-year-old girl who noticed a small lump near the anus. a T1-W MR image shows a well-defined pararectal lesion (arrow). b After intravenous gadolinium administration the lesion shows homogeneous enhancement (arrow) Histopathology: alveolar RMS
Fig. 7.

An 8-year-old girl with haemolysis, fever and a mass underneath the scapula. a Coronal STIR image shows a lesion with mixed signal intensity (open arrow) and multiple enlarged lymph nodes in the neck (solid arrow). b Axial T2-W image shows multiple cystic lesions with fluid-fluid levels (open arrow). Histopathology: stage IV embryonal RMS
For the surgeon, in order to plan surgery, it is of importance to describe the compartment in which the RMS is located [17]. Vascular involvement is considered to be absent if there is a normal tissue plane visible between the tumour and the vessel, or if the tumour has a less then a 180° circumferential relationship. If the tumour surrounds the vessel for more than 180°, it is considered to be encased.
Two studies have addressed the use of whole-body MRI (WB-MRI) in paediatric oncology [18, 19]. In the first study with various malignant tumours WB-MRI had a superior positive predictive value for skeletal metastases compared to bone scintigraphy (94 vs. 76%, respectively), and also a significantly higher sensitivity (99 vs. 26%, respectively) [18]. In the second study, WB-MRI had a higher sensitivity (82%) than skeletal scintigraphy (71%) for the detection of bone marrow metastases, but a lower sensitivity than FDG-PET (90%) [19]. The authors of both studies concluded that WB-MRI can replace bone scintigraphy. One advantage of this approach would be the implementation of a one-stop-shop approach to childhood RMS. A drawback of MRI in young children is, however, the need for general anaesthesia.
Bone scintigraphy
In the current EpSSG protocols, bone scintigraphy is mandatory as part of the work-up in patients with RMS. The finding of an isolated hot spot on the bone scan should be evaluated with conventional radiography or MRI.
Cogswell et al. [20] reported a retrospective series of 40, primarily adult, patients with RMS and found bone metastases in 18%. Bone scintigraphy in their study had a sensitivity of 70% and specificity 95% in the detection of metastatic disease. In contrast to this, in a Dutch study of 109 patients with soft-tissue sarcoma, bone metastases were found in only 8 patients (7%) [21]. However, of these eight patients, six reported bone pain and all had other sites of metastatic disease. The authors conclude that the yield of routine bone scintigraphy is low and that it should be reserved for symptomatic or high-risk patients only.
Positron emission tomography-CT
In PET-CT studies fluorine-18 fluorodeoxyglucose (18F-FDG), a radiolabelled glucose analogue, is used [22]. As 18F-FDG is a glucose analogue, it shows uptake in metabolically active cells, which most malignant tumour cells are. The combination of PET with CT, without moving the relative position of the patient, yields a higher diagnostic accuracy than PET alone (Fig. 8). In general, the CT scan will be low-dose CT scan only meant to identify anatomical structures. However, as the CT scanners in modern PET-CT systems are of high diagnostic quality, it is also possible to combine a diagnostic CT scan, e.g. for the depiction of pulmonary metastases, with a PET scan.
Fig. 8.

A 19-year-old boy with a history of treated metastatic RMS presented with low back pain. The PET-CT image shows intense 18F-FDG uptake in the spinal canal (open arrow). Physiological excretion of the radiopharmaceutical via the kidneys is visible (solid arrows). Histopathology: embryonal RMS
The literature on the use of PET-CT in children with RMS is limited to several case reports or small studies [23, 24]. Although in some cases PET-CT has been shown to be of benefit in individual patients, larger prospective studies are needed.
Staging and follow-up
Staging of RMS is of importance for the individual patient as it gives an indication of prognosis, and thus treatment stratification. From a broader perspective staging makes compiling data on larger patient groups for research purposes possible, enabling evaluation of the outcome of different treatment regimens.
The main staging system is the postsurgical staging system developed by the IRSG. This is currently used by study groups both in the USA and now in Europe also (Table 3). The IRSG was formed in 1972 and consisted of surgeons, pathologists, oncologists, and radiation oncologists. The absence of radiologists is striking, and paediatric radiologists are still infrequently involved in development of paediatric oncology study protocols, although in the EpSSG RMS 2005, paediatric radiologists were involved in the development of the protocol. For staging regional nodes it is important to be familiar with the regional node stations. Lymph node involvement has a negative impact on prognosis, as has been shown in the SIOP Malignant Mesenchymal Tumor 89 trial [25]. Overall 5-year survival was 60% for N1 patients versus 73% in N0 patients (P = 0.03). Distant lymph node involvement upgrades a patient to stage IV disease (Fig. 7), having an adverse impact on prognosis: overall 5-year survival becomes 24% [26].
Table 3.
IRSG classification
| Stage | Characteristics | |
|---|---|---|
| I | Localized disease completely resected (regional nodes not involved) | A: Tumour confined to muscle or organ of origin |
| B: Tumour infiltrating outside organ of (muscle of) origin | ||
| II | Localized or regional disease with total resection of gross tumour | A: Primary tumour grossly resected, with microscopic residual disease (negative findings in local nodes) |
| B: Primary tumour and positive nodes completely resected | ||
| C: Primary tumour and positive nodes resected, with evidence of microscopic residual disease | ||
| III | Incomplete resection of tumour or biopsy, with gross residual disease | |
| IV | Distant metastatic disease present at diagnosis | |
Table 4 lists the regional node stations by primary tumour site. Oval-shape lymph nodes and a short axis <1 cm are considered to be normal [3]. If the node shows peripheral enhancement or is round with a short axis of 1.5–2 cm then the node should be considered probably invaded by tumour. Besides surgical resection or needle-core biopsies, lymph node involvement can also be assessed using fine-needle aspiration (FNA). Klijanienko et al. [27] reported a review of the use of FNA in 180 tumours; 176 (97.8%) were either diagnosed accurately or as round-cell sarcoma.
Table 4.
Regional node stations by primary tumour site. Disease with involvement of other lymph nodes than those specified in the table should be classified as stage IV
| Anatomical site | Node station | |
|---|---|---|
| Extremity | Lower extremity | Inguinal, femoral, popliteal nodes (rare) |
| Upper extremity | Axillary, brachial, epitrochlear, and infraclavicular nodes | |
| Genitourinary | Bladder, prostate, cervix, uterus, paratesticular | Pelvic, retroperitoneal nodes at renal artery level or below |
| Vagina | Retroperitoneal, pelvic nodes at or below common iliac inguinal nodes | |
| Vulva | Inguinal nodes | |
| Thoracic | Intrathoracic | Internal mammary, mediastinal nodes |
| Retroperitoneum/pelvis | Pelvic, retroperitoneal nodes | |
| Trunk | Abdominal wall | Inguinal, femoral nodes |
| Chest wall | Axillary, internal mammary, and infraclavicular nodes | |
| Other | Biliary | Liver hilar nodes |
| Perianal/perineal | Inguinal, pelvic nodes (may be bilateral) | |
Only in patients with intraspinal or suspected meningeal extension (on imaging or clinical assessment) does the EpSSG RMS protocol state that craniospinal MR should also be performed. In the current EpSSG RMS 2005 protocol, risk stratification is based on six criteria that have emerged from the analyses of previous European studies: histology (embryonal vs. alveolar), postsurgical stage (IRSG), tumour site, node stage, tumour size and patient age (Table 1) [28].
During follow-up, tumour size is an important parameter in assessing tumour response. In the current EpSSG protocol complete remission is defined as disappearance of tumour both clinically and on imaging. Minor response is defined as >33% reduction in volume after three courses of chemotherapy; if not reached the patient is eligible for second-line chemotherapy. In recording tumour response the EpSSG uses volumetric evaluation; additionally the presence or absence of a posttherapeutic residue should be mentioned in the radiology report [28].
In studies in adults the use of Response Evaluation Criteria In Solid Tumours (RECIST) has been advocated by the European Organization for Research and Treatment of Cancer (EORTC), National Cancer Institute of Canada Clinical Trials Group and the National Cancer Institute (NCI) of the United States [29]. With respect to implementation in children, RECIST have been a matter of debate [30]. Recently RECIST were retrospectively applied to ten consecutive children with cancer [31]. The authors concluded that tumour size was underestimated and that in disseminated disease many lesions were either calcified or too small to measure and, therefore, that RECIST are not readily applicable in paediatric oncology. Currently the EpSSG has incorporated RECIST to be used alongside the volumetric measurements in their latest protocol in order to prospectively assess the validity of RECIST in a large patient population with a single tumour type.
Tumour relapse in patients most commonly presents with locoregional disease (51%) compared to distant relapse (41%) [32] (Fig. 9). In a retrospective case-based study the use of PET-CT was advocated; this, however, needs to be evaluated in larger prospective studies [33].
Fig. 9.

Two years after initial diagnosis the patient shown in Fig. 6 presented at the outpatient clinic complaining of back pain. a Coronal STIR image of the pelvis shows discrete increased signal intensity in the left ischium (open arrow). b Subsequently acquired PET-CT image confirms the presence of recurrent disease in the same location (open arrow). Note excretion of tracer into the urinary bladder (solid arrow). c PET-CT image also shows a second lesion in the thoracic spine (open arrow). Additional rib and pleural metastases were also visible (not visible on this image)
Tumour locations
Genitourinary
Approximately 25% of all RMS are GU RMS [34, 35]. As mentioned above, GU RMS can simply be subdivided into two subgroups based on different prognosis and subsequent treatment strategy, GU bladder/prostate (GU-BP) being an unfavourable location (Fig. 10). Tumours at other GU non-bladder/prostate (GU-NBP) sites, such as a paratesticular location (testes, epididymis and spermatic cord; Fig. 5), vagina or uterus (Fig. 11), have a favourable prognosis, and thus require less-intensive treatment [36–48]. There is a caveat to MRI of the bladder in RMS: after intravenous contrast medium administration, layering of contrast medium can occur making it difficult to appreciate bladder wall enhancement [49]. T2-W sequences can be particularly useful in this setting to assess bladder wall thickening. Additional cystoscopy is often warranted [50]. At the end of treatment, some residual soft-tissue thickening may persist, and on MRI it is impossible to decide whether this is residual scarring or tumour; in these cases endoscopic biopsy is mandatory.
Fig. 10.
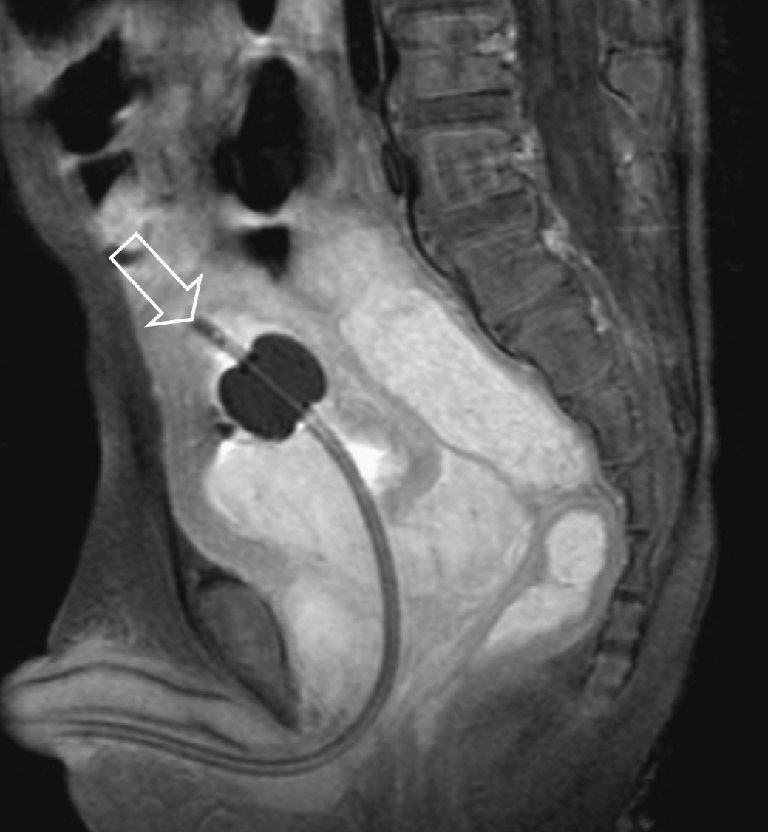
A 3-year-old boy with RMS of the prostate. The sagittal T1-W contrast-enhanced MR image shows the lesion invading the bladder wall. A transurethral catheter has been inserted (open arrow). Histopathology: botryoid RMS
Fig. 11.

A 2-year-old girl presented with a mass in the vagina. a Axial T1-W contrast-enhanced image shows the mass with heterogeneous enhancement. The tumour has both solid (asterisk) and fluid (open arrow) compartments. b Sagittal T2-W MR image shows the mass with mixed signal intensity. The bladder is displaced anteriorly and the uterus cannot be visualized. Histopathology: embryonal RMS
Extremities
RMS of the extremities (Figs. 12 and 13) is almost always of alveolar histology, tends to occur in older children and young adults, is often present with positive regional lymph nodes, and has a propensity to metastasize to unusual sites; these negative prognostic factors contribute to the relatively poor prognosis of RMS in this location [51, 52]. In approximately 12% of patients, nodal involvement is seen on imaging; however, when nodal dissection is performed the rate of nodal involvement increases to almost 50% [53]. This discrepancy between imaging findings and nodal dissection might be reduced by using high-quality state-of-the-art US. In current treatment protocols systematic biopsy of regional nodes is advocated, even if the nodes are not palpable or enlarged on imaging; sentinel node procedures are recommended whenever feasible, although the value of upgrading a patient to a higher risk group based merely on a positive sentinel node has not been studied yet. As in all tumours of the extremities, preoperative imaging plays a vital role in the depiction of the relationship between neurovascular bundles and the tumour.
Fig. 12.

A 4-year-old girl presenting with a mass in the left lower leg. a Axial T1-W contrast-enhanced MR image shows an ill-defined mass circumferential to the fibula. Note the cortical thinning (open arrow) of the fibula. b Sagittal PD-weighted image shows diffuse bone marrow metastases. Histopathology: embryonal RMS
Fig. 13.

A 2-month-old boy with a mass in the third ray of the left foot. T1-W MR image shows a discrete lesion (open arrow) of intermediate signal intensity. Histopathology: embryonal RMS
Other
Chest wall
Chest wall RMS (Fig. 14) is a relatively rare finding with a reported incidence of 3.7% in the IRSG II and IRSG III studies [54]. Most of the reports of chest wall involvement are either case reports or small series [55–57]. In a retrospective analysis of 15 patients, Saenz et al. [57] found a 5-year survival of 67%.
Fig. 14.

A 4-year-old boy presenting with a mass on the right chest wall. a US image shows a heterogeneous mass in the pectoralis major muscle (the pectoralis minor is not involved; asterisk). b T1-W MR image of the chest shows a mass of intermediate signal intensity. c After intravenous contrast medium administration the lesion shows homogeneous enhancement. Histopathology: alveolar RMS
Pulmonary
There is an ongoing debate and controversy whether congenital cystic anomalies predispose children to intralesional development of RMS [58–62]. It has been reported that pleuropulmonary blastoma (PPB) has been mistaken for or classified as RMS arising in congenital cystic adenomatoid malformation (CCAM) on a number of occasions [63]. Despite the fact that the exact incidence in CCAM is unknown, it has prompted paediatric surgeons to resect even small pulmonary cystic lesions (Fig. 15) [64, 65].
Fig. 15.

A 3-year-old boy with dyspnoea. Chest CT image shows displacement of the trachea (open arrow) and oesophagus (solid arrow) to the right due to a large mass (asterisk) with accompanying pleural effusion. Histopathology: embryonal RMS
Biliary tree
RMS is the most common tumour of the biliary tree in childhood, although it only accounts for approximately 0.04% of all childhood tumours [66]. The initial diagnosis will, in most patients, be made on US, which may show a solid or cystic mass situated in the liver hilum, and intrahepatic bile duct dilatation (Fig. 16). MRI is mandatory for presurgical evaluation, where magnetic resonance cholangiopancreatography (MRCP) can depict the biliary tree (Fig. 16). In many patients, however, endoscopic retrograde cholangiopancreatography (ERCP) will have to be performed in order to depict intraductal irregularities (Fig. 16). Biliary tree RMS is a tumour that does not necessarily need to be fully resected in order to achieve long-term survival, as long as adequate radiotherapy is added [67]. Intraperitoneal metastases, which can also be found on follow-up, should also be born in mind.
Fig. 16.

An 8-year-old boy presented with abdominal pain and jaundice. a US image shows a central process in the liver hilum (open arrow) and dilatation of the intrahepatic bile ducts (solid arrow). b T2-W MR image shows a circumscribed lesion with increased signal intensity (open arrow). c MRCP image shows intrahepatic bile duct dilatation. Note that the right and left duct systems do not communicate (open arrow). d ERCP image (ERCP performed in order to insert a stent in the common bile duct). Histopathology: embryonal RMS
Other locations
In extremely rare instances RMS can be found in other organs such as the heart, the diaphragm (Fig. 17), the omentum, the urachus and the digestive tract [68–76].
Fig. 17.

A 4-year-old boy was shown to have a right-sided pleural effusion on a chest radiograph. Balanced FFE sagittal MR image shows a mass (open arrow) arising from the diaphragm (courtesy of S.G.F. Robben, Academic Hospital Maastricht, The Netherlands)
Congenital
RMS has been reported to occur as a congenital tumour (Fig. 18) [77–81]. In congenital alveolar RMS the prognosis is reported to be extremely poor, despite otherwise adequate treatment [79]. Orbach et al. [82] reported the SIOP data on soft-tissue sarcoma in the first year of life. In their study population of 16 newborns, with a follow-up of 1.8–10.0 years, 3 out of 5 newborns with RMS survived. It has been noted that in congenital RMS the disease may be metastatic at the time of birth, with metastases described in a number of organs and in the placenta [81].
Fig. 18.

A 4-day-old girl born with a lump on the left foot. Antenatal ultrasonography at 20 weeks showed no abnormalities. a T1-W MR image shows a large inhomogeneous mass arising from the left foot. b Abdominal US image shows popliteal and inguinal nodal invasion, and hepatic and pancreatic metastases (open arrow). Due to the poor prognosis, no therapy was given, and the child died several weeks later. Histopathology: poorly differentiated soft-tissue sarcoma without distinct translocations
Adult patients
Every once in a while paediatric radiologists and paediatric oncologists receive a request for help in the management of an adult patient (Fig. 19). RMS, although seen as a soft-tissue tumour of childhood, can also occur later in life [83–86]. Compared to childhood RMS, adult RMS has a poor outcome. In a large retrospective study of 171 patients 5-year overall survival was only 40% [85]. However, the patients in this series treated according to the guidelines for treatment of childhood RMS showed survival figures comparable to those seen in children. This suggests that treatment of adult RMS should be based on paediatric protocols tailored to adults, to increase survival in this age group. In the Academic Medical Centre Amsterdam we have a working group on childhood tumours in (often young) adults that specifically deals with this challenging population. This working group consists of medical oncologists, paediatric oncologists, radiation oncologists, (orthopaedic) surgeons and a paediatric radiologist. Imaging features will in general not be of help, as the pretest likelihood of RMS in an adolescent or adult is extremely low.
Fig. 19.
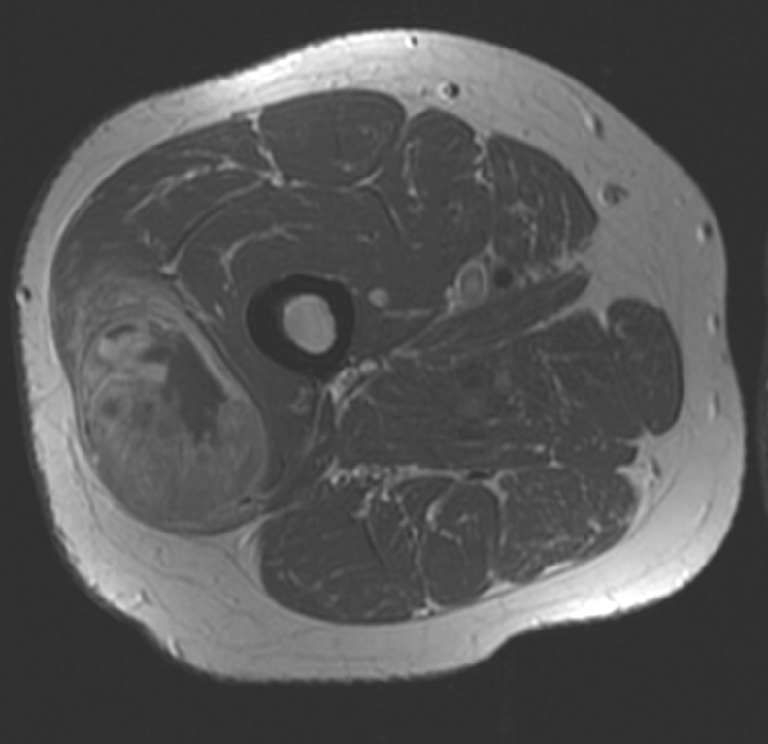
A 45-year-old man with a mass in the thigh. T1-W contrast-enhanced MR image shows a heterogeneous circumscribed mass in the vastus lateralis muscle of the right leg. Histopathology: alveolar RMS
Differential diagnosis
Given the wide variety of locations in which RMS can be found it is difficult to give a concise list of differential diagnoses. The site of the primary lesion determines the differential diagnosis. Keeping location out of the equation there are, however, certain tumours, such as haemangiomas/vascular malformations (Fig. 20), adult-type soft-tissue sarcomas, peripheral neuroectodermal tumours (PNET), infantile fibrosarcoma, aggressive fibromatosis, desmoplastic small round-cell tumours and rhabdoid tumours, and other more even rarer soft-tissue tumours such as nonosseous Ewing sarcoma (Fig. 21), that should be kept in mind when performing US or reading CT or MRI studies of soft-tissue tumours in childhood.
Fig. 20.

A 1-year-old girl with a mass on the left buttock. a Duplex US image shows a highly vascularized, well-delineated heterogeneous mass that was initially thought be a haemangioma. b Coronal STIR image shows a circumscribed solid lesion that invades the pelvis via the greater sciatic foramen (open arrow). c After initial resection, with incomplete margins, tumour recurrence was seen. MR image 2.6 years after initial diagnosis shows progression of disease extending to the abdominal wall (open arrow). Histopathology: alveolar RMS
Fig. 21.
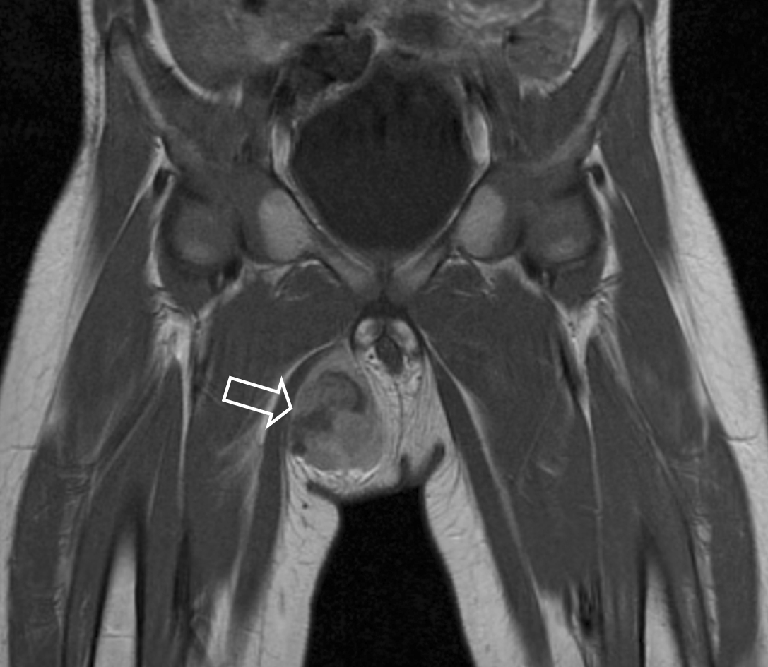
A 22-month-old boy with a mass in the right groin. MRI shows a heterogeneous lesion adjacent to the gracilis muscle (open arrow). Histopathology: extraosseous Ewing sarcoma
Treatment and prognosis
Treatment of RMS requires a multidisciplinary approach, where chemotherapy, surgery and radiotherapy (RT) each has its own specific role.
Chemotherapy
Before the introduction of chemotherapy only 25% of patients with RMS survived, despite adequate local therapy. This indirectly indicates that the vast majority of patients had at least minimal disseminated disease at diagnosis, whereas, with state-of-the-art imaging techniques and bone marrow investigations, only 15% of patients with RMS present with overt metastatic disease. Besides treating minimal disseminated disease, chemotherapy enables local therapy to be more conservative. In most patients, surgery at diagnosis consists of biopsy only. Chemotherapy is given to all patients; it reduces tumour size and extension and often allows a delay in surgery. The tumour is more likely to be completely resected and mutilating surgery avoided. Most international protocols use vincristine and dactinomycin as standard drugs. The choice of alkylating agent differs between Europe (ifosfamide) and North America (cyclophosphamide). These drugs have proved to be equally effective, but differences in toxicity exist: ifosfamide is more nephrotoxic, whereas cyclophosphamide more gonadotoxic [87]. The addition of other antineoplastic agents to vincristine, dactinomycin and alkylators in the treatment of RMS has been investigated, but no significant improvement in outcome has been found [88].
Surgery
In children surgery often starts during the diagnostic phase with biopsy for histological studies, although here interventional radiology plays an increasing role. Excisional biopsy is not advocated except for paratesticular tumours. Most patients end up with postsurgical stage IRS group III. Surgery is generally delayed until after tumour reduction by chemotherapy. The surgical treatment of RMS is site-specific, but the current paradigm is complete wide excision of the primary tumour with a margin of uninvolved tissue whenever possible. Debulking and mutilating procedures should be avoided.
Radiotherapy
The philosophies underlying the treatment strategies in North America and Western Europe have differed in the past. North American protocols involved aggressive surgery and routine RT, except for those tumours that were microscopically radically resected at diagnosis, followed by prolonged chemotherapy regimens for up to 2 years. The SIOP-MMT (International Society for Paediatric Oncology–Malignant Mesenchymal Tumour) group advocated the use of chemotherapy and surgery in order to achieve complete remission in as many patients as possible in order to avoid RT in these often very young patients. RT often has devastating effects in growing children, potentially leading to significant cosmetic and functional problems. Radiation tolerance of growing bone is ≤20 Gy, but the radiation dose from the treatment of RMS ranges from 36 to 50.4 Gy. For certain sites this policy worked out well (e.g. orbital RMS), and in many patients RT could be avoided [89]. At most other sites, relapse rates were high, and salvage rates after relapse were low. The treatment protocols in Germany (Co-operative Weichteilsarcom Studie, CWS) and Italy (Italian Cooperative Soft Tissue sarcoma Group, ICG) were between that in North America and that of the SIOP-MMT group. In 2005, the former SIOP-MMT, CWS and ICG joined to form the EpSSG. As a result of extensive cooperation between the European and the North American groups, the similarities in treatment strategies now outnumber the differences [90]. Although RT is still deemed essential in many patients to achieve cure, the possibility of radiation-induced second malignant neoplasms must be kept in mind [91, 92].
European approach
Today more than 70% of nonmetastatic RMS are cured, but survivors may suffer from sequelae [93–98]. Therefore, the SIOP-MMT group has tried in their RMS75, MMT84, MMT89 and MMT95 studies to avoid mutilating surgery and RT [93, 99, 100]. Only children ≥3 years of age with parameningeal RMS at high risk of meningeal extension and children not achieving complete remission after chemotherapy and surgery were irradiated after intensive chemotherapy. Of all survivors, 49% were treated without significant local therapy [93]. Locoregional relapse occurred in 34%. This has lead to an event-free survival (EFS) of 57% and an overall survival (OS) of 71% [93]. Alveolar histology was associated with a significantly higher risk of relapse and a much higher risk of metastases.
North American approach
In the studies performed by the IRSG the need for RT is based on surgical radicality, localization, and size of the primary tumour. Only completely resected tumours (IRS group I) with a favourable location (stage I; Table 5), and small tumours (≤5 cm) at unfavourable locations (stage II) did not receive RT in the IRS-IV study [87]. Grossly resected tumours with microscopic residual disease (IRS group II), and incompletely resected tumours or tumours with gross residual disease after biopsy (IRS group III) were irradiated [87, 88, 101, 102]. With this approach in the IRS-IV study an EFS of 78% and an OS of 84% were obtained. Between the IRSG and SIOP-MMT group a significant difference in OS in alveolar RMS was seen (71% vs. 38%) [90]. The salvage rate after relapse of an alveolar RMS was low, and therefore in the current European trial, RT is mandatory for all patients with alveolar histology.
Table 5.
Pediatric Oncology Group and Children’s Cancer Group for the Study of Rhabdomyosarcoma classification [28]
| Stage | Site | Primary tumoura | Sizeb | Regional nodesc | Distant metastasisd |
|---|---|---|---|---|---|
| I | Orbit | T1 or T2 | a or b | N0, N1 or NX | M0 |
| Head and neck (excluding parameningeal) | |||||
| Genitourinary, nonbladder nonprostate | |||||
| II | Bladder and prostate | T1 or T2 | a | N0 or NX | M0 |
| Extremity | |||||
| Cranial parameningeal | |||||
| Other (including trunk, retroperitoneum, etc) | |||||
| III | Bladder and prostate | T1 or T2 | a | N1 | M0 |
| Extremity | |||||
| Cranial parameningeal | T1 or T2 | b | N0, N1 or NX | M0 | |
| Other (including trunk, retroperitoneum, etc) | |||||
| IV | All sites | T1 or T2 | a or b | N0 or N1 | M1 |
aT1 tumour limited to original muscle or organ, T2 tumour has extension or fixation to the surrounding tissue.
ba tumour equal to or less then 5 cm in greatest dimension, b tumour larger than 5 cm in greatest dimension.
cN0 no clinical involvement of regional nodes, N1 clinical involvement of regional nodes, NX status of regional nodes unknown.
dM0 no distant metastasis, M1 distant metastasis.
As described above, European and American approaches, although historically different, have converged based on the results of successive international trials and extensive cooperation between the European and North American groups [90]. As approaches now are very similar, in this review the current EpSSG approach for the different clinical groups is discussed.
Current EpSSG approach
IRS group I In the IRS-I study, a randomized trial in clinical IRS group I patients, no difference was seen in survival between patients treated with chemotherapy and those treated with chemotherapy and RT [101]. A subsequent retrospective study based on the subsequent IRS-I to IRS-III trials confirmed that for embryonal RMS although there was a small difference in failure-free survival (FFS) with and without RT, OS was not significantly different [103]. On the other hand, for alveolar RMS and undifferentiated RMS, 10-year FFS was 73% vs. 44%, and OS 82% vs. 52% for patients treated with and without RT, respectively [103]. Therefore the current EpSSG study recommends RT in non-embryonal RMS only.
IRS group II Evaluation of the role of RT in IRS group II patients in subsequent Cooperative Soft Tissue Sarcoma studies (CWS 81, 86, 91 and 96) showed that for embryonal RMS, EFS was significantly different for patients treated with and without RT [104]. However, OS at 5 years was not significantly different (84% vs. 77%). For patients with tumours of unfavourable histology (independent of site and size), EFS and OS were significantly better when RT was part of the treatment [104]. Therefore, in IRS group II patients, RT is recommended. It is compulsory in patients with high-risk features, but may be omitted in patients with favourable histology in whom RT may be considered too toxic when considering patient age and site of the tumour.
IRS group III In patients with IRS group III tumours, RT is the only available therapy in patients who cannot receive a secondary complete resection. Patients who do receive a delayed complete resection benefit from additional RT. In the CWS trials 81–96 5-year EFS was 77% and 58% for those treated with and without additional RT, respectively [28]. OS, however, was not significantly different between the two groups: 84% and 79%, respectively. RT is, therefore, usually indicated except in patients with a favourable site and histology.
Prognosis
As malignant tumours in childhood are rare diseases, most children with cancer have been included in international treatment protocols. This way survival for localized disease (85% of patients) has improved from 25% in the early 1970s to 75% in the most recent international trials [101, 105–107]. Although results for localized RMS have improved dramatically in the past decades, patients with disseminated disease still have a dismal prognosis, with a 5-year OS of 24% [26].
A way to reduce long-term sequelae of RT may be by using brachytherapy, especially in girls with genital tract RMS and patients with bladder-prostate, extremity, and head and neck RMS [100, 108–111]. Other options are the use of modulated RT (IMRT) and proton therapy [112, 113].
The postoperative patient
Image interpretation and management of the patient after surgery and often RT are challenges (Fig. 22). Most important is proper knowledge of the surgical procedure and/or the radiation field. The following postoperative changes can be encountered in children treated for RMS:
Haematoma
Oedema
Soft-tissue infection/abscess
Calcification
Foreign bodies
Muscle flaps/fat pads
Distorted anatomy
Radiation effect
Figure 23 shows a possible decision tree to manage postoperative findings on follow-up imaging.
Fig. 22.
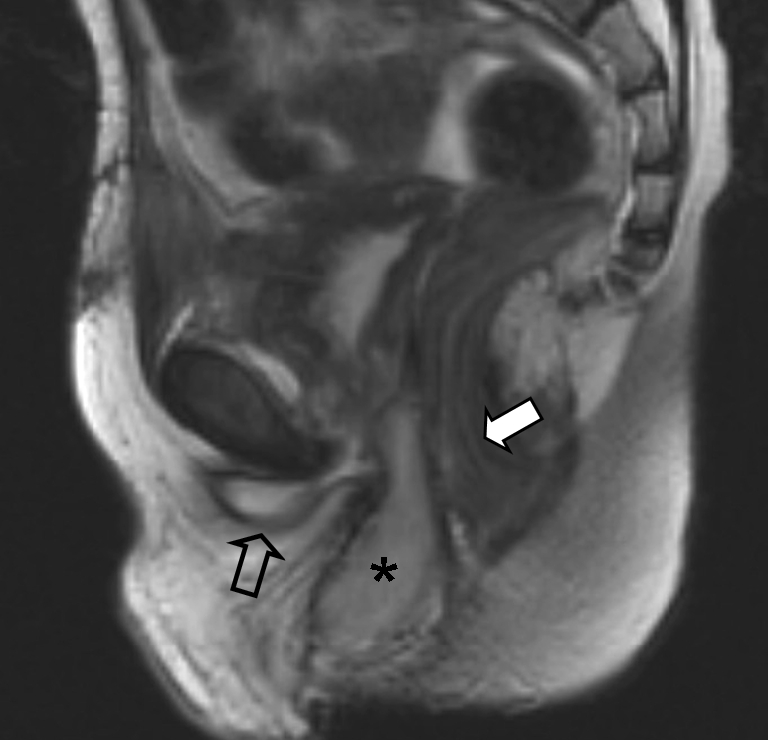
A 6-year-old boy with a history of treated bladder RMS. At surgical resection the urethra was damaged leading to a persistent urinoma in, after RT, nonvital tissue. MRI image after treatment shows a mass (asterisk) between the urethra (open arrow) and the rectum (solid arrow). This mass is a vascularized gracilis muscle flap used to repair the defect. Without proper knowledge of the surgical history of the patient, this might have been interpreted as tumour recurrence. Histopathology: embryonal RMS
Fig. 23.

Flow chart for posttreatment lesions found on MRI. SI signal intensity, CE contrast-enhanced [114]
Conclusion
In this review we have discussed the findings of RMS outside the craniofacial region. The treatment of RMS requires a multidisciplinary approach, in which paediatric oncologists, radiologists, paediatric surgeons, pathologists and radiation oncologists all play a vital role. Although they are the most common soft-tissue tumour of childhood, these still rare tumours should be evaluated and treated in specialized centres.
Acknowledgments
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Miller RW, Young JL Jr, Novakovic B (1995) Childhood cancer. Cancer 75(1 Suppl):395–405 [DOI] [PubMed]
- 2.Stiller CA, Parking DM (1994) International variations in the incidence of childhood soft-tissue sarcomas. Paediatr Perinat Epidemiol 8:107–119 [DOI] [PubMed]
- 3.Brisse H, McHugh K, Scaramuzza D (2005) RMS and non-RMS soft tissue sarcomas. Radiological guidelines. In: EpSSG RMS and NRSTS therapeutic protocols
- 4.Weiss SW, Goldblum JR (2001) Enzinger and Weiss’s soft tissue tumors. Mosby, St. Louis, pp 785–836
- 5.Fletcher CD, Unni KK, Mertens F (2002) WHO classification of tumours: pathology and genetics of tumours of soft tissue and bone. IARC Press, Lyon
- 6.Cavazzana AQ, Schmidt D, Nifo V et al (1992) Spindle cell rhabdomyosarcoma. A prognostic favourable variant of rhabdomyosarcoma. Am J Surg Pathol 16:229–235 [DOI] [PubMed]
- 7.Leuschner I, Newton WA Jr, Schmidt D et al (1993) Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol 17:221–230 [DOI] [PubMed]
- 8.Sorensen PH, Lynch JC, Qualman SJ et al (2002) PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol 20:2672–2679 [DOI] [PubMed]
- 9.Kodet R, Newton WA Jr, Hamoudi AB et al (1991) Rhabdomyosarcomas with intermediate-filament inclusions and features of rhabdoid tumors. Light microscopic and immunohistochemical study. Am J Surg Pathol 15:257–267 [DOI] [PubMed]
- 10.Kodet R, Newton WA Jr, Hamoudi AB et al (1993) Childhood rhabdomyosarcoma with anaplastic (pleomorphic) features. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol 17:443–453 [DOI] [PubMed]
- 11.Folpe AL, McKenney JK, Bridge JA et al (2002) Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol 26:1175–1183 [DOI] [PubMed]
- 12.Newton WA Jr, Gehan EA, Webber BL et al (1995) Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification – an Intergroup Rhabdomyosarcoma study. Cancer 76:1073–1085 [DOI] [PubMed]
- 13.Sebire NJ, Roebuck DJ (2006) Pathological diagnosis of paediatric tumours from image-guided needle core biopsies: a systematic review. Pediatr Radiol 36:426–431 [DOI] [PubMed]
- 14.Paterson A, Frush DP, Donnelly L (2001) Helical CT of the body: are settings adjusted for pediatric patients? AJR 176:297–301 [DOI] [PubMed]
- 15.Brenner DJ, Elliston CD, Hall EJ et al (2001) Estimated risks of radiation induced fatal cancer from pediatric CT. AJR 176:289–296 [DOI] [PubMed]
- 16.Kim EE, Valenzuela RF, Kumar AJ et al (2000) Imaging and clinical spectrum of rhabdomyosarcoma in children. Clin Imaging 24:257–262 [DOI] [PubMed]
- 17.Enneking WF, Spanier SS, Goodman MA (2003) The classic – a system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res 415:4–18 [DOI] [PubMed]
- 18.Goo HW, Choi SH, Ghim T et al (2005) Whole-body MRI of paediatric malignant tumours: comparison with conventional oncological imaging methods. Pediatr Radiol 35:766–773 [DOI] [PubMed]
- 19.Drup-Link HE, Franzius C, Link TM et al (2001) Whole-body MR imaging for detection of bone metastases in children and young adults: comparison with skeletal scintigraphy and FDG PET. AJR 177:229–236 [DOI] [PubMed]
- 20.Cogswell A, Howman-Giles R, Bergin M (1994) Bone and gallium scintigraphy in children with rhabdomyosarcoma: a 10-year review. Med Pediatr Oncol 22:15–21 [DOI] [PubMed]
- 21.Jager PL, Hoekstra HJ, Leeuw J et al (2000) Routine bone scintigraphy in primary staging of soft tissue sarcoma; is it worthwhile? Cancer 89:1726–1731 [DOI] [PubMed]
- 22.Jadvar H, Connolly LP, Fahey FH (2007) PET and PET/CT in pediatric oncology. Semin Nucl Med 37:316–331 [DOI] [PubMed]
- 23.Ben Arush MW, Bar SR, Postovsky S et al (2006) Assessing the use of FDG-PET in the detection of regional and metastatic nodes in alveolar rhabdomyosarcoma of extremities. J Pediatr Hematol Oncol 28:440–445 [DOI] [PubMed]
- 24.McCarville MB, Christie R, Daw NC et al (2005) PET/CT in the evaluation of childhood sarcomas. AJR 184:1293–1304 [DOI] [PubMed]
- 25.Stevens MC, Rey A, Bouvet N et al (2005) Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology – SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol 23:2618–2628 [DOI] [PubMed]
- 26.Carli M, Colombatti R, Oberlin O et al (2004) European intergroup studies (MMT4-89 and MMT4-91) on childhood metastatic rhabdomyosarcoma: final results and analysis of prognostic factors. J Clin Oncol 22:4787–4794 [DOI] [PubMed]
- 27.Klijanienko J, Caillaud JM, Orbach D et al (2007) Cyto-histological correlations in primary, recurrent and metastatic rhabdomyosarcoma: the Institut Curie’s experience. Diagn Cytopathol 35:482–487 [DOI] [PubMed]
- 28.Bisogno G, Bergeron C, Jenney M et al (2005) EpSSG RMS 2005; a protocol for non metastatic rhabdomyosarcoma. 1 June 2005, Padova
- 29.Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216 [DOI] [PubMed]
- 30.McHugh K, Kao S (2003) Can paediatric radiologists resist RECIST (response evaluation criteria in solid tumours)? Pediatr Radiol 33:739–743 [DOI] [PubMed]
- 31.Barnacle AM, McHugh K (2006) Limitations with the response evaluation criteria in solid tumors (RECIST) guidance in disseminated pediatric malignancy. Pediatr Blood Cancer 46:127–134 [DOI] [PubMed]
- 32.Pappo AS, Anderson JR, Crist WM et al (1999) Survival after relapse in children and adolescents with rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 17:3487–3493 [DOI] [PubMed]
- 33.Peng F, Rabkin G, Muzik O (2006) Use of 2-deoxy-2-[F-18]-fluoro-D-glucose positron emission tomography to monitor therapeutic response by rhabdomyosarcoma in children: report of a retrospective case study. Clin Nucl Med 31:394–397 [DOI] [PubMed]
- 34.Castellino SM, McLean TW (2007) Pediatric genitourinary tumors. Curr Opin Oncol 19:248–253 [DOI] [PubMed]
- 35.Agrons GA, Wagner BJ, Lonergan GJ et al (1997) From the archives of the AFIP. Genitourinary rhabdomyosarcoma in children: radiologic-pathologic correlation. Radiographics 17:919–937 [DOI] [PubMed]
- 36.Mak CW, Chou CK, Su CC et al (2004) Ultrasound diagnosis of paratesticular rhabdomyosarcoma. Br J Radiol 77:250–252 [DOI] [PubMed]
- 37.Sanghvi DA, Purandare NC, Jambhekar NA et al (2004) Primary rhabdomyosarcoma of the seminal vesicle. Br J Radiol 77:159–160 [DOI] [PubMed]
- 38.Moroni M, Nesi G, Travaglini F et al (2003) Rhabdomyosarcoma of the spermatic cord. A case report with review of the literature. Urol Int 71:114–117 [DOI] [PubMed]
- 39.Ciftci AO, Bingol-Kologlu M, Senocak ME et al (2001) Testicular tumors in children. J Pediatr Surg 36:1796–1801 [DOI] [PubMed]
- 40.Solomon LA, Zurawin RK, Edwards CL (2003) Vaginoscopic resection for rhabdomyosarcoma of the vagina: a case report and review of the literature. J Pediatr Adolesc Gynecol 16:139–142 [DOI] [PubMed]
- 41.Martelli H, Oberlin O, Rey A et al (1999) Conservative treatment for girls with nonmetastatic rhabdomyosarcoma of the genital tract: a report from the Study Committee of the International Society of Pediatric Oncology. J Clin Oncol 17:2117–2122 [DOI] [PubMed]
- 42.Arul GS, Porter H, Spicer RD (2000) Nonrhabdomyosarcomatous botyroid tumour of the cervix uteri: is it a new entity? Med Pediatr Oncol 35:147–149 [DOI] [PubMed]
- 43.Lowry DL, Guido RS (2000) The vulvar mass in the prepubertal child. J Pediatr Adolesc Gynecol 13:75–78 [DOI] [PubMed]
- 44.Bond SJ, Seibel N, Kapur S et al (1994) Rhabdomyosarcoma of the clitoris. Cancer 73:1984–1986 [DOI] [PubMed]
- 45.Chan YF, Leung CS, Ma L (1989) Primary embryonal rhabdomyosarcoma of the ovary in a 4-year-old girl. Histopathology 15:309–311 [PubMed]
- 46.Montag TW, D’ablaing G, Schlaerth JB et al (1986) Embryonal rhabdomyosarcoma of the uterine corpus and cervix. Gynecol Oncol 25:171–194 [DOI] [PubMed]
- 47.Hays DM, Raney RB Jr, Lawrence W Jr et al (1981) Rhabdomyosarcoma of the female urogenital tract. J Pediatr Surg 16:828–834 [DOI] [PubMed]
- 48.Ghushe ND, Drugas GT (2007) Rhabdomyosarcoma of the clitoris. Pediatr Radiol 37:1179 [DOI] [PubMed]
- 49.McHugh K, Boothroyd AE (1999) The role of radiology in childhood rhabdomyosarcoma. Clin Radiol 54:2–10 [DOI] [PubMed]
- 50.Heyn R, Newton WA, Raney RB et al (1997) Preservation of the bladder in patients with rhabdomyosarcoma. J Clin Oncol 15:69–75 [DOI] [PubMed]
- 51.Miller DV, Coffin CM, Zhou H (2004) Rhabdomyosarcoma arising in the hand or foot: a clinicopathologic analysis. Pediatr Dev Pathol 7:361–369 [DOI] [PubMed]
- 52.Rao BN, Rodriguez-Galindo C (2003) Local control in childhood extremity sarcomas: salvaging limbs and sparing function. Med Pediatr Oncol 41:584–587 [DOI] [PubMed]
- 53.Breneman JC, Wiener ES (2000) Issues in the local control of rhabdomyosarcoma. Med Pediatr Oncol 35:104–109 [DOI] [PubMed]
- 54.Andrassy RJ, Wiener ES, Raney RB et al (1998) Thoracic sarcomas in children. Ann Surg 227:170–173 [DOI] [PMC free article] [PubMed]
- 55.Soyer T, Karnak I, Ciftci AO et al (2006) The results of surgical treatment of chest wall tumors in childhood. Pediatr Surg Int 22:135–139 [DOI] [PubMed]
- 56.Watt AJ (2002) Chest wall lesions. Paediatr Respir Rev 3:328–338 [PubMed]
- 57.Saenz NC, Ghavimi F, Gerald W et al (1997) Chest wall rhabdomyosarcoma. Cancer 80:1513–1517 [DOI] [PubMed]
- 58.Doladzas T, Arvelakis A, Karavokyros IG et al (2005) Primary rhabdomyosarcoma of the lung arising over cystic pulmonary adenomatoid malformation. Pediatr Hematol Oncol 22:525–529 [DOI] [PubMed]
- 59.Ozcan C, Celik A, Ural Z et al (2001) Primary pulmonary rhabdomyosarcoma arising within cystic adenomatoid malformation: a case report and review of the literature. J Pediatr Surg 36:1062–1065 [DOI] [PubMed]
- 60.West D, Nicholson AG, Colquhoun I et al (2007) Bronchioloalveolar carcinoma in congenital cystic adenomatoid malformation of lung. Ann Thorac Surg 83:687–689 [DOI] [PubMed]
- 61.Pai S, Eng HL, Lee SY et al (2007) Correction: pleuropulmonary blastoma, not rhabdomyosarcoma in a congenital lung cyst. Pediatr Blood Cancer 48:370–371 [DOI] [PubMed]
- 62.d’Agostino S, Bonoldi E, Dante S et al (1997) Embryonal rhabdomyosarcoma of the lung arising in cystic adenomatoid malformation: case report and review of the literature. J Pediatr Surg 32:1381–1383 [DOI] [PubMed]
- 63.Priest JR, Hill AD, Williams GM (2006) Type I pleuropulmonary blastoma: a report from the international pleuropulmonary blastoma registry. J Clin Oncol 24:4492–4498 [DOI] [PubMed]
- 64.Granata C, Gambini C, Balducci T et al (1998) Bronchioloalveolar carcinoma arising in congenital cystic adenomatoid malformation in a child: a case report and review on malignancies originating in congenital cystic adenomatoid malformation. Pediatr Pulmonol 25:62–66 [DOI] [PubMed]
- 65.Sauvat F, Michel JL, Benachi A et al (2003) Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg 38:548–552 [DOI] [PubMed]
- 66.Roebuck DJ, Yang WT, Lam WW et al (1998) Hepatobiliary rhabdomyosarcoma in children: diagnostic radiology. Pediatr Radiol 28:101–108 [DOI] [PubMed]
- 67.Spunt SL, Lobe TE, Pappo AS et al (2000) Aggressive surgery is unwarranted for biliary tract rhabdomyosarcoma. J Pediatr Surg 35:309–316 [DOI] [PubMed]
- 68.Hill DA, Dehner LP, Gow KW et al (2002) Perianal rhabdomyosarcoma presenting as a perirectal abscess: a report of 11 cases. J Pediatr Surg 37:576–581 [DOI] [PubMed]
- 69.Caty MG, Oldham KT, Prochownik EV (1990) Embryonal rhabdomyosarcoma of the ampulla of Vater with long-term survival following pancreaticoduodenectomy. J Pediatr Surg 25:1256–1258 [DOI] [PubMed]
- 70.Chung CJ, Fordham L, Little S et al (1998) Intraperitoneal rhabdomyosarcoma in children: incidence and imaging characteristics on CT. AJR 170:1385–1387 [DOI] [PubMed]
- 71.Odim J, Reehal V, Laks H et al (2003) Surgical pathology of cardiac tumors. Two decades at an urban institution. Cardiovasc Pathol 12:267–270 [DOI] [PubMed]
- 72.Aksoylar S, Kansoy S, Bakiler AR et al (2002) Primary cardiac rhabdomyosarcoma. Med Pediatr Oncol 38:146 [DOI] [PubMed]
- 73.Raney RB, Anderson JR, Andrassy RJ et al (2000) Soft-tissue sarcomas of the diaphragm: a report from the Intergroup Rhabdomyosarcoma Study Group from 1972 to 1997. J Pediatr Hematol Oncol 22:510–514 [DOI] [PubMed]
- 74.Gupta AK, Mitra DK, Berry M (1999) Primary embryonal rhabdomyosarcoma of the diaphragm in a child: case report. Pediatr Radiol 29:823–825 [DOI] [PubMed]
- 75.Medeiros CW, Kondo W, Baptista I Jr et al (2002) Primary rhabdomyosarcoma of the diaphragm: case report and literature review. Rev Hosp Clin Fac Med Sao Paulo 57:67–72 [DOI] [PubMed]
- 76.Yokoyama S, Hayashida Y, Nagahama J et al (1997) Rhabdomyosarcoma of the urachus. A case report. Acta Cytol 41(4 Suppl):1293–1298 [DOI] [PubMed]
- 77.Brecher AR, Reyes-Mugica M, Kamino H et al (2003) Congenital primary cutaneous rhabdomyosarcoma in a neonate. Pediatr Dermatol 20:335–338 [DOI] [PubMed]
- 78.Matsunaga GS, Shanberg AM, Rajpoot D (2003) Prenatal ultrasonographic detection of bladder rhabdomyosarcoma. J Urol 169:1495–1496 [DOI] [PubMed]
- 79.Grundy R, Anderson J, Gaze M et al (2001) Congenital alveolar rhabdomyosarcoma: clinical and molecular distinction from alveolar rhabdomyosarcoma in older children. Cancer 91:606–612 [DOI] [PubMed]
- 80.Ahmed OA, Hussain A, King DJ et al (1999) Congenital rhabdomyosarcoma. Br J Plast Surg 52:304–307 [DOI] [PubMed]
- 81.White FV, Dehner LP, Belchis DA et al (1999) Congenital disseminated malignant rhabdoid tumor: a distinct clinicopathologic entity demonstrating abnormalities of chromosome 22q11. Am J Surg Pathol 23:249–256 [DOI] [PubMed]
- 82.Orbach D, Rey A, Oberlin O et al (2005) Soft tissue sarcoma or malignant mesenchymal tumors in the first year of life: experience of the International Society of Pediatric Oncology (SIOP) Malignant Mesenchymal Tumor Committee. J Clin Oncol 23:4363–4371 [DOI] [PubMed]
- 83.Hulse N, Raja S, Kumar A et al (2006) Rhabdomyosarcoma of the extremities in adults. Acta Orthop Belg 72:199–203 [PubMed]
- 84.Parham DM, Ellison DA (2006) Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med 130:1454–1465 [DOI] [PubMed]
- 85.Ferrari A, Dileo P, Casanova M et al (2003) Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 98:571–580 [DOI] [PubMed]
- 86.Allen SD, Moskovic EC, Fisher C et al (2007) Adult rhabdomyosarcoma: cross-sectional imaging findings including histopathologic correlation. AJR 189:371–377 [DOI] [PubMed]
- 87.Crist WM, Anderson JR, Meza JL et al (2001) Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol 19:3091–3102 [DOI] [PubMed]
- 88.Crist W, Gehan EA, Ragab AH et al (1995) The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol 13:610–630 [DOI] [PubMed]
- 89.Oberlin O, Rey A, Anderson J et al (2001) Treatment of orbital rhabdomyosarcoma: survival and late effects of treatment – results of an international workshop. J Clin Oncol 19:197–204 [DOI] [PubMed]
- 90.Donaldson SS, Anderson JR (2005) Rhabdomyosarcoma: many similarities, a few philosophical differences. J Clin Oncol 23:2586–2587 [DOI] [PubMed]
- 91.Scaradavou A, Heller G, Sklar CA et al (1995) Second malignant neoplasms in long-term survivors of childhood rhabdomyosarcoma. Cancer 76:1860–1867 [DOI] [PubMed]
- 92.Kalapurakal JA, Thomas PR (1997) Pediatric radiotherapy. An overview. Radiol Clin North Am 35:1265–1280 [PubMed]
- 93.Stevens MC (2005) Treatment for childhood rhabdomyosarcoma: the cost of cure. Lancet Oncol 6:77–84 [DOI] [PubMed]
- 94.Paulino AC (2004) Late effects of radiotherapy for pediatric extremity sarcomas. Int J Radiat Oncol Biol Phys 60:265–274 [DOI] [PubMed]
- 95.Spunt SL, Sweeney TA, Hudxon MM et al (2005) Late effects of pelvic rhabdomyosarcoma and its treatment in female survivors. J Clin Oncol 23:7143–7151 [DOI] [PubMed]
- 96.Geenen MM, Cardous-Ubbink MC, Kremer LC et al (2007) Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA 297:2705–2715 [DOI] [PubMed]
- 97.Raney B, Anderson J, Jenney M et al (2006) Late effects in 164 patients with rhabdomyosarcoma of the bladder/prostate region: a report from the international workshop. J Urol 176:2190–2194 [DOI] [PubMed]
- 98.Anderson J, Gordon A, Pritchard-Jones K et al (1999) Genes, chromosomes, and rhabdomyosarcoma. Genes Chromosomes Cancer 26:275–285 [PubMed]
- 99.Flamant F, Rodary C, Voute PA et al (1985) Primary chemotherapy in the treatment of rhabdomyosarcoma in children: trial of the International Society of Pediatric Oncology (SIOP) preliminary results. Radiother Oncol 3:227–236 [DOI] [PubMed]
- 100.Flamant F, Gerbaulet A, Nihoul-Fekete C et al (1990) Long-term sequelae of conservative treatment by surgery, brachytherapy, and chemotherapy for vulval and vaginal rhabdomyosarcoma in children. J Clin Oncol 8:1847–1853 [DOI] [PubMed]
- 101.Maurer HM, Crist W, Lawrence W et al (1988) The intergroup rhabdomyosarcoma study-I. A final report. Cancer 61:209–220 [DOI] [PubMed]
- 102.Maurer HM, Gehan EA, Beltangady M et al (1993) The Intergroup Rhabdomyosarcoma Study-II. Cancer 71:1904–1922 [DOI] [PubMed]
- 103.Wolden SL, Anderson JR, Crist WM et al (1999) Indications for radiotherapy and chemotherapy after complete resection in rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Studies I to III. J Clin Oncol 17:3468–3475 [DOI] [PubMed]
- 104.Schuck A, Mattke AC, Schmidt B et al (2004) Group II rhabdomyosarcoma and rhabdomyosarcomalike tumors: is radiotherapy necessary? J Clin Oncol 22:143–149 [DOI] [PubMed]
- 105.Breneman JC, Lyden E, Pappo AS et al (2003) Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma – a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 21:78–84 [DOI] [PubMed]
- 106.Koscielniak E, Harms D, Henze G et al (1999) Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17:3706–3719 [DOI] [PubMed]
- 107.Stewart RJ, Martelli H, Oberlin O et al (2003) Treatment of children with nonmetastatic paratesticular rhabdomyosarcoma: results of the Malignant Mesenchymal Tumors studies (MMT 84 and MMT 89) of the International Society of Pediatric Oncology. J Clin Oncol 21:793–798 [DOI] [PubMed]
- 108.Buwalda J, Schouwenburg PF, Blank LE et al (2003) A novel local treatment strategy for advanced stage head and neck rhabdomyosarcomas in children: results of the AMORE protocol. Eur J Cancer 39:1594–1602 [DOI] [PubMed]
- 109.Haie-Meder C, Flamant F, Revillon Y et al (1994) The role of brachytherapy in the therapeutic strategy of vesico-prostatic rhabdomyosarcoma in children. Ann Urol (Paris) 28:302–305 [PubMed]
- 110.Magne N, Haie-Meder C (2007) Brachytherapy for genital-tract rhabdomyosarcomas in girls: technical aspects, reports, and perspectives. Lancet Oncol 8:725–729 [DOI] [PubMed]
- 111.Merchant TE, Parsh N, del Valle PL et al (2000) Brachytherapy for pediatric soft-tissue sarcoma. Int J Radiat Oncol Biol Phys 46:427–432 [DOI] [PubMed]
- 112.Timmermann B, Schuck A, Niggli F et al (2007) Spot-scanning proton therapy for malignant soft tissue tumors in childhood: first experiences at the Paul Scherrer Institute. Int J Radiat Oncol Biol Phys 67:497–504 [DOI] [PubMed]
- 113.Wolden SL, Wexler LH, Kraus DH et al (2005) Intensity-modulated radiotherapy for head-and-neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys 61:1432–1438 [DOI] [PubMed]
- 114.Vanel D, Shapeero LG, De Baere T et al (1994) MR imaging in the follow-up of malignant and aggressive soft-tissue tumors: results of 511 examinations. Radiology 190:263–268 [DOI] [PubMed]