

Abstract
TRPML3 belongs to the TRPML subfamily of the transient receptor potential (TRP) channels. The A419P mutation in TRPML3 causes the varitint-waddler phenotype as a result of gain-of-function mutation (GOF). Regulation of the channels and the mechanism by which the A419P mutation leads to GOF are not known. We report here that TRPML3 is a Ca2+-permeable channel with a unique form of regulation by extracytosolic (luminal) H+ (H+e-cyto). Regulation by H+e-cyto is mediated by a string of three histidines (H252, H273, H283) in the large extracytosolic loop between transmembrane domains (TMD) 1 and 2. Each of the histidines has a unique role, whereby H252 and H273 retard access of H+e-cyto to the inhibitory H283. Notably, the H283A mutation has the same phenotype as A419P and locks the channel in an open state, whereas the H283R mutation inactivates the channel. Accordingly, A419P eliminates regulation of TRPML3 by H+e-cyto, and confers full activation to TRPML3(H283R). Activation of TRPML3 and regulation by H+e-cyto are altered by both the α-helix-destabilizing A419G and the α-helix-favouring A419M and A419K. These findings suggest that regulation of TRPML3 by H+e-cyto is due to an effect of the large extracytosolic loop on the orientation of fifth TMD and thus pore opening and show that the GOF of TRPML3(A419P) is due to disruption of this communication.
Keywords: extracytosolic loop, extracytosolic pH, histidines string, TRPML3, varitint-waddler phenotype
Introduction
TRPML3 is an ion channel that belongs to the TRPML subfamily of the transient receptor potential (TRP) cation channels superfamily (Pedersen et al, 2005). The TRPML subfamily has three members, TRPML1, TRPML2 and TRPML3. Mutations in TRPML1 cause the lysosomal storage disease mucolipidosis type IV (Bargal et al, 2000; Kiselyov et al, 2007). TRPML1 is a lysosomal channel (Manzoni et al, 2004) that when mutated or deleted impairs mainly lysosomal recycling (Fares and Greenwald, 2001; Hersh et al, 2002).
We have recently reported that TRPML3 functions as an inwardly rectifying cation channel, and that the A419P mutation, which causes the varitint-waddler phenotype, is a gain-of-function mutation (GOF) (Kim et al, 2007). Similar GOF by TRPML3(A419P) was reported by three additional studies (Grimm et al, 2007; Xu et al, 2007; Nagata et al, 2008), although these studies did not examine the properties of wild-type TRPML3 or its regulation. TRPML3 may participate in some form of membrane trafficking, as the varitint-waddler phenotype is characterized by aberrant melanocyte function and defective inner ear stereocilia with eventual degeneration of hair cells in the organ of Corti (Cable and Steel, 1998). Indeed, the native TRPML3 is found in unidentified vesicular compartments and in the stereocilia outer membrane in the hair cells of the cochlea (Di Palma et al, 2002). All the groups that reported the function of TRPML3(A419P) found that, similar to the native TRPML3, recombinant TRPML3 is expressed at the plasma membrane and intracellular vesicles (Grimm et al, 2007; Kim et al, 2007; Xu et al, 2007; Nagata et al, 2008). The report that TRPML3 is expressed exclusively in the endoplasmic reticulum (Venkatachalam et al, 2006) is probably the result of mistargeting of the channel due to masking of C-terminal targeting motifs (Song et al, 2006).
A central question regarding TRPML3 and the varitint-waddler phenotype is how the A419P mutation causes GOF. As proline introduces a kink or a break in α-helical transmembrane spans, and the A419G mutation that may disrupt α-helical structures results in a GOF in TRPML3 (Grimm et al, 2007; Kim et al, 2007), it was postulated that the GOF by TRPML3(A419P) is due to destabilization of the fifth transmembrane domain (TMD) of TRPML3. This was further concluded based on GOF by substitution to proline of the analogous A419 position in several TRP channels (Grimm et al, 2007). However, another possibility is that the A419P mutation disrupts a critical form of channel regulation that inhibits channel activity. To distinguish between these possibilities, it is necessary to understand how TRPML3 function is regulated and how the A419P mutation affects channel regulation.
To better understand the function and dysfunction of TRPML3, we provide the first characterization of wild-type TRPML3 channel properties and its regulation by extracytosolic H+ (H+e-cyto). As TRPML3 is also present in intracellular organelles, and intracellular organelles are acidic (Paroutis et al, 2004; Sun-Wada et al, 2004), regulation by H+e-cyto will determine its function and its activity in the organelles. Here, we report that wild-type TRPML3 is a Ca2+-permeable channel that is inhibited by H+e-cyto when conducting either Na+ or Ca2+. The regulation by pH is mediated by a string of three histidines, each having a unique role in the regulation of TRPML3 by H+e-cyto. Notably, elimination of TRPML3 regulation by H+e-cyto has the same functional and cellular phenotype as the A419P mutation. Accordingly, regulation by H+e-cyto is eliminated by the A419P mutation, accounting for the constitutive activity of TRPML3 and the varitint-waddler phenotype.
Results and discussion
Channel properties of wild-type TRPML3
So far, our work is the only one, to the best of our knowledge, to characterize the channel properties of wild-type TRPML3. Recently, we reported that TRPML3 functions as an inwardly rectifying cation channel and that channel activity requires incubation in Na+-free media (Kim et al, 2007). In this study, we sought to determine the mechanism of this effect. The effect of extracytosolic cations on the activity of wild-type TRPML3 is further illustrated in Figure 1 for both Na+ and Ca2+ currents. TRPML3 current was recorded either by applying 400 ms rapid alterations of membrane potentials (RAMPs) between −100 and +100 mV from a holding potential of 0 mV every 5 s (as in Figures 1A and D), or by holding the membrane potential at −100 mV throughout the experiment (as in Figure 1C). Bathing HEK cells expressing wild-type TRPML3 in a standard bath solution containing 140 mM Na+ resulted in a small current (Figures 1A and C). Incubation of the cells in Na+-free solution, pH 7.4, for a short period and re-addition of Na+ resulted in a large inward current that slowly inactivated in the continuous presence of Na+o. The inactivated channel can be readily and maximally reactivated by a short exposure to Na+-free solution, pH 7.4 (Figure 1A and others). The current/voltage (I/V) relationship of the monovalent cation current showed strong inward rectification with K+ (Figure 1B), Na+, Cs+ or NMDG+ (not shown) as the intracellular cation and with Na+, K+ and Cs+ as the extracellular permeating cation. Measurement of reversal potentials with the different external cations and intracellular K+ indicated TRPML3 monovalent cation selectivity of Na+>K+≫Cs+ (Figure 1F).
Figure 1.

Channel properties of TRPML3. (A) Whole-cell current was measured in HEK cells transfected with GFP–TRPML3. The major cation in the pipette solution is 150 mM K+ (pH 7.3) and Ca2+ is buffered close to 0 with 10 mM BAPTA. The major cation in the bath solution is either140 mM Na+ (dark bars) or 0 Na+, 150 mM NMDG+ (light grey bars) (pH 7.4). TRPML3 was activated by exposing the extracytosolic face to a bath solution containing 0 mM Na+, pH 7.4. The cells were sequentially perfused with solutions containing 140 or 0 mM Na+, as indicated by the dark and light grey bars. The current was measured by the RAMPs protocol at −100 (closed circles) and +100 mV (open circles). (B) I/V relationships of the TRPML3 current recorded at the times shown in the large filled circles in (A). (C) TRPML3 current was measured by holding the membrane potential at −100 mV. The pipette solution is identical to that in (A) and the major ion in the bath solution is either 140 mM Na+ or 150 mM K+, as indicated above the dark bars. The dark dashed line here and in all experiments indicates the 0 current level. (D) Inward Ca2+ current by TRPML3 was measured with a pipette solution containing 150 mM K+ and 10 mM BAPTA. After measurement of the maximal Na+ current, the cells were exposed to 0 Na+ solution containing 1, 10 or 45 mM Ca2+. The Ca2+-containing bath solutions were prepared by isosmotic replacement of NMDG-Cl with CaCl2. (E) The I/V relationships are of the TRPML3 current recorded with 140 mM Na+ (dark trace) or 10 mM Ca2+ (light grey trace) at the times indicated by the large circles in (D). (F) The reversal potential with the indicated cations in the bath solutions. K+, Na+ and Cs+ were at 150 mM. The results are the mean±s.e.m. of 3–5 experiments. A full-colour version of this figure is available at The EMBO Journal Online.
Almost all TRP channels function as non-selective, Ca2+-permeable channels (Pedersen et al, 2005; Nilius et al, 2007), and TRPML3(A419P) is permeable to Ca2+ (Grimm et al, 2007) and conducts Ca2+ at isotonic extracellular Ca2+ (Ca2+o) (Xu et al, 2007). Notably, Figures 1D and E show that wild-type TRPML3 is a permeable and a selective Ca2+ channel. As with monovalent cations, Ca2+ current by wild-type TRPML3 required preincubation in Na+-free solution. As Ca2+ influx by TRPML3(A419P) can be measured at low Ca2+o (Grimm et al, 2007), we measured Ca2+ current with 1 mM Ca2+o. Significant inward Ca2+ current can be measured in the presence of 150 mM intracellular K+ (K+in), 1 mM Ca2+o and no Na+o. Further increase in Ca2+o to 10 and 45 mM resulted in increased Ca2+ current, but the increase in current between 10 and 45 mM Ca2+o was small, if any, and developed slowly. This may be because of an effect of Ca2+o on the extracytoplasmic inhibitory site (see below for effect of Ca2+ on the inhibition by H+e-cyto). With 150 mM K+in and 1 mM Ca2+o, the inward current was 14±0.5% (n=7) of that measured with 140 Na+o. The reversal potential with 1 mM Ca2+o was 23±4 mV (n=5) and with 10 mM Ca2+o it was 57±4 mV (n=6). The results with 1 mM Ca2+o translate to an approximate Ca2+:K+ permeability ratio of 350:1. This ratio is comparable to that measured with the Ca2+-selective TRPV5 and TRPV6 (Owsianik et al, 2006), indicating that TRPML3 is a Ca2+ channel that is also permeable to monovalent cations. Because of the potential inhibition of TRPML3 by high Ca2+o (Figure 1D), it was not possible to determine whether TRPML3 displays anomalous mole fraction behaviour. However, this is not likely to be the case, as after activation by 0 Na+o and exposure to media containing 100 mM Na+ and 1 nM, 10 μM or 1 mM Ca2+, the current increased with increasing [Ca2+o] (not shown). TRPML3 has low permeability to Sr2+, Ba2+ (Figure 1F) and Mg2+ (not shown), and the currents generated by Sr2+, Ba2+ and Mg2+ are small and develop slowly (not shown).
Regulation of TRPML3 by H+
Part of TRPML3 is localized in intracellular organelles (Di Palma et al, 2002; Grimm et al, 2007; Xu et al, 2007; Nagata et al, 2008; not shown) that maintain acidic pH (Paroutis et al, 2004; Sun-Wada et al, 2004). Inhibition of TRPML3 by Na+o (Figures 1 and 2) may relate to the regulation of TRPML3 by extracytosolic H+. To study the regulation of TRPML3 by pH, we measured the effect of intracellular (pHi) and extracellular pH (pHo) on channel function. The effect of pHi reports the effect of H+ on the cytosolic side of TRPML3 and the effect of pHo reports the effect of H+ on the intravesicular, extracytosolic side of TRPML3 (H+e-cyto). The protocol for measuring the effect of pH on TRPML3 activity is shown in Figures 2A and B for Na+ current and in Figures 2D and E for Ca2+ current. Cells transfected with TRPML3 are incubated in Na+-free solution, pH 7.4, and then in Na+-containing solution, pH 7.4, to determine the maximal current. Then, the cells are incubated in Na+-free solution of a desired pHo (6.0 in Figure 2A) before incubation in Na+-containing solution, pH 7.4, to evaluate the extent of current inhibition by preincubation at the desired pH. Finally, the reversal of inhibition is determined by washout with Na+-free solution, pH 7.4, and re-incubation in Na+-containing solution, pH 7.4.
Figure 2.

Regulation of TRPML3 activity by extracytosolic pH (H+e-cyto). (A) Na+ current was measured as in Figure 1A, except that at the period marked by the grey bar the cells were incubated in Na+-free bath solution of pH 6.0. (B) The same conditions as in (A), except that the pH of the pipette solution (pHi) was set at 5.0. (C) H+e-cyto-dependent inhibition of TRPML3-mediated Na+ current. The number of experiments at different H+e-cyto is given next to the symbols. (D) TRPML3-mediated Ca2+ current was measured at 10 mM Ca2+o as in Figure 1D, except that the cells were incubated in Na+-free bath solution of pH 5.0 (grey line) before measurement of Ca2+ current at pH 7.4. (E) The same conditions as in (D) were used to measure the Ca2+ current, except that the pH of the Ca2+-containing solution was 6.0. (F) H+e-cyto-dependent inhibition of TRPML3-mediated Ca2+ current. A full-colour version of this figure is available at The EMBO Journal Online.
Figure 2A shows that preincubating the extracytosolic face of TRPML3 at pHo 6.0 almost completely inhibited TRPML3-mediated Na+ current recorded at pHo 7.4 and the inhibition was readily reversed by re-incubating the cells in Na+-free solution, pH 7.4. Similar experiments at various pHo revealed that the inhibition has an IC50 of pH 6.42, close to the pH of several intracellular organelles (Paroutis et al, 2004), except that of the lysosomes, which have lower pH. Figure 2B shows that the inhibition is specific to H+e-cyto. Exposing the cytoplasmic domains of TRPML3 to pH as low as 5.0 does not inhibit TRPML3 current and does not affect the inhibition by H+e-cyto.
Figures 2D and E show that H+e-cyto also inhibits the TRPML3-mediated Ca2+ current. However, in this case, it was not sufficient to preincubate TRPML3 with H+e-cyto. Even when the pHo of the preincubation solution was lowered to 5.0, substantial Ca2+ current can be measured by subsequent incubation with a solution containing 10 mM Ca2+ at pH 7.4 (Figure 2D). To observe inhibition of the Ca2+ current by H+e-cyto, it is necessary to lower the pH of the Ca2+-containing solution used to measure the current (Figure 2E). With this protocol, H+e-cyto inhibited the Ca2+ current with an IC50 of 6.33 (Figure 2F). The ability of Ca2+o to reduce inhibition by preincubation with H+e-cyto (Figure 2D) suggests that Ca2+ may influence interaction of H+e-cyto with TRPML3 and may explain the inhibition of the current by high Ca2+o (Figure 1D).
H+e-cyto domain inhibition by H+e-cyto is mediated by H283
The inhibition of TRPML3 Na+ current by pretreatment with H+e-cyto and its persistence after returning to pH 7.4 indicate that H+e-cyto does not act on the TRPML3 pore, but rather interacts with a distinct inhibitory site to inactivate the channel. Regulation by H+ is commonly mediated by histidines, which have a pKa of 6.0. Therefore, we performed an alanine scan of all the histidines in the extracytosolic domain of TRPML3 (depicted in Figure 3A). The current and the regulation by H+e-cyto were not affected by the mutations of seven of the histidines shown in black letters in Figure 3A. The mutation of histidines H252, H273 and H283 differentially affected the inhibition of TRPML3 by H+e-cyto. Figure 3B shows that H252A, H273A, the double mutants H252A+H273A, H252A+H283A, H273A+H283A and the triple mutant H252A+H273A+H283A slightly increased the total and surface expression of TRPML3. By contrast, the H283A mutation markedly reduced the total and surface expression of TRPML3. Interestingly, when combined with the H283A mutant, H252A and H273A reversed the inhibition of the total and surface expression observed with the H283A mutant, consistent with their effect on channel activity.
Figure 3.

Inhibition of TRPML3 by H+e-cyto is mediated by histidine 283. (A) The position of all extracytosolic histidines in TRPML3. (B) Analysis of total and surface expression of the indicated individual, double and triple histidine mutants. (C) Na+ current in cells expressing TRPML3 (□) or TRPML3(H283A) (•) was measured as in Figure 2A. Note that TRPML3(H283A) is spontaneously active, does not inactivate and is not inhibited by H+e-cyto. (D) The H283A mutation does not change the I/V relationship of TRPML3. (E) Effect of the H283A and A419P mutations on cell death in the presence and absence of Ca2+o. Cells transfected with TRPML3, TRPML3(H283A) and TRPML3(A419P) were cultured for 24 h in DMEM media containing 1.8 mM CaCl2 or the same media supplemented with 2.5 mM EGTA (marked 0 Ca2+). All cells (floating and attached) were collected to prepare cell extracts and the extracts were probed for expression of the channels. Note that the H283A and A419P mutations similarly reduced the level of TRPML3 as a result of cell death and cell death could be reduced by removal of Ca2+o. (F) Ca2+ influx was measured in HEK cells transfected with eGFP (○), TRPML3 (▵), TRPML3(H283A) (•) and TRPML3(A419P) (▴). The cells were incubated in Ca2+-free medium and plasma membrane Ca2+ permeability was measured by increasing Ca2+o to 1 mM. A full-colour version of this figure is available at The EMBO Journal Online.
Figure 3C shows that the H283A mutation causes GOF by eliminating inhibition of TRPML3 activity by H+e-cyto. Thus, application of −100 to +100 mV RAMPs revealed that it is not necessary to incubate TRPML3(H283A) in Na+-free media to activate the channel and the current does not inactivate in the continuous presence of Na+o. Moreover, the TRPML3(H283A) current is not inhibited by H+e-cyto. The I/V relationship in Figure 3D shows that the H283A mutation did not change the channel properties. To further establish a role for H283 in the regulation of TRPML3 by H+e-cyto, H283 was mutated to arginine, which has a pKa of 12.5. Because of the rapid inactivation of TRPML3(H283R), its activity was recorded by holding the membrane potential at −100 mV. Figure 4A shows that stepping the membrane potential to −100 mV did not activate TRPML3(H283R). After incubation in Na+-free solution, pH 7.4, re-application of Na+o results in a transient, rapidly inactivating current by TRPML3(H283R), which is inhibitable by H+e-cyto.
Figure 4.

Effect of H252, H273 and H283R on regulation of TRPML3 by H+e-cyto. (A) The H283R mutation markedly increases the rate of TRPML3 inactivation and TRPML3(H283R) is inhibited by H+e-cyto. The dark dashed line in this and the other panels indicates the 0 current level. The grey line marks the period of incubation with 0 Na+, pH 6. (B) Na+ current was measured in cells expressing wild-type TRPML3 (black trace), TRPML3(H252A) (dashed trace) or TRPML3(H273A) (grey trace). Current measurement was initiated by clamping the membrane potential to −100 mV. Preincubation of wild-type, H252A and H273A TRPML3 in Na+-free solution resulted in full activation of the current. However, the TRPML3 current inactivated along a bi-exponential time course, with fast inactivation that was completed in 10–20 s and a slow inactivation that took a longer time (minutes). The current of the H252A and H273A mutants showed only fast inactivation. (C) The spontaneously active TRPML3(H283A) current was measured in cells continuously bathed in solution containing 140 mM Na+. Current is observed when switching the holding membrane potential from 0 to −100 mV. (D) TRPML3(H252A) (dashed trace) and TRPML3(H273A) (grey trace) activity is inhibited by H+e-cyto. (E) Dependence of the inhibition of the TRPML3(H252A) and TRPML3(H273A) current on H+e-cyto was measured at a holding potential of −100 mV. The cells were exposed to Na+-free medium, pH 7.4, to determine the control current and then to Na+-free medium of the indicated pHo. After exposure to different Na+-free media of the indicated pHo, the current was measured in media containing 140 mM Na+, pH 7.4. (F) Properties of the double mutants TRPML3(H252,283A) (grey trace) and TRPML3(H273,283A) (dark trace). The spontaneous activity was measured by clamping the membrane potential to −100 mV. The cells were then incubated in Na+-free solution, pH 7.4, and the current was measured by clamping the membrane potential to −100 mV in Na+-containing solution, pH 7.4. Note that the H283A mutation markedly reduces and slows down channel inactivation of H252A and H273A. A full-colour version of this figure is available at The EMBO Journal Online.
These TRPML3(H283A) properties are the same as those reported for TRPML3(A419P) (Kim et al, 2007). To further compare between the two mutants, we measured their effect on cell survival and plasma membrane Ca2+ permeability. Expression of TRPML3(H283A) and TRPML3(A419P) resulted in massive cell death, as evident from the large number of floating cells in the culture. Cell survival was estimated from the total level of TRPML3 in alive and dead cells. Figure 3E shows that expression of TRPML3 was markedly reduced by both mutants. Moreover, culturing the cells in Ca2+-free medium had minimal effect on cells expressing wild-type TRPML3, but reduced the number of dead cells expressing the TRPML3 mutants, as evident from the increased level of TRPML3(H283A) and TRPML3(A419P). Finally, Figure 3F shows that expression of wild-type TRPML3 only slightly increased the plasma membrane Ca2+ permeability, whereas expression of TRPML3(H283A) and TRPML3(A419P) markedly increased the plasma membrane Ca2+ permeability. Massive cell death due to the expression of TRPML3(A419P) was also reported by Xu et al (2007).
Role of H252 and H273 in the H+e-cyto domain
The results in Figures 3 and 4A make it clear that H283 has a major role in determining regulation of TRPML3 by H+e-cyto. Figure 4B shows that the H252A and H273A mutations also alter regulation of TRPML3 by H+e-cyto. Both mutations increased, rather than decreased, the rate of channel inactivation, when compared to the rate of inactivation measured with TRPML3 (Figure 4B) and TRPML3(H283A) (Figure 4C). Moreover, TRPML3(H252A) and TRPML3(H273A) are inhibited by H+e-cyto (Figure 4D). Figure 4E shows that the H252A and H273A mutations do not alter the dependence of the inhibition on H+e-cyto. This suggests that H252 and H273 do not participate in the formation of the binding site for H+e-cyto. Rather, the effects of H252 and H273 are consistent with H252 and H273 impairing access of H+e-cyto to H283 and their mutations to alanine improve access of H+e-cyto to H283. This interpretation predicts that when combined with the H252A and H273A mutations, the H283A mutation should reduce inhibition of TRPML3 by H+e-cyto. Figure 4F shows that this is the case. The first part of Figure 4F shows that TRPML3(H252,283A) and TRPML3(H273,283A) retained 1.6±0.9% (n=3) and 29±7% (n=4), respectively, of the spontaneous activity observed with TRPML3(H283A). Exposing the cells to Na+-free solution, pH 7.4, further activated a current that only slowly inactivated with TRPML3(H252,283A) and only partially inactivated with TRPML3(H273,283A). We could not test the effect of the double mutant TRPML3(H252,273A) and of the triple mutant TRPML3(H252,273,283A), as both are not active (not shown), although both traffic normally to the plasma membrane (see Figure 3B). Hence, the results in Figure 4 suggest that H252 and H273 control the access of H+e-cyto to H283 by retarding its access. They may also affect the interaction of H+e-cyto with H283 to account for the slow and partial inactivation of the double mutants.
Molecular mechanism of the GOF by TRPML3(A419P)
Recently we (Kim et al, 2007) and others (Grimm et al, 2007; Xu et al, 2007; Nagata et al, 2008) reported that the varitint-waddler phenotype is caused by a GOF that makes TRPML3(A419P) spontaneously active, as found with TRPML3(H283A). Moreover, both mutants similarly increase plasma membrane Ca2+ permeability and reduce cell survival in a Ca2+-dependent manner (Figure 3E and F). Therefore, we examined the relationship between A419P mutation and regulation of TRPML3 by H+e-cyto. Figures 5A and C show that TRPML3(A419P) is spontaneously active and is completely resistant to inhibition by H+e-cyto. Moreover, Figure 5B shows that even when H283R mutation, which strongly inhibits TRPML3 (Figure 4A), is combined with A419P, the double mutant TRPML3(A419P,H283R) remains spontaneously active and resistant to inhibition by H+e-cyto.
Figure 5.

Effect of the varitint-waddler phenotype-causing mutation A419P on regulation of TRPML3 by H+e-cyto and on cation selectivity. In all experiments, open symbols are the current at +100 mV and closed symbols are the current at −100 mV. (A) Na+ current of TRPML3(A419P) and its lack of inhibition by H+e-cyto. (B) Na+ current of the double mutant TRPML3(A419P/H283R) and its lack of inhibition by H+e-cyto. (C) Mean±s.e.m. of six experiments on the effect of pHo 5.0 of the current by TRPML3, TRPML3(H283A) and TRPML3(A419P). (D) Current by wild-type TRPML3 measured at 140 mM Na+ and 10 mM Ca2+ and the I/V relationships at peak current. (E) Current of TRPML3(A419P) measured at 140 mM Na+ and 10 mM Ca2+ and the I/V relationships at peak current. (F) Mean±s.e.m of the Ca2+/Na+ selectivity of wild-type TRPML3 (n=7) and TRPML3(A419P) (n=4). A full-colour version of this figure is available at The EMBO Journal Online.
Another effect of the A419P mutation is that it alters the ionic selectivity of TRPML3. In Figures 5D and F, we compared the relative permeability of wild-type TRPML3 and TRPML3(A419P) to Na+ and Ca2+ by measuring the current carried by 140 mM Na+ and 10 mM Ca2+. The Ca2+/Na+ permeability ratio of TRPML3 is 0.51±0.06 and it is reduced to 0.06±0.02 by the A419P mutation. The reduction in the extent of the Ca2+ current was accompanied by a change in the reversal potential for Ca2+ from 30.4±2.7 to 15.4±7.0 mV for TRPML3 and TRPML3(A419P), respectively. This suggests that in addition to eliminating the regulation of TRPML3 by H+e-cyto, the A419P mutation also affects the structure of TRPML3 pore.
The mechanism of GOF by TRPML3(A419P) is not known, but it was postulated that the A419P mutation destabilizes the fifth TMD of TRPML3. Another possibility is that the orientation of the fifth TMD of TRPML3 and thus the properties of the pore are influenced by the H+e-cyto regulatory domain in the large extracytosolic loop between TMD 1 and 2, and the A419P mutation disrupts this communication. The two possibilities can be differentiated by testing the effect of substituting A419 with either glycine, which tends to destabilize α-helices, or lysine, methionine, glutamate or leucine, which tends to favour α-helix formation. The A419E and A419L mutations resulted in inactive channels (not shown). Figure 6A shows that TRPML3(A419G) is spontaneously active and is resistant to inhibition by H+e-cyto. However, A419M and A419K also affected channel function and eliminated regulation by H+e-cyto. About 25% of the TRPML3(A419M) current was spontaneously active and the current increased on 3–4 consecutive incubations in Na+-free solutions to attain the current that was measured with TRPML3(A419P). The TRPML3(A419M) current lost regulation by H+e-cyto and was not inhibited by incubating the extracytosolic face of this mutant even at pH 5 (Figure 6B). The activity of the TRPML3(A419K) mutant was initially minimal, but increased on repeated applications of −100 to +100 mV RAMPs in the continuous presence of Na+o, until it became almost completely spontaneously active. The activated TRPML3(A419K) current is not regulated by H+e-cyto and is not inhibited by incubation at pH as low as 5 (Figure 6C). Hence, the altered regulation of TRPML3 by H+e-cyto by both the α-helix-disrupting A419G and the α-helix-favouring A419M and A419K mutants suggests that disruption of the fifth TMD by A419P is not the only mechanism for the GOF by TRPML3(A419P). Rather, the fifth TMD appears to communicate with the H+e-cyto domain to determine TRPML3 activity and the A419P mutation disrupts this regulation.
Figure 6.

Mutants that destabilize and favour α-helices prevent regulation of TRPML3 by H+e-cyto. In all experiments, open symbols are the current at +100 mV and closed symbols are the current at −100 mV. (A) Na+ current of the α-helix-destabilizing mutant A419G and its lack of inhibition by H+e-cyto. (B) The spontaneous current of TRPML3(A419M), the increase in current by repeated incubation in Na+-free medium and the lack of regulation of TRPML3(A419M) by H+e-cyto. (C) Development of the TRPML3(A419K) current by continuous application of −100 to +100 mV RAMPs in the presence of Na+o and the lack of regulation of TRPML3(A419M) by H+e-cyto. The results in (B) and (C) are representative of four similar experiments. A full-colour version of this figure is available at The EMBO Journal Online.
Conclusions
To understand the function of TRPML3 in cell physiology and in the varitint-waddler phenotype, we characterized the function and regulation of wild-type TRPML3 and of the varitint-waddler phenotype causing mutant TRPML3(A419P). The present results together with our previous studies (Kim et al, 2007) provide the first characterization of the channel properties of wild-type TRPML3. We (Kim et al, 2007) and others (Grimm et al, 2007; Xu et al, 2007; Nagata et al, 2008) reported the properties of TRPML3(A419P), which are similar to those of wild-type TRPML3 but also display important differences. Wild-type TRPML3 can conduct monovalent and divalent cations with high selectivity for Ca2+. It is difficult to determine the exact selectivity of TRPML3 for Ca2+, as Ca2+ appears to influence inhibition by H+e-cyto (Figure 2D). Nevertheless, when 1 mM Ca2+ is used to measure the current, the Ca2+:K+ selectivity ratio is about 350. This is similar to the selectivity of the established Ca2+ channels TRPV5 and TRPV6 (den Dekker et al, 2003; Owsianik et al, 2006). Xu et al (2007) reported Ca2+ permeability of TRPML3(A419P) using isotonic Ca2+ solution. Interestingly, we found that the A419P mutation alters channel function by markedly reducing the Ca2+ permeability of TRPML3 (Figure 5E), suggesting that A419 participates in determining the conductance of the channel pore.
The regulation of TRPML3 by H+e-cyto interacting with H283 is not likely to involve a block of the channel pore, as the channel remains inactive after preincubation at pH 6.0 when the current is measured at pH 7.4. Regulation by H+e-cyto is different from the partial inhibition of TRPML3 current by low pHo (about 25% at pHo 4.6; Xu et al, 2007), as a similar inhibition is observed with TRPML3(H283A) (not shown). Regulation of TRPML3 by H+e-cyto is unique, being mediated by three histidines each of which has a distinct role. H252, H273 and H283 in the large extracytosolic loop between transmembrane spans 1 and 2 form an H+ regulatory domain, with H252 and H273 controlling the access of H+ to H283 to regulate TRPML3 function. This interpretation is based on the finding that inactivation of TRPML3 proceeds along a fast and a slow time course and mutations of the histidines differently affect each time course. TRPML3(H283A) is not inhibited by Na+o or H+e-cyto and show only the slow inactivation time course, whereas TRPML3(H252A) and TRPML3(H273A) show only the fast inactivation time course. Significantly, the H252A and H273A mutations do not change the dependence of the inhibition of TRPML3 on H+e-cyto. This is most simply explained by H252 and H273 hindering access of H+ to H283 and mutations of these residues to alanines remove this hindrance to facilitate access of H+ to H283, resulting in a rapid inhibition of the channel. This interpretation is supported by the finding that the H283A mutation in the context of TRPML3(H252A) and TRPML3(H273A) perceptibly reduces the rate of channel inactivation.
It is likely that the H+e-cyto regulatory domain influences the orientation of the fifth TMD and binding of H+e-cyto to H283 exerts a long-range conformational change that affects pore opening. The GOF by the varitint-waddler A419P mutation is due, at least in part, to the disruption of the regulation of TRPML3 by H+e-cyto that locks the channel in an open state. This conclusion is based on the findings that (a) TRPML3(H283A) and TRPML3(A419P) have identical properties, (b) TRPML3(A419P) is not regulated by H+e-cyto, (c) the H283R mutation does not inhibit the activity of TRPML3(A419P) and (d) the A419M and A419K are not regulated by H+e-cyto. These findings also provide a molecular mechanism for the GOF of TRPML3(A419P).
Materials and methods
Plasmid construction and mutagenesis
The full-length human TRPML3 was amplified by PCR from a human placenta mRNA preparation and cloned in-frame into the pEGFPC1 (Clontech). This construct was used as template to prepare mutants using the QuikChange mutagenesis kit (Stratagene). All mutations were confirmed by sequencing the entire DNA inserts to verify the presence of the desired mutation and the absence of extraneous mutations. All experiments in the current studies were performed with eGFP-tagged TRPML3.
Cell culture, transfection and surface biotinylation assay
HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum in a humidified incubator at 37°C and 5% CO2/air. Cells were transfected with Lipofectamine 2000 (Invitrogen) and used for current measurement or immunoblotting 24–48 h after transfection. In the experiments of Figure 3E, 6 h after transfection, the Ca2+ concentration in the media of part of the cells was reduced below 1 μM by the addition of 2.5 mM EGTA. For the biotinylation assay, cells were washed with PBS and incubated in 1 mg/ml sulpho-NHS-LC-biotin (Pierce) in PBS for 30 min on ice. Free biotin was quenched by the addition of 100 mM glycine in PBS and the cells were washed once with PBS. Lysates were prepared by extracting cells with 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 5 mM MgCl2, complete protease inhibitor mixture tablet (Roche Applied Science) and 0.5% Triton X-100, passing 7–10 times through a 30-gauge needle and sonication. The lysates were centrifuged at 14 000 r.p.m. for 10 min at 4°C and protein concentration in the supernatants was determined. Volume and protein content were equalized and 12.5% avidin beads (Pierce) were added to each sample. After incubation for 2 h at 4°C, the beads were collected by centrifugation, washed three times with 0.5% Triton X-100 in PBS and the proteins were extracted in sample buffer. The proteins collected were blotted with anti-GFP antibody (Cell Signaling).
Measurement of [Ca2+]i
Transfected HEK cells grown on glass coverslips were loaded with Fura2 by incubation with 5 μM Fura2/AM for 50 min at 37°C. The cells were washed with Ca2+-free bath solution and transferred to a perfusion chamber. The cells were continuously perfused first with Ca2+-free media and then with media containing 1 mM Ca2+. Fura2 fluorescence was measured at excitation wavelengths of 340 and 380 nm and the results are given as the 340/380 fluorescence ratio.
Current recordings and solutions
The tight seal, whole-cell configuration of the patch-clamp technique was used to record the whole-cell current at room temperature. The current was measured with an Axopatch 200B patch-clamp amplifier and filtered at 5 kHz. Data were collected using pClamp V.9.0 and Digidata-1322 (all from Axon Instruments) and the application of command pulses. The current was measured in response to voltage ramps (−100 to +100 mV, 400 ms) applied every 5 s from a holding potential of 0 mV or by holding the membrane potential at −100 mV throughout the recording. Results were analysed with pClamp 9.0 and Origin software (Microcal Software). Pipettes had a resistance of 2–4 MΩ. The standard pipette solution contained (in mM) 150 KCl, 10 HEPES, 1.13 MgCl2, 5 Mg-ATP and 10 BAPTA adjusted to pH 7.3 with KOH. The standard bath solution consisted of (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 glucose and 10 HEPES, adjusted to pH 7.4 with NaOH. Na+-free solution contained (in mM) 150 NMDG-Cl and 10 HEPES or 10 MES, adjusted to pH 7.4 or the desired pH with HCl. Divalent ion-containing solutions were prepared by replacing NMDG+ with Ca2+, Sr2+ or Ba2+ in the Na+-free solution. Monovalent solutions contained 150 mM NaCl, KCl or CsCl and 10 HEPES, adjusted to pH 7.4 with NaOH, KOH or CsOH. The permeability ratio of Ca2+:K+ was calculated using the equation 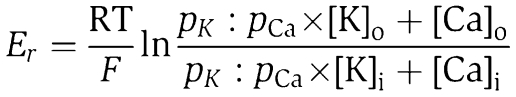
Acknowledgments
This work was supported by National Institutes of Health Grants DE12309 and DK38938 and by the Ruth S Harrell Professorship in Medical Research.
References
- Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G (2000) Identification of the gene causing mucolipidosis type IV. Nat Genet 26: 118–123 [DOI] [PubMed] [Google Scholar]
- Cable J, Steel KP (1998) Combined cochleo-saccular and neuroepithelial abnormalities in the Varitint-waddler-J (VaJ) mouse. Hear Res 123: 125–136 [DOI] [PubMed] [Google Scholar]
- den Dekker E, Hoenderop JG, Nilius B, Bindels RJ (2003) The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium 33: 497–507 [DOI] [PubMed] [Google Scholar]
- Di Palma F, Belyantseva IA, Kim HJ, Vogt TF, Kachar B, Noben-Trauth K (2002) Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proc Natl Acad Sci USA 99: 14994–14999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares H, Greenwald I (2001) Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet 28: 64–68 [DOI] [PubMed] [Google Scholar]
- Grimm C, Cuajungco MP, van Aken AF, Schnee M, Jors S, Kros CJ, Ricci AJ, Heller S (2007) A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc Natl Acad Sci USA 104: 19583–19588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh BM, Hartwieg E, Horvitz HR (2002) The Caenorhabditis elegans mucolipin-like gene cup-5 is essential for viability and regulates lysosomes in multiple cell types. Proc Natl Acad Sci USA 99: 4355–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Li Q, Tjon-Kon-Sang S, So I, Kiselyov K, Muallem S (2007) Gain-of-function mutation in TRPML3 causes the mouse varitint-waddler phenotype. J Biol Chem 282: 36138–36142 [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Soyombo A, Muallem S (2007) TRPpathies. J Physiol 578: 641–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni M, Monti E, Bresciani R, Bozzato A, Barlati S, Bassi MT, Borsani G (2004) Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett 567: 219–224 [DOI] [PubMed] [Google Scholar]
- Nagata K, Zheng L, Madathany T, Castiglioni AJ, Bartles JR, Garcia-Anoveros J (2008) The varitint-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward rectifying currents and causes cell degeneration. Proc Natl Acad Sci USA 105: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA (2007) Transient receptor potential cation channels in disease. Physiol Rev 87: 165–217 [DOI] [PubMed] [Google Scholar]
- Owsianik G, Talavera K, Voets T, Nilius B (2006) Permeation and selectivity of TRP channels. Annu Rev Physiol 68: 685–717 [DOI] [PubMed] [Google Scholar]
- Paroutis P, Touret N, Grinstein S (2004) The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda) 19: 207–215 [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G, Nilius B (2005) TRP channels: an overview. Cell Calcium 38: 233–252 [DOI] [PubMed] [Google Scholar]
- Song Y, Dayalu R, Matthews SA, Scharenberg AM (2006) TRPML cation channels regulate the specialized lysosomal compartment of vertebrate B-lymphocytes. Eur J Cell Biol 85: 1253–1264 [DOI] [PubMed] [Google Scholar]
- Sun-Wada GH, Wada Y, Futai M (2004) Diverse and essential roles of mammalian vacuolar-type proton pump ATPase: toward the physiological understanding of inside acidic compartments. Biochim Biophys Acta 1658: 106–114 [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Hofmann T, Montell C (2006) Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J Biol Chem 281: 17517–17527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Delling M, Li L, Dong X, Clapham DE (2007) Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint waddler mice. Proc Natl Acad Sci USA 104: 18321–18326 [DOI] [PMC free article] [PubMed] [Google Scholar]