

Abstract
Telomere stability plays an important role in the preservation of genomic stability and is maintained through the coordinated actions of telomere specific proteins and DNA repair and replication proteins [1, 2]. Flap Endonuclease 1 (FEN1) is a protein that plays a role in lagging strand DNA replication, base excision repair, homologous recombination, and re-initiation of stalled replication forks [3, 4]. Here, we demonstrate that FEN1 depletion leads to telomere dysfunction characterized by the presence of γH2AX and sister telomere loss. Expression of catalytically active telomerase, the reverse transcriptase that adds telomeric repeats to chromosome ends, was sufficient to rescue telomere dysfunction upon FEN1 depletion. Strikingly, FEN1 depletion exclusively abrogates telomeres replicated by lagging strand DNA replication. Genetic rescue experiments utilizing FEN1 mutant proteins that retained the ability to localize to telomeric repeats revealed that FEN1’s nuclease activity and ability to interact with the Werner protein (WRN) and telomere binding protein, TRF2 were required for FEN1 activity at the telomere. Given FEN1’s role in lagging strand DNA replication and re-initiation of stalled replication forks, we propose that FEN1 contributes to telomere stability by ensuring efficient telomere replication.
Results and Discussion
High fidelity replication of telomeres is critical to maintain telomere stability, and is confounded by both the end replication problem and repetitive G-rich nature of telomeric DNA [5]. Repetitive DNA sequences such as those found in the telomere present a challenging template for the replication machinery due to a propensity to form secondary structures that can lead to stalled replication forks [6, 7]. Due to the importance and difficulty of high fidelity replication through the telomere, recent studies have focused on the role DNA replication/repair proteins play in telomere stability [8-11]. Rad27, the FEN1 homolog is one such replication and repair protein that plays a role at Sachharomyces cerevisiae telomeres [8, 12]. Here, we demonstrate that FEN1 plays a critical role in mammalian telomere stability.
Previous work demonstrated that FEN1 localized to the telomere in a cell cycle dependent manner [13]. We confirmed this observation by chromatin immunoprecipitation (ChIP) from cells 1) synchronized with thymidine and aphidicolin (Figure S1 in Supplemental Data available online) and 2) enriched in different phases of the cell cycle by centrifugal elutriation (Figure S2). In agreement with previous work, we found that FEN1 localized to the telomere in the S and G2/M phases of the cell cycle. Purified FEN1 has been shown to interact directly with TRF2 through both the basic and myb domains of TRF2 [14]. Utilizing antibodies specific for endogenous FEN1 and TRF2, we demonstrate that these proteins interact in vivo (Figure S3).
FEN1’s presence at the telomere and its interaction with TRF2 raised the intriguing possibility that it played a role in telomere biology. To address this directly, lentiviral expressed RNA interference (RNAi) hairpins targeting FEN1 (shFEN) or a scrambled hairpin (negative control, shSCR) were introduced into BJ fibroblasts (Figure 1A). Upon transduction, FEN1 protein expression was virtually undetectable compared to control cells (Figure 1B). To determine whether FEN1 depletion resulted in telomere dysfunction, we analyzed telomeres for the presence of γH2AX (an indicator of DNA damage) by ChIP. Lysates from cells expressing shSCR or shFEN were subject to immunoprecipitation using an antibody to γH2AX, followed by quantitation of isolated telomeric and genomic DNA (ALU). We found that upon FEN1 depletion, immunoprecipitation of γH2AX resulted in a significant increase in the amount of isolated telomeric DNA compared to control cells (1.39 fold greater than control; P<0.05; Figure 1C and 1D). In contrast, no significant increase was observed in γH2AX associated with ALU DNA (1.09 fold; P=0.59), indicating that there is increased DNA damage upon FEN1 depletion at telomeric sequences compared to the genome at large. A similar increase in γH2AX associated telomeric and genomic DNA was observed when cells were treated with the ribonucleotide reductase inhibitor, hydroxyurea (data not shown). Together these results indicate that FEN1 depletion results in telomere dysfunction similar to that observed upon replication stress following hydroxyurea treatment.
Fig. 1. FEN1 depletion leads to telomere dysfunction.
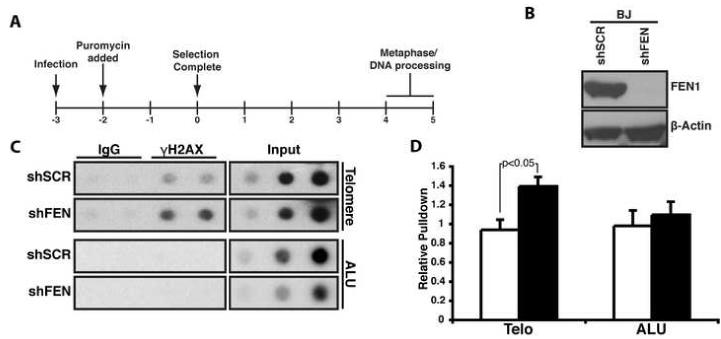
(A) Timeline of experimental procedure given in days. (B) Short hairpins against FEN1 (shFEN) or a scrambled sequence (shSCR) were expressed in BJ fibroblasts. FEN1 (upper panel) and β-Actin (lower panel) protein levels were assessed by Western blot analysis. (C) Representative ChIP assay of cells expressing shSCR or shFEN. ChIPs were conducted as described in the supplemental materials. The inputs indicate 0.2%, 0.1% and 0.04% of the total protein extract. (D) Quantification of six independent ChIP assays. The graph indicates the relative amount of telomere (Telo) or ALU repeat (ALU) DNA isolated from cells expressing shSCR (white) or shFEN (black). The error bars represent standard error of the mean (SEM).
We next assessed the telomeres directly upon FEN1 depletion. FEN1 was depleted in BJ fibroblasts expressing the SV40 early region (BJL) (the presence of the early region facilitated isolation of metaphase chromosomes) (Figure 2A). Following FEN1 depletion, we utilized fluorescence in situ hybridization (FISH) to visualize telomeres. We found that FEN1 depletion resulted in increased sister telomere loss (STL) (Figure 2B and 2C). On average, 9.4% of the chromosomes isolated from control cells displayed STLs (Figure 2C). Upon FEN1 depletion, the percentage of chromosomes displaying STLs increased nearly two-fold (16.8%, P<0.0001; Figure 2C), indicating that FEN1 depletion impacted telomere stability.
Fig. 2. Increased sister telomere loss (STL) upon FEN1 depletion.
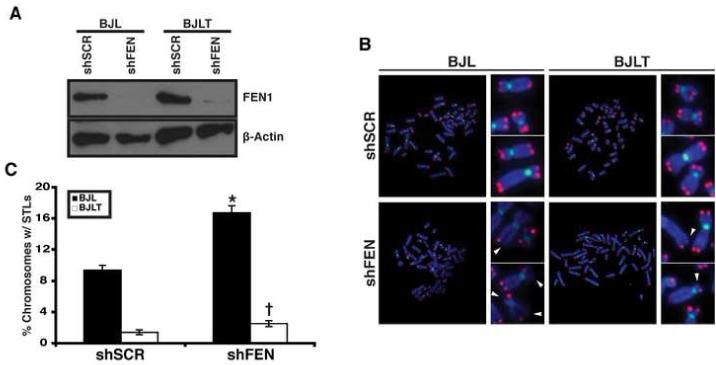
(A) Short hairpins against a scrambled sequence (shSCR) or FEN1 (shFEN) were expressed in BJ fibroblasts expressing SV40 early region, in the absence (BJL) or presence of telomerase (BJLT). FEN1 (upper panel) and β-Actin (lower panel) protein levels were assessed by Western blot analysis. (B) Representative metaphases from BJL and BJLT cells following shRNA expression. FISH analysis was conducted using Cy3-[CCCTAA]3 (red) and FLU-labeled centromere probes (green). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Arrowheads indicate missing sister telomeres. The side panels show higher magnification images of the metaphase chromosomes. (C) Quantification of chromosomes displaying STLs following shRNA expression in BJL (black bars) and BJLT (white bars) cells. A minimum of 60 metaphases, from two independent experiments, was analyzed per treatment in a blinded fashion (* P<0.0001; †, P<0.05). The error bars represent SEM.
Depletion of FEN1 leads to sister telomere loss (Figure 2) resulting in recognition of telomeres by the DNA damage machinery (Figure 1). Several papers have demonstrated that telomerase is preferentially recruited to the shortest telomeres [15-18], raising the possibility that telomerase may compensate for FEN1 depletion at the telomere. Therefore, we expressed shSCR or shFEN in BJL cells expressing telomerase (BJLT; Figure 2A). We found that in the presence of telomerase, STLs were significantly reduced upon FEN1 depletion. Indeed, in BJLT cells only 2.6% of chromosomes displayed STLs upon FEN1 depletion (P<0.05; Figure 2B and C), which was significantly lower than the 16.8% STLs observed in BJL cells devoid of telomerase activity. Together, these results demonstrate that telomerase compensates for FEN1 depletion at the telomere.
The above observation was reminiscent of a report demonstrating that mutations in WRN, a known FEN1 binding protein, led to STLs that were limited to telomeres replicated by lagging strand DNA synthesis [19]. Given FEN1’s known role in lagging strand DNA replication and its interaction with the WRN protein [20, 21], we investigated whether FEN1 depletion compromised lagging strand DNA synthesis of the telomere. To carry out these studies, we employed chromosome orientation fluorescent in situ hybridization (CO-FISH), which is capable of distinguishing between telomeres replicated by leading versus lagging strand DNA synthesis (Figure 3A). CO-FISH analysis revealed that reduction in FEN1 protein levels led to a specific loss of the lagging strand telomere (Figure 3B and 3C). BJL cells expressing the control hairpin (shSCR) had similar levels of telomere loss of both leading and lagging strands (4.4% and 3.8%, Figure 3C). Strikingly, cells expressing shFEN exhibited a significant 2-fold increase in loss of the lagging strand sister telomeres (9.5% versus 3.8%, P<0.0001; Figure 3C), with no change in the number of leading strand STLs observed. Together, these data demonstrate that FEN1 depletion exclusively compromises lagging strand DNA replication at the telomere.
Fig. 3. FEN1 depletion leads to loss of telomeres replicated by lagging strand DNA synthesis.
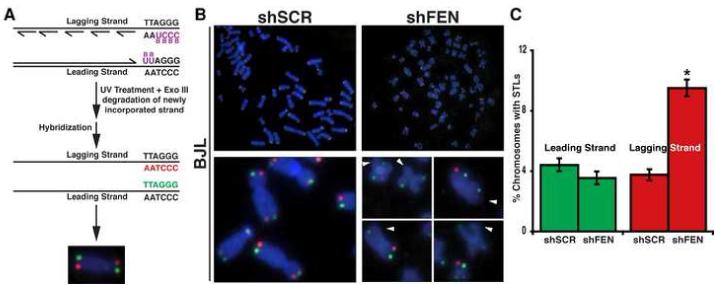
(A) CO-FISH schematic. Newly synthesized strands incorporate BrdU and BrdC. UV and ExoIII treatment results in degradation of newly synthesized DNA containing BrdU and BrdC, and the template strands are hybridized with Cy3-[CCCTAA]3 (red, lagging strand) and FLU-[TTAGGG]3 (green, leading strand) probes. (B) Representative CO-FISH of metaphases from BJL cells expressing the indicated hairpins. Color schemes are as described in (A). The arrowheads indicate missing telomeres. (C) Quantification of (B). A minimum of 60 metaphases from two independent experiments was analyzed per treatment in a blinded fashion (*P<0.0001). The error bars represent SEM.
Several biochemical functions have been ascribed to FEN1 [3, 4]. To determine whether FEN1 nuclease activity or its interaction with the WRN protein is necessary for telomere stability we created a novel vector, pResQ, capable of expressing both an shRNA and a cDNA (Figure S4), and conducted genetic rescue experiments. We also designed a second shRNA targeted to the FEN1 3′ UTR (shFEN3), which facilitated our analysis by allowing us to deplete endogenous protein, while having no effect on mRNA produced from a cDNA devoid of the 3′UTR sequence. The FEN1 mutants utilized were D181A (DA), which completely lacks nuclease activity [22] and delta C (ΔC; 20 amino acid deletion on the C-terminus), which retains partial ability to process flap structures with the replication clamp, PCNA, but is unable to bind the WRN protein [23, 24]. Cells transduced with the indicated vector confirmed that endogenous FEN1 protein was significantly reduced and the wild-type and mutant proteins were expressed, albeit to varying levels (Figure 4A).
Fig. 4. The nuclease activity and C-terminal region of FEN1 are essential for its role at the telomere.

(A) Western blot analysis of endogenous and ectopically expressed FEN1 proteins following transduction of BJL cells (upper panel). The ectopically expressed FEN1 proteins carry a triple-flag tag (3XF), which produces a larger protein. Abbreviations are as follows: Ctrl indicates control cells in which GFP was ectopically expressed, 3XF-F indicates the ectopically expressed wildtype and DA mutant, 3XF-FC indicates the ΔC mutant, and Endog indicates the endogenous FEN1 protein (*, Non-specific band). β-Actin (lower panel) is shown as a loading control. (B) Quantification of STLs after CO-FISH on metaphase chromosomes following depletion of the endogenous protein and expression of the indicated FEN1 protein, depicted as percentage of chromosomes with missing leading and lagging strand telomeres. A minimum of 60 metaphases from two independent experiments was analyzed per treatment in a blinded fashion (†, P<0.0001). The error bars represent SEM. (C) 293T cells transduced with flag-tagged FEN1 mutants, DA and ΔC and subjected to immunoprecipitation (IP) with an anti-TRF2 antibody as described in the supplemental. The presence of TRF2 and the FEN1 mutants were detected by immunoblot (IB) using anti-TRF2 and anti-Flag antibodies, respectively. The input lane indicates 10% of total protein used per immunoprecipitation. (D) FEN1 mutants localize to the telomere. Representative ChIP analysis of 293T cells (Ctrl) or 293T cells transduced with wildtype (WT) or a FEN1 mutant (DA and ΔC), subjected to immunoprecipitation with the M2 flag antibody. Precipitated DNA was probed for the presence of telomeric sequences as described in the supplemental information. The inputs indicate 0.2%, 0.1% and 0.04% of the total protein extract.
We next determined whether the wildtype or mutant FEN1 proteins could rescue the telomere dysfunction observed upon FEN1 depletion. No significant change in leading strand STLs was observed following expression of any of the FEN1 proteins (Figure S5 and Figure 4B). Reduction of FEN1 protein with a second independent hairpin (shFEN3) led to a 2-fold increase in lagging strand STLs (Figure 4B). Importantly, expression of wild-type FEN1 (WT) rescued the lagging strand STL phenotype, indicating that the observed phenotype was specific to the depletion of FEN1 (Figure 4B). In contrast, ectopic expression of either the DA nuclease deficient mutant or the ΔC mutant was unable to rescue the lagging strand STL phenotype upon FEN1 depletion (Figure S5 and Figure 4B). These observations suggest that both the nuclease activity and FEN1’s interaction with WRN is critical for its role at the telomere.
To rule out that failure of the mutants to rescue FEN1 depletion resulted from an inability of the mutants to interact with TRF2 or localize to the telomere, we conducted immunoprecipitation and ChIP experiments. As shown in Figure 4C, the DA mutant, but not the ΔC mutant retained the ability to interact with TRF2, indicating that the C-terminal 20 amino acids are critical for FEN1 binding to TRF2. This also suggests that the phenotype of the ΔC mutant may be compounded by the combined loss of TRF2 and WRN interactions. To determine whether the mutant FEN1 proteins retained the ability to localize to the telomere, we also carried out ChIP analysis on lysates from 293T cells ectopically expressing epitope-tagged proteins. We found that all three FEN1 proteins associated with telomeric DNA (Figure 4D). These results, together with the functional data presented above, demonstrate that failure to rescue sister telomere loss was not due to an inability of the mutants to localize to the telomere.
FEN1 is a DNA replication and repair protein [4, 25]. To explore the possible impact of FEN1 depletion on the genome at large, we carried out karyotypic analysis of BJL and BJLT cells. Upon FEN1 depletion, BJL cells displayed a mild increase in genomic instability as evidenced by a modest increase in the number of chromatid breaks and chromosome gaps observed (Table S1). Because telomerase rescues the telomere phenotype observed upon FEN1 depletion (Figure 2), any chromosomal abnormalities observed in BJLT cells depleted of FEN1 would be attributed to a non-telomeric effect. Karyotypic analysis of BJLT cells revealed no significant differences between cells expressing shSCR or shFEN (Table S1), indicating that the impact of FEN1 depletion on the genome is the result of telomere dysfunction.
FEN1 is a structure specific endonuclease that acts in DNA replication and repair. Here, we assessed FEN1’s role in telomere stability. In agreement with previous work [13, 14], we found that FEN1 is present at mammalian telomeres in a cell cycle dependent manner, and that it interacts with TRF2. This interaction requires the C-terminal region of FEN1. FEN1 depletion led to telomere dysfunction characterized by an increase in γH2AX at telomeres and sister telomere loss (STL). The latter was repressed by telomerase expression. CO-FISH analysis revealed that STLs were limited to telomeres replicated by lagging strand DNA synthesis. We further demonstrated that FEN1 nuclease activity and its C-terminal region are critical for its function at the telomere. FEN1 depletion revealed only a mild increase in genomic instability that was completely abolished in the presence of telomerase. Collectively, these data demonstrate that FEN1 is important for telomere stability and suggest that FEN1 is required for proficient replication and/or repair of telomeres.
Telomere repeat binding proteins interact with DNA replication and repair proteins to maintain telomere stability [1, 5]. Abrogation of these protein-protein interactions in both yeast and mammalian systems can have profound effects on telomere stability [2]. These observations raise the possibility that the telomere represents a specialized structure whose replication and stability is ensured by the coordinated efforts of numerous redundant systems [5]. Highly repetitive sequences such as those present in the telomere can adopt complex secondary structures that are challenging to replicate and have the potential to lead to stalled replication forks [5, 7]. If left unresolved, these can result in double strand breaks [26]. Given FEN1’s potential role in the reinitiation of stalled replication forks [27, 28], its absence is likely to compound the ability of the replication machinery to successfully transit the G-rich TTAGGG tracks. In support of this, our data demonstrate that FEN1 depletion results in specific loss of lagging strand-replicating sister telomeres. We propose that FEN1 is recruited to the telomere to facilitate replication and in its absence the replication machinery has a propensity to stall and/or inefficiently re-initiate stalled replication forks within the telomeric repeats. This hypothesis is particularly attractive in light of work demonstrating that loss of the Werner protein, which localizes with FEN1 at stalled replication forks thereby facilitating processing of branch migrating structures [28], phenocopies FEN1 depletion at the telomere [19]. In both the case of FEN1 depletion (this report) and WRN loss [19], telomerase rescues the telomere phenotype. Because telomerase is recruited to, and extends the shortest telomeres [15-18], its presence would be expected to rescue STLs by lengthening shortened telomeres created after a stalled-fork-induced break. Interestingly, the ΔC FEN1 mutant that does not interact with the WRN [24] or TRF2 protein is unable to rescue the telomere defect observed upon FEN1 depletion despite its ability to localize to the telomere. Because the ΔC mutant retains a partial ability to interact with PCNA [23, 24], this result suggests that it is FEN1’s repair function that is critical for its activity at the telomere.
Supplementary Material
Acknowledgments
This work was supported by the Sidney Kimmel Foundation for Cancer Research and the Edward Mallinckrodt, Jr. Foundation, and the National Centers of Research Resources of the National Institutes of Health (P41-RR00954). A.S. was supported by the Lucille P. Markey Program. S.A.S. is a Sidney Kimmel Scholar. We are grateful to Y. Tao for statistical analyses, G. Sharma for advice on metaphase analysis, R. Veile for expert assistance with cytogenetics, R. Verdun, J. Karlseder, and S. Bailey for helpful comments on ChIP and FISH, Y. Feng and G. Longmore for the pFLRu vector, members of the Stewart Laboratory for valuable discussions and W. Hahn, H. True, J. Weber, L. Michel, S. Gonzalo, Z. Nahle, and H. Piwnica-Worms for critical reviews.
The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 2.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 4.Shen B, Singh P, Liu R, Qiu J, Zheng L, Finger LD, Alas S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. Bioessays. 2005;27:717–729. doi: 10.1002/bies.20255. [DOI] [PubMed] [Google Scholar]
- 5.Gilson E, Geli V. How telomeres are replicated. Nat Rev Mol Cell Biol. 2007;8:825–838. doi: 10.1038/nrm2259. [DOI] [PubMed] [Google Scholar]
- 6.Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 7.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 8.Parenteau J, Wellinger RJ. Differential processing of leading- and lagging-strand ends at Saccharomyces cerevisiae telomeres revealed by the absence of Rad27p nuclease. Genetics. 2002;162:1583–1594. doi: 10.1093/genetics/162.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choe W, Budd M, Imamura O, Hoopes L, Campbell JL. Dynamic localization of an Okazaki fragment processing protein suggests a novel role in telomere replication. Mol Cell Biol. 2002;22:4202–4217. doi: 10.1128/MCB.22.12.4202-4217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomita K, Kibe T, Kang HY, Seo YS, Uritani M, Ushimaru T, Ueno M. Fission yeast Dna2 is required for generation of the telomeric single-strand overhang. Mol Cell Biol. 2004;24:9557–9567. doi: 10.1128/MCB.24.21.9557-9567.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng Z, Dheekollu J, Broccoli D, Dutta A, Lieberman PM. The origin recognition complex localizes to telomere repeats and prevents telomere-circle formation. Curr Biol. 2007;17:1989–1995. doi: 10.1016/j.cub.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 12.Parenteau J, Wellinger RJ. Accumulation of single-stranded DNA and destabilization of telomeric repeats in yeast mutant strains carrying a deletion of RAD27. Mol Cell Biol. 1999;19:4143–4152. doi: 10.1128/mcb.19.6.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–720. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 14.Muftuoglu M, Wong HK, Imam SZ, Wilson DM, 3rd, Bohr VA, Opresko PL. Telomere Repeat Binding Factor 2 Interacts with Base Excision Repair Proteins and Stimulates DNA Synthesis by DNA Polymerase {beta} Cancer Res. 2006;66:113–124. doi: 10.1158/0008-5472.CAN-05-2742. [DOI] [PubMed] [Google Scholar]
- 15.Teixeira MT, Arneric M, Sperisen P, Lingner J. Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell. 2004;117:323–335. doi: 10.1016/s0092-8674(04)00334-4. [DOI] [PubMed] [Google Scholar]
- 16.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/s0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 17.Grobelny JV, Kulp-McEliece M, Broccoli D. Effects of reconstitution of telomerase activity on telomere maintenance by the alternative lengthening of telomeres (ALT) pathway. Hum Mol Genet. 2001;10:1953–1961. doi: 10.1093/hmg/10.18.1953. [DOI] [PubMed] [Google Scholar]
- 18.Perrem K, Colgin LM, Neumann AA, Yeager TR, Reddel RR. Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol Cell Biol. 2001;21:3862–3875. doi: 10.1128/MCB.21.12.3862-3875.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Li J, Harrington J, Lieber MR, Burgers PM. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J Biol Chem. 1995;270:22109–22112. doi: 10.1074/jbc.270.38.22109. [DOI] [PubMed] [Google Scholar]
- 21.Brosh RM, Jr., von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. Embo J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen B, Nolan JP, Sklar LA, Park MS. Essential amino acids for substrate binding and catalysis of human flap endonuclease 1. J Biol Chem. 1996;271:9173–9176. doi: 10.1074/jbc.271.16.9173. [DOI] [PubMed] [Google Scholar]
- 23.Stucki M, Jonsson ZO, Hubscher U. In eukaryotic flap endonuclease 1, the C terminus is essential for substrate binding. J Biol Chem. 2001;276:7843–7849. doi: 10.1074/jbc.M008829200. [DOI] [PubMed] [Google Scholar]
- 24.Sharma S, Sommers JA, Gary RK, Friedrich-Heineken E, Hubscher U, Brosh RM., Jr. The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–6781. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi ML, Purohit V, Brandt PD, Bambara RA. Lagging strand replication proteins in genome stability and DNA repair. Chem Rev. 2006;106:453–473. doi: 10.1021/cr040497l. [DOI] [PubMed] [Google Scholar]
- 26.Branzei D, Foiani M. The DNA damage response during DNA replication. Curr Opin Cell Biol. 2005;17:568–575. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Zheng L, Zhou M, Chai Q, Parrish J, Xue D, Patrick SM, Turchi JJ, Yannone SM, Chen D, Shen B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, Kao HI, Bambara RA, Brosh RM., Jr. WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol Biol Cell. 2004;15:734–750. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.