


Abstract
Lsr2 is a small, basic protein present in Mycobacterium and related actinomycetes. Recent studies suggest that Lsr2 is a regulatory protein involved in multiple cellular processes including cell wall biosynthesis and antibiotic resistance. However, the underlying molecular mechanisms remain unknown. In this article, we performed biochemical studies of Lsr2–DNA interactions and structure–function analysis of Lsr2. Analysis by atomic force microscopy revealed that Lsr2 has the ability to bridge distant DNA segments, suggesting that Lsr2 plays a role in the overall organization and compactness of the nucleoid. Mutational analysis identified critical residues and selection of dominant negative mutants demonstrated that both DNA binding and protein oligomerization are essential for the normal functions of Lsr2 in vivo. These results provide strong evidence that Lsr2 is a DNA bridging protein, which represents the first identification of such proteins in bacteria phylogenetically distant from the Enterobacteriaceae. DNA bridging by Lsr2 also provides a mechanism of transcriptional regulation by Lsr2.
INTRODUCTION
The bacterial chromosome is highly organized and forms a compact structure called the nucleoid. A number of factors, including DNA supercoiling, macromolecular crowding, and nucleoid-associated proteins (NAPs) contribute to nucleoid compaction (1–3). At least 12 polypeptides in Escherichia coli have been described as NAPs (4). The best characterized and most abundant members are the histone-like nucleoid structuring (H-NS) protein, its homolog StpA, the factor for inversion stimulation (Fis), the histone-like protein from E. coli U93 (HU) and its near relative, integration host factor (IHF) (5). H-NS interacts non-specifically with DNA but has a preference for intrinsically curved DNA (6). HU also binds to DNA non-specifically but has a higher affinity to distorted DNA such as kinked, gapped and cruciform structures (7). These two proteins employ different mechanisms contributing to nucleoid compaction: HU reduces the effective volume occupied by the DNA molecule by inducing local bends (8), whereas H-NS has the ability to bridge adjacent tracts of DNA (9). In addition, these proteins act as transcriptional regulators. H-NS is considered a global transcription repressor that silences a large number of genes involved in stress response and virulence pathways (9–12). The global regulatory role of H-NS can be explained by the capacity of the H-NS protein to bridge DNA, to interact with RNA and proteins in highly ordered nucleoprotein complexes (13), the post-translational modifications observed on H-NS proteins (14), and the demonstrated link with metabolites such as ppGpp (15). HU has an important role in the initiation of replication and gene regulation (16,17).
Despite its important role, on the basis of sequence similarity, H-NS-related proteins have only been found in α-, β-, and γ-proteobacteria thus far (18), in contrast to proteins such as HU, which are present in all bacteria (8) including mycobacteria (19–21).
Mycobacterium tuberculosis, the causative agent of tuberculosis, is one of the most successful and deadly pathogens, owing in part to its ability to adapt to and persist in diverse host environments (22). Host adaptation requires controlled regulation of key genes that allow M. tuberculosis to alter its physiology in response to changes in environmental stimuli. Identification of regulatory proteins involved in these processes will help to understand how M. tuberculosis achieves this goal.
Lsr2 is a small (∼12 kDa) and basic protein found in all mycobacterial genomes that have been sequenced so far (23,24). Lsr2 homologs are also present in related actinomycetes including Streptomyces, Nocardia and Rhodococcus. Despite the early identification of Lsr2 in M. leprae as an immunodominant T-cell antigen (25), Lsr2 shows no significant sequence homology to any known proteins and its biological functions remained unknown until our recent studies. We showed by genetic experiments that Lsr2 is involved in the biosynthesis of mycolyl-diacylglycerols, an apolar lipid in the cell wall of M. smegmatis (23). We proposed that Lsr2 is a DNA-binding protein playing a regulatory role (23). This premise is further supported by two recent studies. Colangeli et al. (24)showed that Lsr2 negatively regulates the iniBAC operon, which encodes a multidrug efflux system in M. tuberculosis. Recently, we have further shown by genetic experiments that Lsr2 participates in the negative regulation of the mps operon, the biosynthetic locus of glycopeptidolipids in M. smegmatis (Kocíncová et al., submitted for publication). Comparative transcriptome analysis also revealed a number of genes that are potentially regulated by Lsr2 (24). Colangeli et al. also showed that Lsr2 binds to DNA non-specifically and hypothesized that Lsr2 is a histone-like protein (24). Together, these studies indicate that Lsr2 is a regulatory protein involved in multiple cellular processes. However, the molecular mechanisms by which these activities are accomplished by Lsr2 protein and the nature of the structural alteration imposed on the DNA by Lsr2 remain unknown. Here, we provide experimental evidence that Lsr2 is a DNA-bridging protein that likely influences the organization of bacterial chromatin and gene regulation. Analysis by atomic force microscopy (AFM) revealed that Lsr2 has the ability to bridge DNA, which provides a direct explanation for the mechanism of transcriptional regulation by Lsr2. We show that Lsr2 exhibits preferential binding to AT-rich sequences irrespective of DNA topology. Furthermore, we have identified residues that are critical for Lsr2 functions and demonstrated that the abilities of Lsr2 to bind DNA and to form oligomers are both essential for the function of Lsr2 in vivo. Together, our study provides strong evidence that Lsr2 is a DNA-bridging protein in mycobacteria and related actinomycetes, and represents the first identification of a H-NS like protein outside of Enterobacteriaceae.
MATERIALS AND METHODS
Purification of Lsr2 protein
The open reading frame of the lsr2 gene of M. tuberculosis was PCR amplified using the forward primer 5′-CAGAAGCTTATGGCGAAGAAAGTAACCGTCACC-3′ and the reverse primer 5′-CGCCTCGAGGGTCGCCGCGTGGTATGC-3′. The amplified DNA fragment was digested with HindIII and XhoI and then cloned into pET21d pre-treated with the same enzymes, to produce an expression plasmid pLSR2. The pLSR2 was transformed into E. coli BL21 (DE3) and protein expression induced at 37°C with 1 mM IPTG at OD600 = 0.6 for 3 h. The recombinant Lsr2 protein (His-tagged at the C-terminal) was purified by affinity chromatography on Ni-NTA agarose using the standard protocols (Qiagen). The purity of the Lsr2 protein was estimated to be >95%.
Oligomers, fragments and plasmids
Large DNA fragments used for the gel shift experiments were generated by PCR amplification using the following appropriate primer pairs: 5′-TGCTGGTCAATTAGGCACTG-3′ (forward) and 5′-GCTCTCCTCGAGTGGAAGTG-3′ (reverse) for 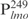 ; 5′- TGCTGGTCAATTAGGCACTG-3′ (forward) and 5′-GGGAGCCAGCGGATAGAT-3′ (reverse) for
; 5′- TGCTGGTCAATTAGGCACTG-3′ (forward) and 5′-GGGAGCCAGCGGATAGAT-3′ (reverse) for 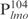 ; 5′- CAGAGAAGTGGCGTCGGCAA-3′ (forward) and 5′- GTTTCTCCCGGCTGTTCAGC-3′ (reverse) for
; 5′- CAGAGAAGTGGCGTCGGCAA-3′ (forward) and 5′- GTTTCTCCCGGCTGTTCAGC-3′ (reverse) for 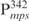 . Oligonucleotides were purchased from Operon (listed in Table 1). For small DNA fragments (<100 bp), pairs of single-stranded oligonucleotides at equal concentrations in 20 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 50 mM NaCl were annealed by incubation in a thermocycler at 95°C for 5 min and then cooled at a rate of 0.5°C/min to room temperature. Double-stranded DNA probes were gel purified and quantified before using in the gel shift assays. To make biotinylated
. Oligonucleotides were purchased from Operon (listed in Table 1). For small DNA fragments (<100 bp), pairs of single-stranded oligonucleotides at equal concentrations in 20 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 50 mM NaCl were annealed by incubation in a thermocycler at 95°C for 5 min and then cooled at a rate of 0.5°C/min to room temperature. Double-stranded DNA probes were gel purified and quantified before using in the gel shift assays. To make biotinylated 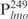 , biotinylated forward primer (biotin labeled at the 5′ end) described above was used in PCR amplifications.
, biotinylated forward primer (biotin labeled at the 5′ end) described above was used in PCR amplifications.
Table 1.
Short sequences used for the EMSA experiments
| Name | Bp | Sequence (5′–3′) |
|---|---|---|
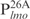 |
26 | TGCTGGTCAATTAGGCACTGCGTGAC |
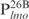 |
26 | AATTTATCGAGTCGCACAACCGGATC |
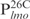 |
26 | ATCCATCCGTTATTTACGTCAATTTA |
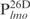 |
26 | AACGGATGATCTATCCGCTGGCTCCC |
| C42 | 42 | AAAAATCTCTAAAAAATCTCTAAAAA TCTCTAAAAAATCTCT |
| NC42 | 42 | TCTAATCTCTCTCTAATCTCTTCTAATC TCTCTCTAATCTCT |
All the sequences used were double stranded.
Electrophoretic mobility shift assays (EMSAs)
DNA was incubated for 30 min at room temperature at indicated amounts of Lsr2 in a total volume of 20 µl buffer (10 mM Tris–HCl, pH 7.5, 50 mM KCl, 1 mM DTT, 5 mM MgCl2, 0.05% NP-40 and 2.5% glycerol). The DNA–protein mix was then loaded on polyacrylamide gels (4% for large DNA fragments, 8% for small fragments) or 1% agarose gels and run in TBE buffer. The gel was stained with SYBR green (Molecular Probes) in TBE, and detected by a Typhoon system (GE Healthcare) or a Geldoc scanner (Biorad).
For competition assays, biotinylated DNA was used as the probe in gel shift experiments and competed with excess non-specific DNA as indicated. The gel was detected using the chemiluminescent nucleic acid detection kit (Pierce), following manufacturer's instructions. Briefly, the gel was electrotransferred to Hybond-N+ nylon membrane (Amersham Pharmacia), UV-crosslinked using Stratalinker (Stratagene) and developed.
Site-directed mutagenesis of Lsr2
Substitutions of individual amino acids by alanine were performed using the QuickChange site-directed mutagenesis kit according to the recommendations of the manufacturer (Stratagene). Each mutation was confirmed by DNA sequencing. For in vivo complementation experiments, plasmid pLSR2-HIS was used as the template for mutagenesis, which contains the lsr2 gene of M. smegmatis fused with a histidine tag at the C-terminal and was capable of complementing the morphological phenotype of the Δlsr2 mutant (23). Each mutant construct was transformed into the Δlsr2 mutant by electroporation and transformants selected on Middlebrook 7H11 agar containing hygromycin (75 μg/ml). For expression and purification of Lsr2 mutant proteins, pLSR2 plasmid described above was used as the template for mutagenesis, which contains the M. tuberculosis lsr2 gene. Each mutant construct was transformed into E. coli BL21 (DE3), and Lsr2 mutant protein was expressed and purified as described above. The Lsr2 of M. smegmatis contains two extra amino acids than that of M. tuberculosis. For conveniences, we used the numbering of amino acids from M. smegmatis Lsr2 throughout the article.
In vitro cross-linking of Lsr2
Cross-lining experiments were performed with purified Lsr2 WT and mutant proteins (6 μg per sample) using the protocol described in our previous publication (23). The samples were separated on 14% Tricine SDS gels and analyzed by western blot using an anti-His antibody.
Atomic force microscopy
Linear pMPS was generated by cloning the 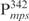 fragment into pDrive, and then linearized by digestion with HindIII. Linear pDrive was also prepared by digestion with HindIII. Relaxed, circular pDrive was obtained from closed circular (supercoiled) molecules by partial digestion with DNase I as described previously (26). For AFM experiments, complexes of Lsr2 and DNA were formed by incubation of 175 ng DNA with 16, 32 or 64 ng of Lsr2, which corresponds to a protein/DNA ratio of 1 dimer: 260 bp; 1 dimer: 130 bp and 1 dimer: 65 bp respectively, in 20 µl reaction volumes containing 10 mM Tris–HCl, pH 7.5, 50 mM KCl, 1 mM DTT, 5 mM MgCl2, 0.05% NP-40 and 2.5% glycerol for 30 min at room temperature. This mixture was diluted 2.5 times with water and deposited on freshly cleaved SPI-3 mica (SPI, West Chester, PA, USA). DNA without protein was deposited under the same buffer conditions. After 2 min the mica disc was rinsed with filtered HPLC-grade water (Sigma), excess water removed with absorbent tissue paper and dried under a stream of filtered nitrogen. Tapping mode AFM (TMAFM) images were acquired at ambient temperature on a Digital Instruments Nanoscope IIIa Multimode scanning probe microscope (SPM, Digital Instruments/Veeco, Santa Barbara, CA, USA) equipped with an ‘E’ scanner having a maximum lateral scan area of 14.6 μm × 14.6 μm, using 125 μm long silicon diving-board TESP cantilevers. The AFM cantilevers were irradiated with UV light prior to use to remove any adventitious organic contaminants. All AFM images were captured as 512 × 512 pixel images at scan rates of between 2 and 3 Hz using a tip oscillation frequency of ∼270 kHz. AFM image analysis was conducted using the Digital Instruments Nanoscope software (version 4.42r9, Veeco, Santa Barbara, CA, USA).
fragment into pDrive, and then linearized by digestion with HindIII. Linear pDrive was also prepared by digestion with HindIII. Relaxed, circular pDrive was obtained from closed circular (supercoiled) molecules by partial digestion with DNase I as described previously (26). For AFM experiments, complexes of Lsr2 and DNA were formed by incubation of 175 ng DNA with 16, 32 or 64 ng of Lsr2, which corresponds to a protein/DNA ratio of 1 dimer: 260 bp; 1 dimer: 130 bp and 1 dimer: 65 bp respectively, in 20 µl reaction volumes containing 10 mM Tris–HCl, pH 7.5, 50 mM KCl, 1 mM DTT, 5 mM MgCl2, 0.05% NP-40 and 2.5% glycerol for 30 min at room temperature. This mixture was diluted 2.5 times with water and deposited on freshly cleaved SPI-3 mica (SPI, West Chester, PA, USA). DNA without protein was deposited under the same buffer conditions. After 2 min the mica disc was rinsed with filtered HPLC-grade water (Sigma), excess water removed with absorbent tissue paper and dried under a stream of filtered nitrogen. Tapping mode AFM (TMAFM) images were acquired at ambient temperature on a Digital Instruments Nanoscope IIIa Multimode scanning probe microscope (SPM, Digital Instruments/Veeco, Santa Barbara, CA, USA) equipped with an ‘E’ scanner having a maximum lateral scan area of 14.6 μm × 14.6 μm, using 125 μm long silicon diving-board TESP cantilevers. The AFM cantilevers were irradiated with UV light prior to use to remove any adventitious organic contaminants. All AFM images were captured as 512 × 512 pixel images at scan rates of between 2 and 3 Hz using a tip oscillation frequency of ∼270 kHz. AFM image analysis was conducted using the Digital Instruments Nanoscope software (version 4.42r9, Veeco, Santa Barbara, CA, USA).
RESULTS
Lsr2 forms multiple complexes with DNA
Our previous study suggested that Lsr2 functions as a regulator involved in the biosynthesis of cell wall lipids in M. smegmatis (23). To identify potential target genes of Lsr2, we performed a comparative two-dimensional (2D)-SDS electrophoretic analysis of cell lysate proteins prepared from the Δlsr2 mutant and its parental, wild-type (WT) strain M. smegmatis mc2155. Several proteins were differentially expressed between the Δlsr2 mutant and the WT (Supplementary Figure S1). One protein produced at a higher level in the Δlsr2 mutant than the WT was identified as lactate 2-monoxygenase (LMO) (MSMEG_3962 in the recent annotation of M. smegmatis genome at TIGR center http://tigrblast.tigr.org/), by trypsin digestion followed by mass spectrometric (MALDI-TOF) analysis (data not shown). Although the functional significance of this observation remains unknown, this result suggests that Lsr2 negatively regulates the lmo gene. We have recently shown by genetic experiments that Lsr2 down-regulates the expression of mps operon, which encodes the biosynthetic machinery of glycopeptidolipids in M. smegmatis (Kocíncová et al., submitted for publication). However, it remained to be determined if Lsr2 directly regulates these genes. To address this question, we examined the interaction of purified Lsr2 protein with the promoter sequences of lmo and mbtH genes (mbtH is the first gene of the mps operon) using electrophoretic mobility shift assays (EMSAs). We chose to study the Lsr2 of M. tuberculosis since it is functionally interchangeable with Lsr2 of M. smegmatis (Kocíncová et al., submitted for publication). The Lsr2 of M. tuberculosis was expressed and purified from E. coli as a C-terminal His-tagged protein (data not shown). Double-stranded DNA fragments upstream of the start codon of lmo and mbtH, 249 bp (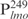 ) and 342 bp (
) and 342 bp (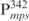 ), respectively, were PCR amplified and used as probes in the gel shift experiments. The protein–DNA complexes were resolved in 4% polyacrylamide gels, detected by staining with SYBR green and scanning by a Typhoon system. Our results showed that Lsr2 bound both DNA fragments in a concentration-dependent manner (Figure 1A and Supplementary Figure S2). A number of protein–DNA complexes were formed between Lsr2 and both DNA fragments, which could not be resolved on the 4% polyacrylamide gel. To investigate this further, a shorter DNA fragment, 104 bp upstream of the lmo starting codon (
), respectively, were PCR amplified and used as probes in the gel shift experiments. The protein–DNA complexes were resolved in 4% polyacrylamide gels, detected by staining with SYBR green and scanning by a Typhoon system. Our results showed that Lsr2 bound both DNA fragments in a concentration-dependent manner (Figure 1A and Supplementary Figure S2). A number of protein–DNA complexes were formed between Lsr2 and both DNA fragments, which could not be resolved on the 4% polyacrylamide gel. To investigate this further, a shorter DNA fragment, 104 bp upstream of the lmo starting codon (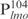 ), was amplified by PCR and used in the EMSAs. Lsr2 bound to
), was amplified by PCR and used in the EMSAs. Lsr2 bound to 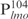 at apparently the same affinity as for
at apparently the same affinity as for 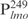 (lanes 6–10, Figure 1A). With saturating amounts of Lsr2, ∼11 distinct Lsr2-
(lanes 6–10, Figure 1A). With saturating amounts of Lsr2, ∼11 distinct Lsr2-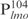 complexes were formed (C1–C11, Figure 1A). Because lmo is a target gene of Lsr2, we wondered if there is a high-affinity binding site within its promoter sequence. For this purpose, we further divided the
complexes were formed (C1–C11, Figure 1A). Because lmo is a target gene of Lsr2, we wondered if there is a high-affinity binding site within its promoter sequence. For this purpose, we further divided the 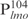 into four even-length fragments each containing 26 bp, which together cover the entire 104 bp of
into four even-length fragments each containing 26 bp, which together cover the entire 104 bp of 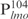 . These fragments, named
. These fragments, named 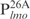 ,
, 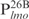 ,
, 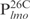 and
and 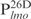 , were chemically synthesized, annealed and their interactions with Lsr2 examined by performing EMSAs. Lsr2 protein bound all four 26 bp fragments, each forming three protein–DNA complexes (Figure 1B and C). The affinity of Lsr2 for
, were chemically synthesized, annealed and their interactions with Lsr2 examined by performing EMSAs. Lsr2 protein bound all four 26 bp fragments, each forming three protein–DNA complexes (Figure 1B and C). The affinity of Lsr2 for 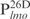 appears to be lower than for the other three DNA fragments. These results suggest Lsr2 either has multiple binding sites or does not display a sequence-specific site on DNA. To address this question, we performed EMSAs using biotinylated
appears to be lower than for the other three DNA fragments. These results suggest Lsr2 either has multiple binding sites or does not display a sequence-specific site on DNA. To address this question, we performed EMSAs using biotinylated 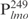 as the probe and competed with unlabeled specific and non-specific DNA (Figure 1D). The Lsr2-
as the probe and competed with unlabeled specific and non-specific DNA (Figure 1D). The Lsr2-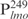 interaction was efficiently competed by excessive poly (dI-dC) and salmon sperm DNA but less efficiently by poly (dG-dC) (Figure 1D). Two conclusions can be drawn from these experiments:
interaction was efficiently competed by excessive poly (dI-dC) and salmon sperm DNA but less efficiently by poly (dG-dC) (Figure 1D). Two conclusions can be drawn from these experiments:
Figure 1.

Detection of Lsr2–DNA complexes by EMSAs. (A) DNA fragment (50 ng each) 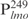 (lanes 1–5) or
(lanes 1–5) or 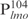 (lanes 6–10) was incubated with the indicated amounts of Lsr2 and analyzed on 4% polyacrylamide gel and stained with SYBR green. C: protein–DNA complex; (B) DNA fragment (50 ng each)
(lanes 6–10) was incubated with the indicated amounts of Lsr2 and analyzed on 4% polyacrylamide gel and stained with SYBR green. C: protein–DNA complex; (B) DNA fragment (50 ng each) 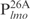 (lanes 1–5) or
(lanes 1–5) or 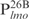 (lanes 6–10) was incubated with the indicated amounts of Lsr2 and analyzed on 8% polyacrylamide gel and stained with SYBR green. (C) The same as (B) except probes
(lanes 6–10) was incubated with the indicated amounts of Lsr2 and analyzed on 8% polyacrylamide gel and stained with SYBR green. (C) The same as (B) except probes 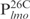 (lanes 1–5) and
(lanes 1–5) and 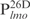 (lanes –610) were used. (D) Biotinylated
(lanes –610) were used. (D) Biotinylated 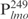 (0.05 ng) was incubated with the indicated amounts of Lsr2 (lanes 1–6) and then competed with non-specific DNA (lanes 7–10). Lanes 7–10: biotinylated
(0.05 ng) was incubated with the indicated amounts of Lsr2 (lanes 1–6) and then competed with non-specific DNA (lanes 7–10). Lanes 7–10: biotinylated 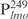 (0.05 ng) pre-incubated with 160 ng Lsr2 was competed with 12.5 ng of unlabeled
(0.05 ng) pre-incubated with 160 ng Lsr2 was competed with 12.5 ng of unlabeled 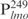 (lane 7), salmon sperm (SS) DNA (lane 8), poly (dI-dC) (lane 9), and poly (dG-dC). The samples were run on 4% polyacrylamide gel and detected by chemiluminescence kit. (E) A 1 kb DNA ladder (50 ng, Fermentas) with size ranging from 0.25 to 10 kb was incubated with the indicated amounts of Lsr2 and analyzed on 1% agarose gel. (F) Fragments (50 ng each) NC42 (lanes 1–5) and C42 (lanes 6–10) were incubated with the indicated amounts of Lsr2 and analyzed on 6% polyacrylamide gel. C: protein–DNA complex.
(lane 7), salmon sperm (SS) DNA (lane 8), poly (dI-dC) (lane 9), and poly (dG-dC). The samples were run on 4% polyacrylamide gel and detected by chemiluminescence kit. (E) A 1 kb DNA ladder (50 ng, Fermentas) with size ranging from 0.25 to 10 kb was incubated with the indicated amounts of Lsr2 and analyzed on 1% agarose gel. (F) Fragments (50 ng each) NC42 (lanes 1–5) and C42 (lanes 6–10) were incubated with the indicated amounts of Lsr2 and analyzed on 6% polyacrylamide gel. C: protein–DNA complex.
First, Lsr2 binds to DNA in a relatively sequence-independent manner and forms multiple protein–DNA complexes. The number of Lsr2–DNA complexes obtained with a given double-stranded DNA fragment appears to correlate with the length of DNA. Thus, 11 Lsr2–DNA complexes were formed with 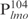 and three complexes formed with the 26 bp DNA fragments. Importantly, the ability of Lsr2 to form multiple complexes with DNA is not restricted to the lmo promoter sequences. The same result was obtained with the mps promoter sequences (Supplementary Figure S2), as well as non-specific DNA sequences like a random internal fragment of Lsr2 open-reading frame or a cloning vector pDrive (data not shown), thus reflecting an intrinsic property of Lsr2 protein. However, it appears that Lsr2 protein has a higher affinity for longer DNA fragments. This is evident by comparing the binding of Lsr2 protein to
and three complexes formed with the 26 bp DNA fragments. Importantly, the ability of Lsr2 to form multiple complexes with DNA is not restricted to the lmo promoter sequences. The same result was obtained with the mps promoter sequences (Supplementary Figure S2), as well as non-specific DNA sequences like a random internal fragment of Lsr2 open-reading frame or a cloning vector pDrive (data not shown), thus reflecting an intrinsic property of Lsr2 protein. However, it appears that Lsr2 protein has a higher affinity for longer DNA fragments. This is evident by comparing the binding of Lsr2 protein to 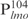 and the four smaller 26 bp fragments
and the four smaller 26 bp fragments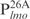 ,
, 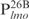 ,
, 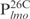 and
and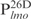 . Nearly all
. Nearly all 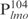 was shifted at the highest Lsr2 concentration tested (Figure 1A). In contrast, only a small fraction of the 26 bp fragments were shifted even though the same concentrations of DNA and protein used (Figure 1B and C). This is further confirmed by EMSAs shown in Figure 1E, in which a 1 kb DNA ladder was incubated with Lsr2 protein. Larger DNA fragments were shifted with less Lsr2 protein than the smaller ones (lane 5, Figure 1E). The interpretations and implications of these results are described in ‘Discussion’ section.
was shifted at the highest Lsr2 concentration tested (Figure 1A). In contrast, only a small fraction of the 26 bp fragments were shifted even though the same concentrations of DNA and protein used (Figure 1B and C). This is further confirmed by EMSAs shown in Figure 1E, in which a 1 kb DNA ladder was incubated with Lsr2 protein. Larger DNA fragments were shifted with less Lsr2 protein than the smaller ones (lane 5, Figure 1E). The interpretations and implications of these results are described in ‘Discussion’ section.
Second, Lsr2 has a preference for AT-rich DNA sequences. Although Lsr2 was able to bind all four 26 bp fragments at the lmo promoter region, its binding affinity for 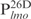 appears lower than for the other three DNA sequences of equal length, i.e.
appears lower than for the other three DNA sequences of equal length, i.e. 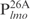 ,
, 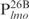 and
and 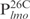 . Inspection of individual probes reveal that,
. Inspection of individual probes reveal that, 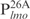
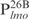 and
and 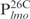 are relatively AT-rich, with 46, 54 and 65% of A+T, respectively, whereas
are relatively AT-rich, with 46, 54 and 65% of A+T, respectively, whereas 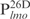 contains only 38% A+T (Table 1). In addition, consecutive AT sequences are present in
contains only 38% A+T (Table 1). In addition, consecutive AT sequences are present in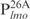 ,
, 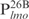 and
and 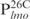 but not in
but not in 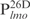 (Table 1, underlined). These results suggest that Lsr2 preferentially binds to AT-rich sequences. This is further supported by the competition experiment (Figure 1D). As described above, the Lsr2–
(Table 1, underlined). These results suggest that Lsr2 preferentially binds to AT-rich sequences. This is further supported by the competition experiment (Figure 1D). As described above, the Lsr2–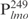 complexes were abrogated when competed with excessive poly (dI-dC) and salmon sperm DNA, similar to the effect by the unlabeled specific probe (Figure 1D). Interestingly, at equivalent concentrations, poly (dI-dC) was the most effective competitor, even more effective than the unlabeled specific probe. In contrast, poly (dG-dC) was the least effective competitor; majority of the Lsr2-
complexes were abrogated when competed with excessive poly (dI-dC) and salmon sperm DNA, similar to the effect by the unlabeled specific probe (Figure 1D). Interestingly, at equivalent concentrations, poly (dI-dC) was the most effective competitor, even more effective than the unlabeled specific probe. In contrast, poly (dG-dC) was the least effective competitor; majority of the Lsr2-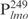 still remained in the presence of 250-fold excess poly (dG-dC) (lane 10, Figure 1D). It is well-known that I-C base pairs contribute a pattern of hydrogen bond donors and acceptors that is identical to G-C pairs in the major groove and A-T pairs in the minor groove. Because of this, poly (dI-dC) has been used as a specific competitor for studying interactions of transcription factors, such as TFIID and HMG, with their specific DNAs (27,28). Efficient competition of protein–DNA complexes by poly (dI-dC) in EMSAs indicate the binding of these proteins to minor grooves of AT-rich sequences such as the TATA box (27,28). Taken together, our results suggest that Lsr2 has a preference for AT-rich DNA and likely binds to the minor groove of A-T pairs. Our results are consistent with the observation made by Colangeli et al. (24), who found that the promoter regions of 15 genes presumably regulated directly by Lsr2 have on average a higher percentage of A+T composition than other regions of M. smegmatis genome.
still remained in the presence of 250-fold excess poly (dG-dC) (lane 10, Figure 1D). It is well-known that I-C base pairs contribute a pattern of hydrogen bond donors and acceptors that is identical to G-C pairs in the major groove and A-T pairs in the minor groove. Because of this, poly (dI-dC) has been used as a specific competitor for studying interactions of transcription factors, such as TFIID and HMG, with their specific DNAs (27,28). Efficient competition of protein–DNA complexes by poly (dI-dC) in EMSAs indicate the binding of these proteins to minor grooves of AT-rich sequences such as the TATA box (27,28). Taken together, our results suggest that Lsr2 has a preference for AT-rich DNA and likely binds to the minor groove of A-T pairs. Our results are consistent with the observation made by Colangeli et al. (24), who found that the promoter regions of 15 genes presumably regulated directly by Lsr2 have on average a higher percentage of A+T composition than other regions of M. smegmatis genome.
Equivalent affinity of Lsr2 for curved and non-curved DNA
To further investigate the structural basis for the preferential binding of Lsr2 to AT-rich DNA, we compared the binding of Lsr2 to curved and non-curved DNA. H-NS binds with higher affinity to strongly curved DNA when compared with non-curved or moderately curved DNA (29,30). The DNA fragment to which H-NS binds with the highest affinity contains in phase repeated A5 and A6 tracts, which induce a bend with a planar orientation (31). Recently, it was also shown that H-NS recognizes a 10 bp defined high-affinity site (32), which was actually also present in the previously used fragments with A5 and A6 tracts, providing an additional explanation for the preferential binding of Lsr2 to AT-rich DNA. We wondered if Lsr2 also exhibits preferential binding to fragments with A5 and A6 tracts. For this purpose, a pair of 42 bp double-stranded oligonucleotides (C42 and NC42) previously used by other groups to compare the binding to curved and non-curved DNA (33) were used for gel shift experiments. C42 contains repeats of A5 every 11 bp, which is naturally curved in the absence of proteins (33,34), whereas NC42 serves as a non-curved DNA control containing the same nucleotide composition but with the sequence scrambled (33,34) (Table 1). Interestingly, Lsr2 bound C42 and NC42 with equivalent affinities (Figure 1F). No marked preference was observed for the curved DNA as opposed to non-curved DNA, which is a distinctive feature of Lsr2 compared with H-NS.
Identification of residues critical for the function of Lsr2 in vivo
To gain insight into the structure–function relationship of Lsr2, we constructed a set of Lsr2 mutants each containing a single amino acid substitution by alanine. The experimental strategy was based on the following rationales. We previously showed that the Δlsr2 mutant of M. smegmatis exhibited a dramatic change of colony morphology: the Δlsr2 mutant colonies are very smooth, wet and round, in contrast to the dry, rough and rugose morphology of the WT mc2155 strain (23). This phenotype of the mutant was fully complemented by intact lsr2 gene from either M. smegmatis or M. tuberculosis [(23); Kocíncová et al., submitted for publication]. Taking advantage of this simple phenotype, we assessed the effects of each mutation on Lsr2 function by examining the ability of individual lsr2 mutant alleles to complement the colony morphology of the Δlsr2 strain. The plasmid pLSR2-HIS, which expresses the His-tagged Lsr2 protein of M. smegmatis and is able to complement the Δlsr2 phenotype (23), was used as the template to construct individual lsr2 mutations. Individual lsr2 alleles were transformed into the Δlsr2 strain and examined for their ability to complement the morphological phenotype. For those alleles that failed to complement the Δlsr2 phenotype, the protein level of the Lsr2 mutant in each recombinant strain was examined by immunoblotting analysis with an anti-His monoclonal antibody before further detailed characterization. These experiments allowed us to examine the roles of individual residues for the function of Lsr2 in vivo.
We first focused on replacing charged residues of Lsr2 for the following reasons. Analysis of the primary sequence reveals that Lsr2 is a basic protein rich in Arg and Lys (15 Arg + 5 Lys) with a calculated pI of 10.7. We hypothesized that Lsr2 could employ these positively charged residues, especially those that are surface exposed, to form salt bridges with the phosphate backbone of DNA, which would explain the non-specific binding of Lsr2 to DNA. To test this, we systematically substituted each positively charged residue with Ala and successfully obtained 16 mutants (Table 2). The Lsr2 protein also contains 17 negatively charged residues (10 Asp + 7 Glu), which could form intra- or inter-molecular salt bridges with the positively charged residues and indirectly affect DNA binding. We also successfully engineered nine Ala substitutions of these residues (Table 2). Mutagenesis of other charged residues were either unsuccessful or the results of the complementation assay ambiguous (e.g. D11A and D12A), and set aside for future investigation. In addition to the charged residues, we also engineered three other mutants, P103A, G26A and G29A for reasons described later. Each mutation of lsr2 was confirmed by DNA sequencing.
Table 2.
Lsr2 mutants and their ability to complement the mutant colony morphological phenotype and to exhibit dominant-negative phenotype in the wild-type strain
| Lsr2 and mutants | Complementation of the colony morphology of Δlsr2 mutant | Dominant negative mutants |
|---|---|---|
| Parent Lsr2 | + | NA |
| R45A | − | – |
| R56A | + | NA |
| R57A | + | NA |
| R61A | + | NA |
| R63A | + | NA |
| R65A | + | NA |
| R72A | + | NA |
| R74A | + | NA |
| R79A | + | NA |
| R86A | − | + |
| R90A | + | NA |
| R91A | + | NA |
| R99A | + | NA |
| R101A | + | NA |
| K39A | + | NA |
| K43A | + | NA |
| D14A | + | NA |
| D20A | + | NA |
| D28A | − | − |
| D35A | + | NA |
| D47A | + | NA |
| D78A | + | NA |
| D108A | + | NA |
| E16A | + | NA |
| E53A | + | NA |
| P103A | + | NA |
| G26A | + | NA |
| G29A | + | NA |
NA: not applicable; +: Yes; −: No.
Table 2 summarizes the results of in vivo complementation experiments. Of the 28 mutants examined, three mutants, R45A, R86A and D28A, failed to complement the colony morphological phenotype of the Δlsr2 strain (data not shown), whereas the rest of the mutants all complemented the Δlsr2 phenotype. Western blot analysis of cell lysates of the recombinant strains showed that all three mutant proteins were expressed in the Δlsr2 strain at levels equivalent to that of the WT protein (data not shown), excluding the possibility that the inability of these mutants to complement the Δlsr2 phenotype was due to inadequate production of mutant Lsr2 protein in the cells. Together, these results indicate that residues R45, R86 and D28 are critical for the function of Lsr2 in vivo. Secondary structure prediction reveals that Lsr2 is mainly α-helical (43%) containing two predicted α-helices (residues 35–54 and 76–90). Residues R45 and R86 are within the two helices, respectively, whereas D28 is in a predicted random coil.
R86A mutant of Lsr2 is a dominant negative mutant
We previously showed that Lsr2 forms a dimer in vivo (23). We reasoned that if mutant forms of Lsr2 with deficient DNA-binding capacity but with oligomerization capacity intact were expressed in a WT background, such mutant proteins could have a dominant negative effect on WT Lsr2 activity. Therefore, we next examined if the three Lsr2 mutants identified above exhibit a dominant negative phenotype in the WT background. Individual plasmids containing R45A, R86A or D28A lsr2 allele was transformed into the WT strain mc2155 and transformants examined for their colony morphology. Interestingly, the WT strain expressing the R86A lsr2 allele on a multicopy plasmid exhibited colony morphology indistinguishable to that of the Δlsr2 mutant (Figure 2), indicating that R86A is a dominant negative mutant. Transformation of the other two mutant lsr2 alleles (R45A and D28A) into the WT strain did not change the colony morphology (data not shown).
Figure 2.

Dominant negative effect of R86A Lsr2 mutant. Colony morphology of the Δlsr2 mutant, the WT strain mc2155 carrying the lsr2 R86A allele on a plasmid, and the WT strain carrying the WT lsr2 gene as control. WT strain carrying the lsr2 R86A allele exhibits colony morphology identical to that of the Δlsr2 mutant.
DNA binding and protein oligomerization are both essential for Lsr2 normal function in vivo
As hypothesized earlier, we reasoned that the R86A mutant protein is defective in DNA binding but retains the ability to oligomerize. As such, the mixed oligomers containing both mutant and WT proteins would have reduced activity since the incorporation of the mutant protein produces an overall reduction in the ability of the protein to carry out an essential function, i.e. DNA binding. To confirm this, we engineered the equivalent R86A mutant of M. tuberculosis Lsr2 using pLSR2 as the template. The M. tuberculosis Lsr2 R86A mutant protein was expressed and purified from E. coli and its ability to bind DNA and to form oligomers were assessed.
Purified WT and R86A mutant proteins were subjected to in vitro cross-linking experiments with glutaraldehyde as previously described (23) and analyzed by western blotting with an anti-His antibody. Consistent with our previous finding (23), the WT Lsr2 protein formed a dimer (Figure 3A). However, three higher order protein complexes were apparent when purified Lsr2 protein was subjected to cross-linking, which includes at least a tetramer (Figure 3A). Our previous cross-linking experiments with cell lysates of M. smegmatis revealed only an Lsr2 dimer (23). This discrepancy is likely due to the much higher Lsr2 protein concentration (6 μg purified protein per sample) used in the current study. Although it is not known if Lsr2 forms higher order complexes in vivo, this result indicates that Lsr2 is capable of forming a series of higher order protein complexes dependent on its local concentration. Consistent with our prediction, R86A mutant protein was able to form the same series of higher order protein complexes including dimer and tetramer, indistinguishable from the WT protein (Figure 3A), indicating that the mutant retains the full capacity to form oligomers.
Figure 3.

In vitro analysis of Lsr2 mutants. (A) Cross-linking of purified Lsr2 proteins. Glutaraldehyde (1%) was added to purified protein (6 μg per sample). Aliquots were removed at the indicated time points and analyzed by western blotting with an anti-His antibody. (B) EMSA experiments. DNA fragment 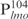 (50 ng) was incubated with the indicated amounts of Lsr2 proteins and analyzed on 4% polyacrylamide gel and stained with SYBR green. The amounts of protein added are: lanes 1–5: 0, 144, 288, 576 and 1152 ng, respectively, for WT Lsr2 protein; lanes 6–9: 144, 288, 576 and 1152 ng, respectively, for P103A mutant protein; lanes 10–13: 144, 288, 576 and 1152 ng, respectively, for R86A mutant protein. (C) DNA ladder (50 ng) was incubated with the indicated amounts of Lsr2 and analyzed on 1% agarose gel. The amounts of protein added are: lanes 1–5: 0, 144, 288, 576 and 1152 ng, respectively, for WT Lsr2 protein; lanes 6–9: 144, 288, 576 and 1152 ng, respectively, for R86A mutant protein.
(50 ng) was incubated with the indicated amounts of Lsr2 proteins and analyzed on 4% polyacrylamide gel and stained with SYBR green. The amounts of protein added are: lanes 1–5: 0, 144, 288, 576 and 1152 ng, respectively, for WT Lsr2 protein; lanes 6–9: 144, 288, 576 and 1152 ng, respectively, for P103A mutant protein; lanes 10–13: 144, 288, 576 and 1152 ng, respectively, for R86A mutant protein. (C) DNA ladder (50 ng) was incubated with the indicated amounts of Lsr2 and analyzed on 1% agarose gel. The amounts of protein added are: lanes 1–5: 0, 144, 288, 576 and 1152 ng, respectively, for WT Lsr2 protein; lanes 6–9: 144, 288, 576 and 1152 ng, respectively, for R86A mutant protein.
We next examined the ability of the R86A mutant to bind DNA. The WT Lsr2 protein was included as a control and 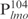 was used as the specific probe in EMSA experiments. As shown in Figure 3B, the R86A mutant protein exhibited defective DNA binding. No protein–DNA complexes were detected for the mutant protein at concentrations ranging from to 144 to 576 ng, and only a moderate level of DNA shifts was observed at the highest protein concentration examined (1152 ng). A similar result was obtained when non-specific DNA i.e. DNA ladder, was used as the probe in EMSA experiments (Figure 3C). These results indicate that R86 of Lsr2 is critical for DNA binding and substitution of this residue with Ala decreased its DNA binding activity. Together, results from the in vitro experiments are consistent with the dominant negative effect of the R86A mutant observed in vivo. Furthermore, these results demonstrate that DNA binding and protein oligomerization are both essential for the normal function of Lsr2 in vivo.
was used as the specific probe in EMSA experiments. As shown in Figure 3B, the R86A mutant protein exhibited defective DNA binding. No protein–DNA complexes were detected for the mutant protein at concentrations ranging from to 144 to 576 ng, and only a moderate level of DNA shifts was observed at the highest protein concentration examined (1152 ng). A similar result was obtained when non-specific DNA i.e. DNA ladder, was used as the probe in EMSA experiments (Figure 3C). These results indicate that R86 of Lsr2 is critical for DNA binding and substitution of this residue with Ala decreased its DNA binding activity. Together, results from the in vitro experiments are consistent with the dominant negative effect of the R86A mutant observed in vivo. Furthermore, these results demonstrate that DNA binding and protein oligomerization are both essential for the normal function of Lsr2 in vivo.
We also engineered the equivalent P103A mutant of M. tuberculosis Lsr2 using the same approach and purified this mutant protein for cross-linking and DNA-binding experiments. We were interested in further characterizations of this mutant because proline residue plays a critical role for the functions of H-NS and HU proteins. In H-NS, changing the Pro at position 116 to Ala or Ser abolished the ability of H-NS to distinguish curved from non-curved DNA, while retaining its non-specific-DNA binding activity (35,36). The Pro at position 63 is the residue in HU that intercalates DNA and is critical for the HU-induced DNA bending (37,38). Lsr2 contains only one highly conserved Pro residue and we were intrigued by the finding that the P103A mutant retained the ability to fully complement the Δlsr2 morphological phenotype (Table 2). This seemed to suggest that in Lsr2, the Pro is dispensible for function. Therefore, we sought to confirm this by performing in vitro characterizations of the purified P103A mutant protein. Cross-linking and EMSA experiments showed that P103A mutant was able to form oligomers (data not shown) and bind to DNA (Figure 3B), in a manner indistinguishable from the WT protein. Moreover, no difference was observed between the WT and P103A mutant proteins in their binding to curved and non-curved DNA (data not shown). Together, results from the in vivo and in vitro experiments indicate that the lone Pro residue is not essential for Lsr2 normal function.
Lsr2 is a DNA bridging protein
In order to obtain insight into the structural effects of Lsr2 binding to DNA and to understand the molecular mechanism of Lsr2 action, we applied AFM to observe directly structures of the Lsr2–DNA complexes. AFM is a useful tool for studying protein–DNA complexes, especially for proteins that do not interact with specific DNA sequences or structures. Individual DNA–protein complexes can be directly visualized and qualitatively categorized. AFM studies of bacterial NAPs including H-NS and HU have provided much insight into the molecular mechanisms of these proteins (2,26,39–41). On the basis of these studies, the major NAPs are classified, according to their structural effect on DNA, as DNA bridging proteins (e.g. H-NS, StpA) and DNA bending proteins (e.g. HU, IHF) (2,40).
The effects of Lsr2 binding were studied on both linear and circular DNA molecules. We found that Lsr2 bound to all three forms of DNA (linear DNA, relaxed and supercoiled circular DNA) at apparently equivalent affinities (Supplementary Figure S3). For AFM analysis, we first examined the effects of Lsr2 binding to linear DNA (Figure 4). Two linear DNA fragments were used in this experiment, pDrive and pMPS. The pMPS was generated by cloning the 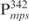 fragment into pDrive, and then linearized by restriction digestion. Figure 4A shows pMPS in the absence of Lsr2. Figure 4B–F show representative images of Lsr2–pMPS complexes at a ratio of 1 Lsr2 dimer per 260 bp (large field images with multiple DNA molecules are shown in Supplementary Figure S4). On the basis of the appearance of the complexes, they can be divided into two classes. Complexes in the first class of molecules, depicted in Figure 4B and C, is characterized by the formation of ‘protrusions’ or ‘branches’ at various locations of the DNA. The DNA inside such a protrusion is apparently folded back to form a hairpin-like configuration. The second class of complexes, depicted in Figure 4D–F, shows large tracts with two distant regions of linear DNA held close together, creating local loops that resemble circular DNA. The formation of these two classes of complexes is not exclusive to each other, which can happen concurrently in the same molecule (Figure 4E and F). Importantly, the formation of these complexes occurs at various positions throughout the contour length of the DNA and appears to be sequence independent, which is consistent with the non-specific binding of Lsr2 to DNA. This is further supported by our observation that similar complexes were formed between Lsr2 protein and the non-specific DNA, pDrive. Representative images of Lsr2–pDrive complexes are shown in Figure 4G and H.
fragment into pDrive, and then linearized by restriction digestion. Figure 4A shows pMPS in the absence of Lsr2. Figure 4B–F show representative images of Lsr2–pMPS complexes at a ratio of 1 Lsr2 dimer per 260 bp (large field images with multiple DNA molecules are shown in Supplementary Figure S4). On the basis of the appearance of the complexes, they can be divided into two classes. Complexes in the first class of molecules, depicted in Figure 4B and C, is characterized by the formation of ‘protrusions’ or ‘branches’ at various locations of the DNA. The DNA inside such a protrusion is apparently folded back to form a hairpin-like configuration. The second class of complexes, depicted in Figure 4D–F, shows large tracts with two distant regions of linear DNA held close together, creating local loops that resemble circular DNA. The formation of these two classes of complexes is not exclusive to each other, which can happen concurrently in the same molecule (Figure 4E and F). Importantly, the formation of these complexes occurs at various positions throughout the contour length of the DNA and appears to be sequence independent, which is consistent with the non-specific binding of Lsr2 to DNA. This is further supported by our observation that similar complexes were formed between Lsr2 protein and the non-specific DNA, pDrive. Representative images of Lsr2–pDrive complexes are shown in Figure 4G and H.
Figure 4.

AFM images of protein–DNA complexes formed by Lsr2 and linear DNA. (A) Linear pMPS in the absence of Lsr2 protein. (B)–(F): Lsr2–pMPS complexes at low protein/DNA ratios (1 dimer per 260 bp). (G) and (H): Lsr2–pDrive complexes at low protein/DNA ratios (1 dimer per 260 bp). (I)–(K): Lsr2–pDrive complexes at high protein/DNA ratios (1 dimer per 65 bp). The dimension for (I) is 5 µm × 5 µm and for (J) is 2 µm × 2 µm. The same color scale ranging from 0.0 to 5.0 nm (from dark to bright) was used for all images.
At higher protein concentrations (1 Lsr2 dimer per 65 bp DNA), extensive intra- and inter-molecular bridging occurred, resulting in a number of large complexes that are arranged more like a DNA ‘network’ (Figure 4I–K). These complexes are characterized by the presence of large, highly condensed foci at the core of the complexes, which are presumably aggregates of Lsr2 protein (Figure 4J and K). These complexes tend to associate with each other, forming clustered complexes involving multiple foci (Figure 4J). We were unable to obtain AFM images of Lsr2–DNA complexes at even higher protein concentrations corresponding to the formation of totally retarded complexes in the EMSAs (i.e. 1 Lsr2 dimer per 4 bp DNA), because Lsr2 formed films (large protein aggregates) on the mica under these conditions.
The effects of Lsr2 on circular DNA were studied on relaxed circular plasmid, which was obtained by introducing a limited number of nicks in circular pDrive by partial digestion with DNase I. Figure 5A shows the representative image of relaxed circular pDrive in the absence of Lsr2 protein, which has an open appearance. Figures 5B–D are representative of Lsr2–DNA complexes at low protein/DNA ratios (1 Lsr2 dimer per 260 bp) (large field images are shown in Supplementary Figure S5). These complexes are characterized by the presence of multiple loops created by the lateral bridging of two tracts of DNA by Lsr2 protein. Novel forms (different from sole lateral bridging) were also observed, in which DNA loops folded back inside other loops, leading to more bridging between DNA tracks in these loops (Figure 5B and C). At higher protein/DNA ratios (1 Lsr2 dimer per 65 bp DNA), extensive bridging within and between individual circular DNA molecules occurred, resulting in large DNA networks that center at Lsr2 aggregates (Figures 5E and F), which is similar to the complexes observed with linear DNA (Figure 4J–K).
Figure 5.

AFM images of protein–DNA complexes formed by Lsr2 and relaxed circular DNA. (A) Relaxed circular pDrive without Lsr2. (B–D) Lsr2–pDrive complexes at low protein/DNA ratios (1 dimer per 260 bp). (E) and (F) Lsr2–pDrive complexes at high protein/DNA ratios (1 dimer per 65 bp). The dimension for (E) is 2 µm × 2 µm. The same color scale ranging from 0.0 to 5.0 nm (from dark to bright) was used for all images.
Together, the AFM results indicate that Lsr2 protein has the ability to bridge DNA, a characteristic shared by H-NS-like proteins (40), suggesting that Lsr2 is functionally similar to H-NS, despite the lack of any sequence homology. Lsr2 has essentially the same structural effect on linear and circular DNA duplexes, both of which can be explained by the DNA bridging property of Lsr2. Although these experiments are carried out at fixed concentrations of Lsr2, they represent snapshots of what is likely a continuum of dynamic Lsr2–DNA structures that occur in vivo, which is dependent on the global distribution and local concentration of Lsr2 protein.
DISCUSSION
The bacterial nucleoid is organized into topologically independent loops, which are in the order of 10 kb in size (42). The loop structure of the nucleoid is maintained in part by the binding of NAPs (2). Such a role has recently been suggested for H-NS (43). Results presented in the current study suggest that despite the lack of sequence homology to any known bacterial NAPs, Lsr2 of M. tuberculosis likely plays a role in the loop formation and contributes to nucleoid compaction. Lsr2 could be one of the structural components directly involved in the formation of patches of bridged DNA segments along the DNA loops. Indeed, due to the relatively non-specific DNA binding, Lsr2 could bridge distant DNA segments and consequently have a large impact on the overall organization and compactness of the nucleoid. Although a similar function has been proposed for H-NS in Gram-negative bacteria (5,43), DNA-bridging proteins have never been reported previously in bacteria phylogenetically distant from the Enterobacteriaceae (18). Our AFM results provide direct evidence that Lsr2 is functionally similar to H-NS. Although its oligomeric state has been highly disputed, in vitro cross-linking studies have revealed that H-NS exists as at least a dimer (5). A characteristic feature of dimeric H-NS is the presence of two DNA-binding domains that can potentially interact with two DNA duplexes simultaneously and results in ‘bridges’ between adjacent DNA duplexes, as demonstrated by AFM studies (26,39,40). The results of our study demonstrate that Lsr2 fits these criteria as a DNA-bridging molecule and could potentially employ the same mechanism as H-NS to bridge DNA (44). At low protein/DNA ratios (1 Lsr2 dimer per 260 bp), Lsr2 causes primarily intramolecular bridging of different tracts of DNA molecules, resulting in the formation of local branches and/or loops, which is similar to the structural effect of H-NS on DNA (26,39,40). With increasing concentrations of Lsr2 protein (1 Lsr2 dimer per 65 bp), extensive intermolecular interactions occur presumably via Lsr2 oligomerization, resulting in the formation of DNA bundles and networks. Together, our study represents the first identification of an H-NS-like, DNA-bridging protein in mycobacteria and related actinomycetes. Our study also supports the hypothesis that H-NS-like proteins could be too divergent to be identified on the sole basis of sequence similarity (18).
The ability of Lsr2 to bridge DNA provides a direct explanation for the mechanism of transcriptional regulation by Lsr2 [(23,24); Kocíncová et al., submitted for publication]. DNA binding and protein oligomerization are both crucial for the normal repressor activity of H-NS (9), consistent with its proposed model of transcriptional silencing in which H-NS directly inhibits transcription by binding to the promoter region and trapping RNA polymerase via DNA looping or bridging (9), or by occlusion of RNA polymerase from the promoter. Like H-NS, Lsr2 exists as a dimer and possibly higher order oligomers in vivo. The functional significance of Lsr2 oligomerization is demonstrated by our successful isolation of a dominant negative mutant, R86A, which exhibits a defective DNA-binding activity and consequently, disrupts the native Lsr2 activity by virtue of heteromeric protein–protein interactions in vivo. Our in vivo and in vitro experiments with the R86A mutant demonstrate that DNA binding and protein oligomerization are both essential for the normal function of Lsr2 in vivo. The two other mutants, R45A and D28A, may have compromised ability to form protein oligomers since they failed to complement the mutant phenotype but did not exhibit a dominant negative effect in the WT background. Together, our mutational analysis and AFM results provide strong evidence that Lsr2 could use the same mechanism as H-NS to mediate transcriptional repression, i.e. by trapping or occlusion of RNA polymerase in the promoter region. The finding that Lsr2 exhibits preferential binding to AT-rich sequences, which is widely associated with many bacterial promoters, could explain the broad spectrum of genes repressed by Lsr2 (24). Lsr2 also appears to positively regulate some genes (24). However, this could be an indirect or secondary effect. Lsr2 could negatively regulate repressors that control these genes, as has been demonstrated for H-NS (9).
On the other hand, Lsr2 also exhibit unique characteristics that distinguish them from H-NS. Our EMSA experiments indicate that Lsr2 forms complexes of different stoichiometries with DNA, which is an essential difference with H-NS (4). Importantly, the formation of these complexes is dependent on the concentration of Lsr2 and is only abolished after denaturation of Lsr2 protein by SDS (data not shown). In addition, the number of Lsr2–DNA complexes appears to correlate with the length of DNA, irrespective of the nucleotide sequence. The simplest interpretation of these data is that Lsr2 can orderly bind, side by side, to a linear DNA fragment. Under this assumption, we deduce that the binding site of Lsr2, based on EMSA results, is 9–14 bp. Thus, 11 distinct Lsr2-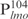 complexes were formed (Figure 1A) and 3 complexes were formed by Lsr2 with C42 or NC42 (Figure 1F). Variation of non-specific DNA binding site between 11 and 25 bp has been reported for H-NS, depending on experimental conditions (44–46). It was concluded, based on experiments on two DNA molecules by optical tweezers, that H-NS binding site can be expressed in multiples of the DNA helical repeat (44), which provides an explanation for the binding site variability. A similar mechanism could explain the length and the variation of DNA binding site of Lsr2.
complexes were formed (Figure 1A) and 3 complexes were formed by Lsr2 with C42 or NC42 (Figure 1F). Variation of non-specific DNA binding site between 11 and 25 bp has been reported for H-NS, depending on experimental conditions (44–46). It was concluded, based on experiments on two DNA molecules by optical tweezers, that H-NS binding site can be expressed in multiples of the DNA helical repeat (44), which provides an explanation for the binding site variability. A similar mechanism could explain the length and the variation of DNA binding site of Lsr2.
Alternatively, the multiple Lsr2–DNA complexes observed in EMSAs may represent various secondary complex structures originated from extensive intermolecular interactions, as revealed by AFM analysis. This model could explain the increased affinity of Lsr2 for longer DNA fragments (Figures 1E and 3C). Longer DNA molecules with more bound Lsr2 at various locations would have a greater chance to form topologically different and more stabilized structures, thus shifting the binding equilibrium towards the bound form. Clearly, future studies including the determination of the 3D structures of Lsr2 and Lsr2-DNA complexes are required to test these two models.
Our results suggest that Lsr2 has a binding preference for AT-rich sequences. However, unlike H-NS, which has a preference for curved DNA, particularly fragments containing in-phase repeats of A5 and A6 tracts (29,30), Lsr2 does not exhibit preferential binding to the same type of DNA substrates. Therefore, Lsr2 has a preference for AT-rich but not necessarily curved DNA. This conclusion is further supported by the characterization of the P103A Lsr2 mutant. Unlike H-NS, in which the proline at position 116 is critical for its ability to distinguish curved from non-curved DNA (35,36), substitution of proline at 103, which is the only proline residue in Lsr2 sequence, did not affect the normal functions of Lsr2 in vivo and in vitro, suggesting that the proline residue is not essential for Lsr2 function. On the other hand, H-NS was recently shown to recognize a defined high-affinity site (32), the lack of high affinity by Lsr2 for a similar site could explain the difference between Lsr2 and H-NS. Lsr2 binding to DNA could be mediated by electrostatic interactions. Lsr2 is characterized by a large number of positively charged residues with a calculated pI close to that of HU (10.1 and 10.2 for the α and β subunit of E. coli HU, respectively). In contrast, H-NS is a neutral protein. Like HU, Lsr2 could employ positively charged surface residues to form salt bridges with the phosphate backbone of DNA, which would explain the non-specific binding of Lsr2. Our finding that R86 is involved in DNA binding is consistent with this hypothesis.
Intriguingly, DNA bundles and networks formed at high Lsr2 concentration (1 Lsr2 dimer per 65 bp), as revealed by AFM (Figures 4 and 5), were not described for H-NS even at a higher protein/DNA ratio (1 H-NS dimer per 12 bp) (26). This probably reflects their differences in DNA binding specificity and affinity, as well as the nature of protein oligomerization. Alternatively, a difference in local DNA concentration (inhomogeneity in the sample before deposition for AFM imaging) could account for the observed difference.
It was recently proposed that H-NS acts as a genome guardian that silences horizontally transferred foreign genes, which are relatively AT-rich and often associated with virulence (11,12). It was speculated that the xenogeneic silencing by H-NS could play a role in maintaining the characteristic GC content of individual bacterial genomes (11,12). Within this context, it is interesting to note that Lsr2 proteins are only present in actinomycetes, which are characterized by the high GC content of their genomes (∼70%). As such, Lsr2 may play a more faithful (stringent) role than H-NS in silencing laterally acquired AT-rich genes and contribute to the high GC bias in these bacteria. This hypothesis is consistent with our finding that Lsr2 has a preference for AT-rich DNA, irrespective of the DNA topology. Furthermore, transcription of lsr2 is regulated by environmental stress conditions including nutrient availability, growth temperature and antibiotic exposures (24,47,48). It is conceivable that under these conditions, lsr2 is activated and acts on a number of genes involved in multiple cellular processes to allow a better adaptation of the bacillus to the environment including within the host.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Greg German for technical assistances with EMSA experiments. This work was supported by funding from Canadian Institutes of Health Research (CIHR) (MOP-15107 and MOP-82772 to J.L.) and from INSEM under the Avenir programme (to J.-M.R.). Funding to pay the Open Access publication charges for this article was provided by CIHR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Dame RT. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol. Microbiol. 2005;56:858–870. doi: 10.1111/j.1365-2958.2005.04598.x. [DOI] [PubMed] [Google Scholar]
- 2.Luijsterburg MS, Noom MC, Wuite GJ, Dame RT. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: a molecular perspective. J. Struct. Biol. 2006;156:262–272. doi: 10.1016/j.jsb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Travers A, Muskhelishvili G. Bacterial chromatin. Curr. Opin. Genet. Dev. 2005;15:507–514. doi: 10.1016/j.gde.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Azam TA, Ishihama A. Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and DNA binding affinity. J. Biol. Chem. 1999;274:33105–33113. doi: 10.1074/jbc.274.46.33105. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RC, Johnson MJ, Schmidt JW, Gardner JF. Major nucleoid proteins in the structure and function of the Escherichia coli chromosome. In: Higgins NP, editor. The Bacterial Chromosome. Washington DC: American Society for Microbiology Press; 2005. pp. 65–132. [Google Scholar]
- 6.Owen-Hughes TA, Pavitt GD, Santos DS, Sidebotham JM, Hulton CS, Hinton JC, Higgins CF. The chromatin-associated protein H-NS interacts with curved DNA to influence DNA topology and gene expression. Cell. 1992;71:255–265. doi: 10.1016/0092-8674(92)90354-f. [DOI] [PubMed] [Google Scholar]
- 7.Bonnefoy E, Takahashi M, Yaniv JR. DNA-binding parameters of the HU protein of Escherichia coli to cruciform DNA. J. Mol. Biol. 1994;242:116–129. doi: 10.1006/jmbi.1994.1563. [DOI] [PubMed] [Google Scholar]
- 8.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr. Opin. Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Dorman CJ. H-NS: a universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2004;2:391–400. doi: 10.1038/nrmicro883. [DOI] [PubMed] [Google Scholar]
- 10.Hommais F, Krin E, Laurent-Winter C, Soutourina O, Malpertuy A, Le Caer JP, Danchin A, Bertin P. Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS. Mol. Microbiol. 2001;40:20–36. doi: 10.1046/j.1365-2958.2001.02358.x. [DOI] [PubMed] [Google Scholar]
- 11.Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2006;2:e81. doi: 10.1371/journal.ppat.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, Fang FC. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science. 2006;313:236–238. doi: 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- 13.McLeod SM, Johnson RC. Control of transcription by nucleoid proteins. Curr. Opin. Microbiol. 2001;4:152–159. doi: 10.1016/s1369-5274(00)00181-8. [DOI] [PubMed] [Google Scholar]
- 14.Reusch RN, Shabalin O, Crumbaugh A, Wagner R, Schroder O, Wurm R. Posttranslational modification of E. coli histone-like protein H-NS and bovine histones by short-chain poly-(R)-3-hydroxybutyrate (cPHB) FEBS Lett. 2002;527:319–322. doi: 10.1016/s0014-5793(02)03236-2. [DOI] [PubMed] [Google Scholar]
- 15.Johansson J, Balsalobre C, Wang SY, Urbonaviciene J, Jin DJ, Sonden B, Uhlin BE. Nucleoid proteins stimulate stringently controlled bacterial promoters: a link between the cAMP-CRP and the (p)ppGpp regulons in Escherichia coli. Cell. 2000;102:475–485. doi: 10.1016/s0092-8674(00)00052-0. [DOI] [PubMed] [Google Scholar]
- 16.Dixon NE, Kornberg A. Protein HU in the enzymatic replication of the chromosomal origin of Escherichia coli. Proc. Natl Acad. Sci. USA. 1984;81:424–428. doi: 10.1073/pnas.81.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flashner Y, Gralla JD. DNA dynamic flexibility and protein recognition: differential stimulation by bacterial histone-like protein HU. Cell. 1988;54:713–721. doi: 10.1016/s0092-8674(88)80016-3. [DOI] [PubMed] [Google Scholar]
- 18.Tendeng C, Bertin PN. H-NS in Gram-negative bacteria: a family of multifaceted proteins. Trends Microbiol. 2003;11:511–518. doi: 10.1016/j.tim.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Yeruva VC, Duggirala S, Lakshmi V, Kolarich D, Altmann F, Sritharan M. Identification and characterization of a major cell wall-associated iron-regulated envelope protein (Irep-28) in Mycobacterium tuberculosis. Clin. Vaccine Immunol. 2006;13:1137–1142. doi: 10.1128/CVI.00125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shires K, Steyn L. The cold-shock stress response in Mycobacterium smegmatis induces the expression of a histone-like protein. Mol. Microbiol. 2001;39:994–1009. doi: 10.1046/j.1365-2958.2001.02291.x. [DOI] [PubMed] [Google Scholar]
- 21.Katsube T, Matsumoto S, Takatsuka M, Okuyama M, Ozeki Y, Naito M, Nishiuchi Y, Fujiwara N, Yoshimura, M. Tsuboi T, et al. Control of cell wall assembly by a histone-like protein in mycobacteria. J. Bacteriol. 2007;189:8241–8249. doi: 10.1128/JB.00550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boshoff HI, Barry CE. Tuberculosis - metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- 23.Chen JM, German GJ, Alexander DC, Ren H, Tan T, Liu J. Roles of Lsr2 in colony morphology and biofilm formation of Mycobacterium smegmatis. J. Bacteriol. 2006;188:633–641. doi: 10.1128/JB.188.2.633-641.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colangeli R, Helb D, Vilcheze C, Hazbon MH, Lee CG, Safi H, Sayers B, Sardone I, Jones MB, Fleischmann RD, et al. Transcriptional regulation of multi-drug tolerance and antibiotic-induced responses by the histone-like protein Lsr2 in M. tuberculosis. PLoS Pathog. 2007;3:e87. doi: 10.1371/journal.ppat.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laal S, Sharma YD, Prasad HK, Murtaza A, Singh S, Tangri S, Misra RS, Nath I. Recombinant fusion protein identified by lepromatous sera mimics native Mycobacterium leprae in T-cell responses across the leprosy spectrum. Proc. Natl Acad. Sci. USA. 1991;88:1054–1058. doi: 10.1073/pnas.88.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dame RT, Wyman C, Goosen N. H-NS mediated compaction of DNA visualised by atomic force microscopy. Nucleic Acids Res. 2000;28:3504–3510. doi: 10.1093/nar/28.18.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Starr DB, Hawley DK. TFIID binds in the minor groove of the TATA box. Cell. 1991;67:1231–1240. doi: 10.1016/0092-8674(91)90299-e. [DOI] [PubMed] [Google Scholar]
- 28.Lokuta MA, Maher J, Noe KH, Pitha PM, Shin ML, Shin HS. Mechanisms of murine RANTES chemokine gene induction by Newcastle disease virus. J. Biol. Chem. 1996;271:13731–13738. doi: 10.1074/jbc.271.23.13731. [DOI] [PubMed] [Google Scholar]
- 29.Zuber F, Kotlarz D, Rimsky S, Buc H. Modulated expression of promoters containing upstream curved DNA sequences by the Escherichia coli nucleoid protein H-NS. Mol. Microbiol. 1994;12:231–240. doi: 10.1111/j.1365-2958.1994.tb01012.x. [DOI] [PubMed] [Google Scholar]
- 30.Jordi BJ, Fielder AE, Burns CM, Hinton JC, Dover N, Ussery DW, Higgins CF. DNA binding is not sufficient for H-NS-mediated repression of proU expression. J. Biol. Chem. 1997;272:12083–12090. doi: 10.1074/jbc.272.18.12083. [DOI] [PubMed] [Google Scholar]
- 31.Rimsky S, Zuber F, Buckle M, Buc H. A molecular mechanism for the repression of transcription by the H-NS protein. Mol. Microbiol. 2001;42:1311–1323. doi: 10.1046/j.1365-2958.2001.02706.x. [DOI] [PubMed] [Google Scholar]
- 32.Bouffartigues E, Buckle M, Badaut C, Travers A, Rimsky S. H-NS cooperative binding to high-affinity sites in a regulatory element results in transcriptional silencing. Nat. Struct. Mol. Biol. 2007;14:441–448. doi: 10.1038/nsmb1233. [DOI] [PubMed] [Google Scholar]
- 33.Bonnefoy E, Rouviere-Yaniv J. HU and IHF, two homologous histone-like proteins of Escherichia coli, form different protein-DNA complexes with short DNA fragments. EMBO J. 1991;10:687–696. doi: 10.1002/j.1460-2075.1991.tb07998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diekmann S. Sequence specificity of curved DNA. FEBS Lett. 1986;195:53–56. doi: 10.1016/0014-5793(86)80128-4. [DOI] [PubMed] [Google Scholar]
- 35.Badaut C, Williams R, Arluison V, Bouffartigues E, Robert B, Buc H, Rimsky S. The degree of oligomerization of the H-NS nucleoid structuring protein is related to specific binding to DNA. J. Biol. Chem. 2002;277:41657–41666. doi: 10.1074/jbc.M206037200. [DOI] [PubMed] [Google Scholar]
- 36.Spurio R, Falconi M, Brandi A, Pon CL, Gualerzi CO. The oligomeric structure of nucleoid protein H-NS is necessary for recognition of intrinsically curved DNA and for DNA bending. EMBO J. 1997;16:1795–1805. doi: 10.1093/emboj/16.7.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka I, Appelt K, Dijk J, White SW, Wilson KS. 3-A resolution structure of a protein with histone-like properties in prokaryotes. Nature. 1984;310:376–381. doi: 10.1038/310376a0. [DOI] [PubMed] [Google Scholar]
- 38.White SW, Appelt K, Wilson KS, Tanaka I. A protein structural motif that bends DNA. Proteins. 1989;5:281–288. doi: 10.1002/prot.340050405. [DOI] [PubMed] [Google Scholar]
- 39.Dame RT, Wyman C, Goosen N. Structural basis for preferential binding of H-NS to curved DNA. Biochimie. 2001;83:231–234. doi: 10.1016/s0300-9084(00)01213-x. [DOI] [PubMed] [Google Scholar]
- 40.Dame RT, Luijsterburg MS, Krin E, Bertin PN, Wagner R, Wuite GJ. DNA bridging: a property shared among H-NS-like proteins. J. Bacteriol. 2005;187:1845–1848. doi: 10.1128/JB.187.5.1845-1848.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Noort J, Verbrugge S, Goosen N, Dekker C, Dame RT. Dual architectural roles of HU: formation of flexible hinges and rigid filaments. Proc. Natl Acad. Sci. USA. 2004;101:6969–6974. doi: 10.1073/pnas.0308230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Postow L, Hardy CD, Arsuaga J, Cozzarelli NR. Topological domain structure of the Escherichia coli chromosome. Genes Dev. 2004;18:1766–1779. doi: 10.1101/gad.1207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noom MC, Navarre WW, Oshima T, Wuite GJ, Dame RT. H-NS promotes looped domain formation in the bacterial chromosome. Curr. Biol. 2007;17:R913–R914. doi: 10.1016/j.cub.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Dame RT, Noom MC, Wuite GJ. Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature. 2006;444:387–390. doi: 10.1038/nature05283. [DOI] [PubMed] [Google Scholar]
- 45.Amit R, Oppenheim AB, Stavans J. Increased bending rigidity of single DNA molecules by H-NS, a temperature and osmolarity sensor. Biophys. J. 2003;84:2467–2473. doi: 10.1016/S0006-3495(03)75051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ono S, Goldberg MD, Olsson T, Esposito D, Hinton JC, Ladbury JE. H-NS is a part of a thermally controlled mechanism for bacterial gene regulation. Biochem. J. 2005;391:203–213. doi: 10.1042/BJ20050453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, Young DB, Butcher PD. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology. 2002;148:3129–3138. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- 48.Wong DK, Lee BY, Horwitz MA, Gibson BW. Identification of fur, aconitase, and other proteins expressed by Mycobacterium tuberculosis under conditions of low and high concentrations of iron by combined two-dimensional gel electrophoresis and mass spectrometry. Infect. Immun. 1999;67:327–336. doi: 10.1128/iai.67.1.327-336.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]