


Abstract
Translational research hinges on the ability to make observations in model systems and to implement those findings into clinical applications, such as the development of diagnostic tools or targeted therapeutics. Tumor cell lines are commonly used to model carcinogenesis. The same tumor cell line can be simultaneously studied in multiple research laboratories throughout the world, theoretically generating results that are directly comparable. One important assumption in this paradigm is that researchers are working with the same cells. However, recent work using high throughput genomic analyses questions the accuracy of this assumption. Observations by our group and others suggest that experiments reported in the scientific literature may contain pre-analytic errors due to inaccurate identities of the cell lines employed. To address this problem, we developed a simple approach that enables an accurate determination of cell line identity by genotyping 34 single nucleotide polymorphisms (SNPs). Here, we describe the empirical development of a SNP panel identification assay (SPIA) compatible with routine use in the laboratory setting to ensure the identity of tumor cell lines and human tumor samples throughout the course of long term research use.
INTRODUCTION
The recognition that cancer is a genomic disease has prompted significant efforts to characterize large numbers of human tumor samples. For example, a pilot project called The Cancer Genome Altas (TCGA) has begun to sequence thousands of samples from three common tumor types (i.e. brain (glioblastoma multiforme), lung (squamous carcinoma) and ovarian (serous cystadenocarcinoma)) with a long-term goal of comprehensive human cancer genome characterization (National Human Genome Research Institute, http://www.genome.gov). Discoveries in human tumor samples will pave the road in our understanding of commonly altered gene pathways and lead to pre-clinical studies to address the biologic significance of these newly identified mutations, amplifications and deletions.
Pre-clinical, functional studies are often conducted in tumor cell lines as model systems for understanding perturbations in primary human tumors. In addition to functional studies, cell lines are now important in the identification of therapeutic targets and in the understanding of molecular pathways related to drug–tumor interactions as recently demonstrated by Lamb et al. (1).
A MedLine search of ‘cell line’ and ‘cancer human’ identified 96 758 articles where human cell lines have been employed to study cancer biology. Bio-resource centers preserve and sell human cell lines. For example, the American Type Culture Collection (ATCC, http://www.lgcpromochem-atcc.com/) provides researchers with a collection of 700 human cancer cell lines. Commonly, cell lines are transferred extensively between investigators and institutes, and may be cultivated over prolonged periods of time. One well-recognized risk in cell line maintenance is human error, either by mislabeling or cross-contamination, the latter ultimately resulting in overgrowth of a contaminating cell line with a shorter doubling time after extended cultivation. In many cases, scientists are unaware that such errors have occurred.
Contamination remains a persistent concern among biomedical researchers. Therefore, in an effort to identify latent cross-contamination or other errors in mislabeling, cell lines included in the NCI60 panel were systematically genotyped at the Sanger Institute using the Affymetrix single nucleotide polymorphism (SNP) array (10K SNP array) (Cancer Genome Project, http://www.sanger.ac.uk/genetics/CGP/Genotyping/synlinestable.shtml). In this study, the presumed breast adenocarcinoma cell line, NCI-ADR-RES (2), and human ovarian carcinoma cell line, OVCAR-8, were determined to share 99% of genotype calls (out of 10 000 SNPs). The Sanger Institute also reported that two glioblastoma cell lines, SNB19 and U-251, exhibited a near-identical genotype. The MDA-MB435 cell line, another presumed breast adenocarcinoma cell line, was determined to be genotypically identical to the M14 melanoma cell line (3).
These errors due to inadvertent cross-contamination or processing remain largely unrecognized by investigators and may have profound adverse effects on experimental results and their interpretation. MDA-MB435 alone is cited in over 400 publications as a breast cancer cell line.
Cross-contamination and mislabeling may represent a significant confounder to experimental interpretation (4–6) and cases of cell line mistaken identity leading to spurious results are a major concern (7). Occasionally researchers adopt techniques to control for this problem, such as short tandem repeats (STR) DNA fingerprinting (8). It has been shown that random dinucleotide markers, the most informative class of microsatellites, are 5–8 times more informative than random SNPs, but 2–12% of SNPs are more informative than the median dinucleotides (9). Indeed, SNPs as DNA markers have been shown to be well suited for different purposes such as animal identification (10), identification of population ancestry (11) and for forensic purposes (12).
The advent of high-throughput genotyping both highlights the challenges of sample identity verification and provides a mechanism for its resolution. The ability of high-density oligonucleotide arrays to accurately genotype hundreds of thousands of SNP loci in parallel provides an unequivocal molecular fingerprint of each sample. Genotypic differences between two individuals along the entire genome would score less than 0.1%. However, when genotyping SNPs the percentage of genotype differences will significantly increase.
Our analysis of several hundred tumor samples, cell lines and xenografts using SNP arrays has uncovered several instances in which samples thought to be distinct were actually genetically identical. This stimulated our interest in genetic-based methods to precisely identify DNA samples a priori. To this end, we have developed an assay that employs 30–50 single loci across the genome and is capable of distinguishing any two DNA samples based on genotype calls. Concomitantly, this assay can correctly identify a given DNA sample by comparing the genotype call set (‘barcode’) within a reference database that contains bar codes of the most commonly used cell lines. Widespread application of this approach may reduce erroneous experimentation and data interpretation associated with inaccurate tumor sample identity, thereby providing a significant benefit to cancer scientists.
MATERIALS AND METHODS
Oligonucleotide SNP Array Analysis
SNP detection on the 50K Xba array was performed as described previously (3,13–15). Arrays were scanned with a GeneChip Scanner 3000. Genotyping calls and signal quantification were obtained with GeneChip Operating System 1.1.1 and Affymetrix Genotyping Tools 2.0 software.
Cell lines
The initial dataset included genotype data of 155 cell lines derived from different organs including: breast (50), colorectal (12), endometrial (4), glioma (11), leukemia (6), lung (27), melanoma (12), ovarian (6), pancreas (3), prostate (4), and renal (18) cell lines. All these were neoplastic cell lines, except for two non-malignant breast cell lines. Seven additional cell lines were used only for the validation step of the study (analysis on Sequenom platform): prostate (5), and lung (2). The NCI60 cancer cell line collection (16–18) is included in the study dataset. A complete annotated list of the cell lines used in this study is included in the Supplementary Material ‘List of Cell Lines used in the SPIA study’.
Establishment of N15C6 and N33B2 cell lines
The N15C6 and N33B2 epithelial cell lines were established from normal prostate tissues explanted from two different patients and immortalized through transduction with the recombinant LXSNE6E7 retrovirus harboring the human papilloma virus E6 and E7 genes as described previously (19). Transduced cells were selected by use of 400 µg/ml geneticin. After an initial round of cell death and crisis, cells resistant to geneticin grew out and were considered immortal after 10 passages. Spectral karyotype analysis demonstrated both cell lines to be pseudo-diploid. The N15C6 (at passage 45, 10 cells analyzed) karyotype was determined as: 44, X, dupinv(Y)(q11q12), i(5)(p10), der(8;19)(q10;p10), der(11;15)(q10;q10), der(13;20)(q10;q10), −16, +20, del(22)(q13) (20). The N33B2 karyotype (at passage 21, 9 cells analyzed) was determined as: 40–44, X,Y, -19 (4/7 cells), -22 (5/7 cells), der(1)t(1;13)(p36;q32) (6/7 cells), i(8q) (5/7 cells), der(13)t(11;13)(p10;q10) (2/7 cells), der(15)t(15;19)(q10;p10) (3/7 cells) (21).
In this study we used eight passages of N15C6 (passages: 48, 50, 52, 54, 56, 58, 60, 63) and six passages of N33B2 (passages: 21, 27, 33, 35, 37, 39).
Genotype distance
To quantitatively evaluate how similar two DNA samples are, we introduce a similarity measure D. D is proportional to the number of genotype mismatches between the samples and is normalized to the number of genotype calls available for both samples.
Given a set of NSNPs individual SNPs, let CL1 and CL2 be the ordered sets of genotype calls of two samples and vNSNPs = Card (T), where 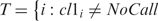
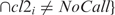 . For vNSNPs > 0, D is defined as:
. For vNSNPs > 0, D is defined as:
Every mismatch counts 1, every match counts 0. The distance is normalized over the number of available calls. Due to technical limitations of the genotyping assay, the genotype calls of some SNPs may not be available for one or the other DNA sample, therefore vNSNPs ≤ NSNPs. In addition to the distance D and to the number of available calls vNSNPs, the algorithm implementing the genotype distance provides summary information on the type of matches and mismatches. It evaluates: (i) the count of mismatches where the two samples are homozygous for different alleles (AA versus BB or vice-versa—like double mutation), (ii) the count of mismatches where one is homozygous and the other is heterozygous (and vice-versa—gain or loss of heterozygosity), (iii) the count of homozygous matches (AA versus AA and BB versus BB) and the count of heterozygous matches (AB versus AB). For each mismatch the algorithm reports the identifier of the sample with largest number of heterozygous calls. The allelic imbalance information (loss of heterozygosity) may be important when handling presumed matched (from the same individual) normal-tumor tissue samples, for example upstream to sequencing. The algorithm would check the consistency of the match and verify which is the tumor sample and which is the normal sample. For specific purposes, the implementation of the distance D can be modified to weight different types of mismatch (see Supplementary Material ‘Genotype Distance with error weighting’).
SNP panel selection procedure
We know that extensive genotype profiles of DNA samples can work as a unique identifier of samples. We hypothesized that by using a small number of SNPs we would still be able to accurately distinguish samples, providing researchers with a convenient way to check the identity of their samples during the course of their use in the laboratory.
Before applying the computational search of the most suitable SNPs, we filtered the 50 K SNPs represented on the Xba Affymetrix SNP Array, using the following rules: (i) SNPs have assigned rs identifier (Reference SNP records); (ii) SNPs are not located in intronic regions and (iii) SNPs are also represented on the 10 K Affymetrix oligonucleotide array (∼8 K). The second rule allows for eventual application of the test on RNA samples and the third rule ensures that the panel will be useful for fingerprint comparison experiments with samples run on the 10 K array.
In order to define and rank a list of suitable SNPs, we applied the following procedure: (i) we randomly divided the cell line dataset into training and testing sets using two-thirds and one-third of the samples, respectively, (ii) on the training set, we evaluated the minor allele frequency, the heterozygosity rate and the call rate for each SNP across all samples; (iii) we then identified the SNPs satisfying the Hardy Weinberg equilibrium applying elastic boundaries (PAA > 0.22, PBB > 0.22, PAB > 0.44) and having SNP call rates greater than 80%, and (iv) on the test set, we evaluated the heterozygosity rate of the identified SNPs. We iteratively ran this procedure 1000 times (repeated hold-out approach). The procedure did not predefine the number of SNPs to select; therefore, at each iteration we obtained a variable number of SNPs. We then ranked the SNPs based on the selection rate on the training set and on the mean value of the heterozygosity rates on the test sets, evaluated on the 1000 iterations. The mean number of SNPs identified at each iteration was 30.15 with a standard deviation of 6.23 (min = 14 and max = 54). The mean value of the heterozygosity on the test set was 0.3636 with a standard deviation of 0.0775 (min = 0.0526 and max = 0.7420). The corresponding distributions are shown in Supplementary Figure 1. The top ranked SNPs represent the best choices.
SPIA probabilistic test on cell line genotype distance
In order to discern when two cell lines are close enough to be called similar and when they are not, we implemented a double probabilistic test to apply on the genotype distance. The test score depends on the number of matches and on the total number of SNPs evaluated for the two cell lines, given a required confidence. The test output reads: ‘similar’, ‘different’ or ‘uncertain’ and relies on the probability of the evaluated distance belonging to the population of real matched pairs or to the population of real non-pairs. If the output test is not clear (depending on the required confidence), the score will be ‘uncertain’ and a second panel of SNPs would need to be investigated.
If we assume the SNPs being independent (call at locus i does not depend on call at locus j ≠ i) and the genotype call probability being the same at each SNP, then the probability of having k matches (successes) out of N SNPs (trials) follows the binomial distribution: 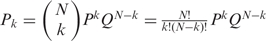 , where P and Q are the probability of match and mismatch, respectively, and N is the number of available SNPs (vNSNPs). We can draw the distributions of real matched pair and of real non-pair, by knowing the probability of match at a single SNP for a real matched pair (PM) and for a non-matched pair (Pnon-M). For a given vNSNPs, we can then define areas corresponding to ‘different’, ‘uncertain’ or ‘similar’. The area limits depend on the level of confidence which is needed for the application. In brief, the mean number of successes kmean is equal to NPM and the standard deviation (sdkmean) is
, where P and Q are the probability of match and mismatch, respectively, and N is the number of available SNPs (vNSNPs). We can draw the distributions of real matched pair and of real non-pair, by knowing the probability of match at a single SNP for a real matched pair (PM) and for a non-matched pair (Pnon-M). For a given vNSNPs, we can then define areas corresponding to ‘different’, ‘uncertain’ or ‘similar’. The area limits depend on the level of confidence which is needed for the application. In brief, the mean number of successes kmean is equal to NPM and the standard deviation (sdkmean) is 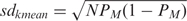 . The probability that a distance measurment falls within m standard deviations from the mean (i.e. within the interval [kmean − m * sdkmean, kmean + m * sdkmean]) is given by the integral of the distribution function. By setting the parameter m one can define the area limits corresponding to ‘different, ‘uncertain’ and ‘similar’. For example, setting m = 2, the integral of the distribution function is 0.954. Figure 1 shows the binomial distributions of real match pair population (red dots) and of non-pair population (blue dots) for N equal to 30; the probabilities PM and Pnon-M, for the real match pair population and the non-pair population, are set to 0.9 and 0.4 respectively and mM and mnon-M are set to 2 and 1, respectively. The red, blue and green bars define regions of ‘different, ‘uncertain’ and ‘similar’ SPIA test calls. The smaller the number of SNPs is, the narrower the region of uncertainty and the higher the probability of making an incorrect call.
. The probability that a distance measurment falls within m standard deviations from the mean (i.e. within the interval [kmean − m * sdkmean, kmean + m * sdkmean]) is given by the integral of the distribution function. By setting the parameter m one can define the area limits corresponding to ‘different, ‘uncertain’ and ‘similar’. For example, setting m = 2, the integral of the distribution function is 0.954. Figure 1 shows the binomial distributions of real match pair population (red dots) and of non-pair population (blue dots) for N equal to 30; the probabilities PM and Pnon-M, for the real match pair population and the non-pair population, are set to 0.9 and 0.4 respectively and mM and mnon-M are set to 2 and 1, respectively. The red, blue and green bars define regions of ‘different, ‘uncertain’ and ‘similar’ SPIA test calls. The smaller the number of SNPs is, the narrower the region of uncertainty and the higher the probability of making an incorrect call.
Figure 1.

Schematic illustration of probabilistic test settings. The figure shows the binomial distributions of real match pair population (red dots) and of non-pair population (blue dots) for N equal to 30 and PM and Pnon-M, for the real match pair population and the non-pair population, equal to 0.9 and 0.4. The red, blue and green bars define regions of ‘different’ (mnon-M set equal to 1), ‘uncertain’ and ‘similar’ (mM set equal to 2) SPIA test calls. The smaller the number of SNPs is, the narrower the region of uncertainty and the higher the probability of making an incorrect call.
In theory, for Hardy–Weinberg SNPs we expect Pnon-M to be equal to 0.375. For exactly the same DNA sample we expect PM being equal to 1. In fact, if we genotype the same DNA sample twice using the same platform, the expected number of mismatches depends on the platform reproducibility error. If, for the same individual, we compare the DNA extracted from normal tissue and from tumor tissue, we expect some variation, most commonly derived from loss of heterozygosity. Similarly, if we compare DNA extracted years apart, some genomic variation can be expected.
To empirically evaluate PM (probability of match for a real match pair) we calculated the mean percentage of matches using seven paired cell line samples, constituted by tumor cell line DNA and blood extracted DNA from the same individual. Using the 100 top ranked SNPs, the value of PM was estimated as 90%.
Sequenom platform for genotyping human cells
We tested a SPIA panel using Sequenom mass spectrometric genotyping technology (22). This MALDI-TOF mass spectrometer system can differentiate SNP alleles given the different molecular weights of the allele-specific products. First, a software package supplied by the manufacturer is used to design a series of primers that enable SNP detection in a multiplexed fashion. To carry out SNP detection, tumor-derived genomic DNA is first subjected to whole genome amplification to generate enough material for a series of multiplexed reactions, which are carried out in parallel on microtitre plates. Next, multiplexed PCR is performed (in 96- or 384-well plate format) on tumor genomic DNA to amplify regions harboring loci of interest, or ‘query’ nucleotides. After denaturation, PCR products are incubated with oligonucleotides that anneal immediately adjacent to the query nucleotide, and a primer extension reaction is performed in the presence of chain-terminating dideoxynucleotides that generate allele-specific DNA products. Primer extension products are spotted onto a specially designed chip and analyzed by MALDI-TOF mass spectrometry to determine the single allele. Since allele calling depends exclusively on the mass of the resulting primer extension product, the Sequenom assay does not require expensive fluorescence primer labeling and has a very low error rate.
SPIA was written in R (23) (the R code is available on request).
RESULTS
Our approach to selection of a SNP panel to distinguish any cell line or tumor pair in use by the research community is shown in Figure 2. SNP computational selection is based on empirical data from 155 cell lines genotyped on 50 K oligonucleotide SNP arrays. The number of independent loci chosen depends on the level of confidence one needs to make a definitive identification. Although the panel is trained on the identification of cell lines, this approach is suitable for additional applications, such as matching human tumor samples with their non-neoplastic normal tissue. The resulting assay, termed the SNP panel identification assay (SPIA), allows investigators to accurately identify known cell lines and tumors from the genotype of extracted DNA.
Figure 2.

Schema of SNP panel identification assay (SPIA) applicability and use modality. One typical scenario would be to check out of the identity of a cell lines (SAMPLE 1) with respect to already fingerprinted cell lines, the data of which are stored in the BARCODING REFERENCE database. Another scenario would be to determine the identity of two cell lines, for example derived from the same patients, one from benign tissue cells and one from cancer tissue cells (SAMPLE 1 and SAMPLE 2). First, the DNA is extracted from the cell line(s) and is experimentally genotyped along the set of specified loci included in the SNP panel. Sequenom mass spectrometric technology, allele-specific PCR or other techniques can be implemented. Afterwards, the allele call output sets (DNA fingerprints) are compared. Comparison is performed pair-wise. Comparison with all the fingerprints stored in the reference database can be performed on demand. The SPIA test provides the genotype distance between the two samples and detailed information on the allele mismatches. It also provides the user with a probability measure of the test output, scoring the tested pair as ‘similar’ or ‘different’. If the test result is uncertain, the assay can be repeated using an additional SNP panel.
SNP panel selection
To define the optimal SNP panel, we reasoned that the ideal SNPs should collectively maximize the probability of obtaining distinct genotype calls on different samples, i.e. exhibiting the greatest heterogeneity across samples. SNPs with two alleles (A and B) give rise to possible genotype calls AA, BB and AB. Hypothetically, genotype frequencies equal to one-third each should maximize the probability of obtaining distinct genotyping calls on different samples, with 9 SNPs being sufficient to distinguish 20 000 samples (see Supplementary Material ‘How many SNPs do we need to have a robust fingerprint’).
However, in a given population, SNPs under neutral selection are in Hardy–Weinberg equilibrium, in which allele frequencies fit the equation P2 + 2PQ + Q2 = 1. Since the vast majority of SNPs in the human genome approximate Hardy–Weinberg equilibrium, the optimal genotype probabilities are PAA = 0.25, PBB = 0.25, PAB = 0.5 for genotype calls AA, BB and AB, respectively.
The SNP panel will have the most power to distinguish individuals when the selected SNPs are independent of one another. This can be accomplished by selecting SNPs from different chromosomes and/or SNPs from the same chromosome that are not in linkage disequilibrium.
If all human cell lines were genotyped, one could rank the SNPs that best distinguish these cell lines from the entire set based on the call frequencies. Iterative analysis would then best define the most accurate and parsimonious panel identifying both which SNPs to use and how many are needed. However, genotype information is not yet available for all cell lines. Instead, we used a computational approach for the selection of candidate SNPs using available empirical genotype data from a broad set of cancer cell lines (n = 155) derived from different organs (e.g. breast, prostate, lung, etc.), and applied an empirical learning approach followed by validation on an independent cell line dataset. The cell lines used in this study are listed in Supplementary Material, Table 1 (‘List of cell lines used for SPIA study’).
Applying general filtering criteria to the 50 K genotype data, as detailed in Materials and Methods, resulted in a set of 5.3 K SNPs, which genotype characteristics graphically represented in Figure 3.
Figure 3.

Genotype characteristics of the filtered 5.3 K SNPs. We filtered the 50 K SNPs from the Affymetrix Xba chip ending with a set of about 5.3 K SNPs. Genotype characteristics were evaluated for this set of SNPs on a collection of 155 cell lines. Panels (A) and (B) show the heterozygosity frequency and the log of the p-value of the Hardy–Weinberg equilibrium test, respectively. In the context of identifying SNPs for the SPIA panel, we expect the best SNPs being the ones with minor allele frequency close to 0.5 and Heterozygosity frequency close to 0.5 (i.e. P-value Hardy–Weinberg = 1).
In brief, we divided the cell line set into training and testing sets and using the set of filtered SNPs, we selected the SNPs that both satisfied Hardy–Weinberg equilibrium constraints and exhibited overall call rates of >80% across the dataset. After 1000 iterations of training and testing, the SNPs selected at each iteration were ranked based on the selection rate at the end of the iterative procedure. The iterative SNP selection procedure is detailed in Methods. Similar results in terms of SNP suitability were obtained when SNPs were selected based on concordance with a random allele distribution (e.g. 1/3 frequency for each 2-SNP allele).
Set of best SNPs
One hundred and thirty three cell lines were used for the selection of the best SNPs through the iterative process, as described in SNP panel selection. Supplementary Table 2 shows the ranked list of SNPs (top list) ordered by selection rate after the 1000 iterations. The table also reports the mean value of heterozygosity frequency for each SNP evaluated on the sampled test set of each iteration. Supplementary Figure 2 represents the location along the genome of all the selected SNPs and highlights the 100 top ranked.
Comparison of pair-wise distances using multiple SNP panels and implementation of statistical test (SPIA)
To quantitatively measure the relatedness of two samples, we determined a genotype distance function D, which is proportional to the number of genotype mismatches between the two samples (see Materials and Methods, ‘Genotype distance’ and Figure 4, Panel A). It also allows to easily compare multiple pairs of samples and to rank them based on relatedness. In our initial cell line data set, using 5.3K SNPs, the pair-wise mean distance D is 0.4735 and the standard deviation 0.0357 (min = 0 and max = 0.5765). Eleven cell line pairs exhibited unusually strong similarity (see Table 1). Some have previously been reported to be similar: M14 and MDA.MB435, NCI.ADR.RES and OVCAR.8, and SNB.19 and U251 (Cancer Genome Project, http://www.sanger.ac.uk/genetics/CGP/Genotyping/synlinestable.shtml).
Figure 4.

Graphical representation of SPIA results on 155 cell lines: pair-wise distances and probabilistic test using SNP panels of different sizes. The number of possible combinations of pairs from a set of 155 samples is 11 935. Black dots represent distances were less than 90% of the genotype calls available. (A) The genotype distance was evaluated on the 5279 SNPs, which were filtered starting from the 50K SNPs represented on the Xba Affymentrix Array. The mean distance is 0.4736 and the standard deviation is 0.0358, accordingly to the confinement of almost all the evaluations. Dots below the dotted gray line represent pairs of cell lines which are genotypically very similar to each other (they share more than 80% of the 5279 loci here considered), suggesting that they share the same ancestral. (B)–(E) Multi-panel distances using 80, 60, 40 and 20 SNPs randomly sampled from the top 100 SNPs selected through the multi-step selection process. The mean distances are significantly higher respect to panel (A). The probability test was applied setting the match probability for the non-pair and for the matched pair population to 0.4 and 0.9 respectively and the parameter for the confidence level to 3 and 2 respectively. Red, blue and green dots indicate ‘different’, ‘uncertain’ and ‘similar’ test scores.
Table 1.
List of 11 cell line pairs detected to have very similar genotype profiles, evaluated on a set of 5.3K SNPs
| No of mismatches | |||||
|---|---|---|---|---|---|
| Cell line name (CL1) | Cell line name (CL2) | Distance D | Percentage of valid calls | Homozygous (AA)– Homozygous (BB) [or vice versa] | Homozygous (AA or BB)– Heterozygous (AB) [or vice versa] |
| M14 | MDA.MB435 | 0.0747 | 0.794 | 2 | 311 |
| MCF7 | BT.20 | 0.0279 | 0.781 | 0 | 115 |
| MCF7 | KPL.1 | 0.0271 | 0.797 | 0 | 114 |
| NCI.ADR.RES | OVCAR.8 | 0.0076 | 0.874 | 0 | 35 |
| NCI.H460 | H2195 | 0.0680 | 0.696 | 0 | 250 |
| SNB.19 | U251 | 0.0394 | 0.866 | 0 | 180 |
| 184A1 | 184B5 | 0.1084 | 0.978 | 37 | 523 |
| BT.20 | KPL.1 | 0.0308 | 0.831 | 1 | 134 |
| H1450 | H2141 | 0.0092 | 0.866 | 0 | 42 |
| H1450 | H220 | 0.0000 | 0.861 | 0 | 0 |
| H2141 | H220 | 0.0088 | 0.857 | 0 | 40 |
Interestingly, the MCF7 breast cancer cell line showed a remarkably short distance from two other breast cancer lines (BT.20 and KPL.1), suggesting a high degree of genetic similarity. This difference is significantly lower than the reproducibility error we evaluated for the same platform (0.02%, data not shown), suggesting that these cell lines may be derived from a single individual. MCF7 and BT.20 have been reported as being ER positive and ER negative, respectively, and as having different phenotypes (24, 25). Similarities between MCF-7 and KPL-1 has been previously reported (http://www.sanger.ac.uk/genetics/CGP/Genotyping/synlinestable.shtml). To our knowledge no report exists on the similarity between MCF7 and BT.20. This finding will require independent validation.
After excluding the 9 cell lines involved in the detected similar pairs (N=11), we were left with 146 distinct cell lines. Of these, 133 were then used to identify and rank the best SNPs through a repeated hold-out training and testing approach, and 13 were used as an independent validation set.
To measure the effect of the SNP selection process on the ability to distinguish different cell lines and to determine the minimum number of SNPs required to identify genetically similar cell lines, we evaluated the pair-wise distances using several sets of SNPs sampled from the 100 top ranked ones. We varied the number of single loci randomly selecting 80, 60, 40 and 20 SNPs. In Table 2 the mean distances as evaluated between all the possible pairs of two subsets of cell lines are reported; the first subset contains the 133 cell lines used in the selection process, whereas the second set is the independent validation set. Table 2 also includes the mean distance values evaluated on the ∼50 K SNPs contained on the 50 K Xba chip and on the ∼5.3 K set of filtered SNPs. We can appreciate how the distance values increase between the first collection of SNPs and the filtered set of 5.3 K SNPs. Comparing the distances obtained for the smaller sets of SNPs (i.e. 80, 60, 40 and 20 SNPs) with the 5.3 K set, we see a significant increment of percentage differences. This result holds on the independent validation set of cell lines, confirming the ability of the selected SNPs to distunguish different cell lines. When comparing the results obtained with 80, 60, 40 and 20 SNPs to each other, we observe that the mean pair-wise distances do not change significantly. As expected, the standard deviations tend to increase when going from 80 to 20 SNPs (see Figure 4).
Table 2.
Summary of pair-wise distances/differences varying the number of SNPs
| Set of CLs (133) used for the SNP selection process | Set of CLs (13) used for independent validation | |||
|---|---|---|---|---|
| Number of SNPs | Mean distance D (SD) | Min–max distance D | Mean distance D (SD) | Min–max distance D |
| ∼58 000a | 0.3832 (0.0347) | 0.2649–0.4891 | 0.4227 (0.0330) | 0.3613–0.4962 |
| 5279b | 0.4723 (0.0328) | 0.3774–0.5765 | 0.4967 (0.0337) | 0.4274–0.5699 |
| 80 | 0.66 (0.06) | 0.44–0.86 | 0.65 (0.06) | 0.49–0.78 |
| 60 | 0.66 (0.07) | 0.37–0.90 | 0.65 (0.06) | 0.50–0.77 |
| 40 | 0.66 (0.09) | 0.28–0.94 | 0.65 (0.08) | 0.46–0.85 |
| 20 | 0.66 (0.12) | 0.20–1 | 0.64 (0.11) | 0.40–0.90 |
CL, cell line.
aSet of SNPs represented on the 50K Xba chip.
bSet of filtered SNPs, used for the selection of the best SNPs.
We then added to the dataset the 9 cell lines identified as being very similar during the preprocessing step and therefore excluded from the SPN selection process and, by using the same sets (i.e. 80, 60, 40 and 20), we ran the distance and the statistical test, as described in the Materials and Methods section. Briefly, this test scores each single pair as ‘different’, ‘uncertain’ and ‘similar’, based on the total number of queried SNPs, on the number of matches and on a set of parameters adjusted on the required level of confidence. The probability test was applied setting the match probability for the non-pair and for the matched pair population to 0.4 and 0.9, respectively, and the parameter for the confidence level to 3 and 2, respectively. With 80 and 60 SNPs all the ‘real pairs’, eligible for the statistical test (10 out of 11), were scored as ‘similar’. No other cell line pair was scored as ‘similar’ (see Figure 4, Panels B and C). With 40 SNPs, 9 real pairs were scored ‘similar’, and one real pair was scored ‘uncertain’ (see Figure 4, Panel D). Using the panel of 20 SNPs a few cell lines were mis-scored as ‘similar’, suggesting that this number of SNPs is insufficient for accurate identity determination. To further prove the efficacy of the SNP selection process with the goal of defining an optimal SNP panel, we ran a ‘baseline experiment’, randomly selecting four sets of SNPs comprised of either 80, 60, 40 or 20 SNPs taken from the 5.3 K filtered set. We measured the pair-wise distances on the two sets of cell lines as reported in Table 2. The mean distance values are similar to the 5.3 K set mean distances but the standard deviations from the means are larger compared to the experiments with the selected SNP panels. Furthermore, in the randomly selected trials unrelated samples were detected as having exactly the same genotype calls for all the considered SNPs (see Supplementary Table 3). These experiments suggest the limitation of using randomly selected SNPs for the development of an identification test.
These results suggest that any set of 40 SNPs selected from the top 100 SNPs, listed in Supplementary Table 2, provides researchers with a good SNP panel for DNA sample identification. However, the more SNPs in the panel, the more confident one can be in the final call.
Distance between different cell line passages
A common concern cited with use of cell lines regards genetic changes that occur during in vitro cultivation. Cell line genomic stability when going from one passage to the next can be assessed by comparing genomic profiles. The ability to determine relatedness between samples suggests that the SPIA distance score may be useful in characterizing genetic drift of cell lines over time in culture. In order to first assess genetic stability over time and secondly to evaluate the capability of SPIA assay to correctly identify them regardless of the passage number, we genotyped and studied two prostate cell lines, N15C6 and N33B2 (21), over multiple passages using 50 K Xba Affimetrix arrays. We ran SPIA on different passages using 40 SNPs out of the top 100 SNPs. The pair-wise distances were all equal to zero (probabilistic test scores ‘similar’), suggesting that our approach can correctly identify one cell line, regardless of the passage. Interestingly, when extensively looking at the 50 K data we noticed that, where the N33B2 was very stable all along the genome, the cell line N15C6 exhibits genetic instability. The distance between passage 48 and passage 63 along chromosome 11 was about 0.2, meaning that up to 20% of the genotype calls on chromosome 11 differ between the two passages. Indeed, these human papillomavirus (HPV) transfected cell lines are known to be affected by the introduction of E7, a HPV gene (26,27), resulting in instability on chromosome 11. This experiment demonstrated that SPIA correctly identifies a cell line regardless of the passage number and that using a dedicated panel of SNPs provides a useful quantitative approach to monitor genetic drift of cell lines, important for characterization of mammalian embryonic cell lines, which may exhibit genetic instability over time.
A genotyping-based assay for tumor sample fingerprinting
One goal of the SPIA panel was to make it small enough to allow it to be exported for routine use in the research laboratory settings. Limited panels of small numbers of microsatellite markers or SNPs are commonly used for identification; however, given concerns regarding error rates and non-informative SNP calls, these panels have only limited value, especially when working with a variety of samples. Therefore with the goal of exporting the SPIA panel to other platforms, we tested the panel using Sequenom mass spectrometric genotyping technology. This MALDI-TOF mass spectrometer system can differentiate SNP alleles given the different molecular weights of the allele-specific products. This system also has the advantage of being fully automated and regularly used in genome centers throughout the world. We evaluated the pair-wise distances on a set of 34 SNPs for a set of 93 cell lines. The 34 SNPs were picked from the ranked list of SNPs. Figure 5 shows the pair-wise distances. The mean and the median distances are 0.656 and 0.594, respectively, and the minimum and maximum distances are 0.059 and 0.941. As expected, the cell lines MCF7 and BT20 were scored as ‘similar’.
Figure 5.

Pair-wise distances of 93 cell lines genotyped on Sequenom platform. With the goal of exporting the SPIA assay to other platforms, we tested a 34 SNP panel using the Sequenom platform on a set of 93 cell lines. The statistical test was applied to get individual score for each distance. The pair made of MCF7 and BT20 was scored as ‘match’ (green dot), accordingly with expectations. One pair out of 4277 was scored as ‘uncertain’ (blue dot). All others 4276 pairs were correctly scored as ‘different’. The list of the 93 cell lines is reported in the Supplementary Material.
DISCUSSION
Accurate tracking and labeling of samples is critical to experimental integrity in the genomic era. This is particularly true for cell lines, which are often cultured over a period of years and are handed from one laboratory to another. Recent reports of inadvertent cross-contamination or labeling mistakes can stay hidden from researchers for years (2,28). Given the large number of published reports that use functional cell line data to make significant claims with respect to cellular mechanisms of tumor biology and that this data may even serve as the basis for pre-clinical therapeutic trials, establishment of the proper identity is critical. While experimentally possible, extensive genome-wide genotyping of cell lines as part of standard operating procedures (SOPs) in a laboratory is not practical or cost-effective. Therefore, we developed the SPIA assay that allows investigators to accurately identify cell lines taking into account both issues of cost and feasibility.
SPIA is based on the selection of highly informative SNPs for the creation of a DNA sample-specific barcode that can be used for a likelihood evaluation of the DNA identity. This assay was demonstrated to be suitable for the identity check of a broad range of cell lines by using 40 SNPs. The more SNPs in the panel, the more confident the final DNA identity determination.
To select an optimized SNP panel we developed an in silico approach by mining 50 K genotype data from more than 150 cancer cell lines, derived from 11 different tumor types. We reasoned that this approach would be more solid for the identification of tumor cell lines and tissue samples than taking SNPs based on their genotype frequencies in normal human populations (29), because genomic alterations, as loss of heterozygosity, gain of heterozygosity and, less frequently, double mutations affect tumor genotypes.
The SNP selection process we presented focused on the task of general cell line identification. However, if a research group works on a specific tumor type (e.g. glioma) the assays could be tailored such that known mutations at specific loci can be added to the panel. By adjusting the model constraints, one can ask for more or less conservative output calls and by increasing the number of SNPs the identity test can become more accurate (30). The general selection rule would still, however, apply to these tailored SPIA assays. The SNPs must still be genetically unlinked and systematically spread across the genome so as to represent all the chromosomes. Regions of known recurring deletion in tumor samples must be avoided. SNP probes with similar annealing temperatures can be selected to facilitate the test implementation in case of multiplex PCR-based assay.
SPIA can track genomic drift across passages, look for replicated samples (in a set of patients) and verify matched tissues from the same individual (for example, normal and tumor tissue of the same patient). In this last case, one can adjust SPIA in order to force false positive identity calls. One current alternative application of SPIA is in the upfront analytical phase of a large genomic study. For example, in the process of performing extensive genomic characterization of prostate cancer cell lines, xenografts and human tissues using SNP array analysis and extensive sequencing for known mutations, SPIA was employed to look for similar samples (unpublished data). Interestingly, SPIA identified a xenograft sample as being genotypically very similar to one of the tumor tissue samples (see Supplementary Material ‘Genotype distance between a rapid autopsy tissue and a xenograft’). Further investigation determined that the xenograft and primary tumor samples were derived from the same individual. Without the SPIA analysis, the relationship between these samples would have gone unnoticed and may have led to over-interpretations of the associated genomic alterations.
Thus, SPIA allows rapid and accurate barcoding of many sample types, including cell lines, embryonic stem cells and DNA from large numbers of clinical samples.
We envision that researchers would run the SPIA assay in their laboratory on each new DNA sample at the beginning of experiments and refer to an on-line publicly available data bank for correct identification of their cell line as a quality assurance measure. Investigators can check the identity of a given cell line over many passages and confirm that no contamination has occurred. This might be critically important when two or more laboratories are comparing results of experiments using the same cell lines. In addition to individual research groups, large organizations or institutes that maintain cell lines such as the ATCC can employ this method to credential the lines that they carry. We also envision that core facilities at research centers may provide this assay as a service. Researchers would send out an aliquot of DNA to external facilities, which using such platforms as the ABI PRISM® SNaPshot™ Multiplex kit (Applied Biosystems) (31), Sequenom mass spectrometer system or other systems, would obtain confirmation of the cell line identity. As a proof of principle, we demonstrated that the Sequenom mass spectrometer system can be used of this purpose by testing 34 SNPs from the SPIA assay. As the NIH and other governmental agencies explore means of making the best use of limited research funds, careful annotation of samples will play a critical role. The SPIA assay and other similar methods should become the standard for validation of sample provenance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
Supported by the NIH Prostate SPORE at the Dana-Farber/Harvard Cancer Center NCI P50 CA090381 (FD, RB, WRS and MAR), and the University of Michigan NCI P50 CA69568 (MAR), R01AG21404 (FD, MAR), R01CA109038 (WRS), the Prostate Cancer Foundation (FD) and the Department of Defense Fellowship Award PC040638 (RB). The authors would like to acknowledge the support of Robert Vessella (NCI/NIH Prostate SPORE, University of Washington and Fred Hutchinson Cancer Research Center grant P50 CA097186), Kirsten D Mertz and Barbara Weir for providing us with cell line genotype data. The authors are grateful to Liuda Ziaugra, Alissa C Baker and to Kelly Lamb for technical support and to Andrea Sboner, Wendy M Winckler and Sunita Setlur for critical discussion on this study. Funding to pay the Open Access publication charges for this article was provided by Prostate Cancer Foundation (FD).
Conflict of interest statement. None declared.
REFERENCES
- 1.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 2.Liscovitch M, Ravid D. A case study in misidentification of cancer cell lines: MCF-7/AdrR cells (re-designated NCI/ADR-RES) are derived from OVCAR-8 human ovarian carcinoma cells. Cancer Lett. 2007;245:350–352. doi: 10.1016/j.canlet.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 4.MacLeod RA, Dirks WG, Matsuo Y, Kaufmann M, Milch H, Drexler HG. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int. J. Cancer. 1999;83:555–563. doi: 10.1002/(sici)1097-0215(19991112)83:4<555::aid-ijc19>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 5.Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl Acad. Sci. USA. 2001;98:8012–8017. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson EW, Waltham M, Ramus SJ, Hutchins AM, Armes JE, Campbell IG, Williams ED, Thompson PR, Rae JM, Johnson MD, et al. LCC15-MB cells are MDA-MB-435: a review of misidentified breast and prostate cell lines. Clin. Exp. Metastasis. 2004;21:535–541. doi: 10.1007/s10585-004-4117-z. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee R. Cell biology. Cases of mistaken identity. Science. 2007;315:928–931. doi: 10.1126/science.315.5814.928. [DOI] [PubMed] [Google Scholar]
- 8.Parson W, Kirchebner R, Muhlmann R, Renner K, Kofler A, Schmidt S, Kofler R. Cancer cell line identification by short tandem repeat profiling: power and limitations. FASEB J. 2005;19:434–436. doi: 10.1096/fj.04-3062fje. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg NA, Li LM, Ward R, Pritchard JK. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet. 2003;73:1402–1422. doi: 10.1086/380416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heaton MP, Harhay GP, Bennett GL, Stone RT, Grosse WM, Casas E, Keele JW, Smith TP, Chitko-McKown CG, Laegreid WW, et al. Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm. Genome. 2002;13:272–281. doi: 10.1007/s00335-001-2146-3. [DOI] [PubMed] [Google Scholar]
- 11.Lao O, van Duijn K, Kersbergen P, de Knijff P, Kayser M. Proportioning whole-genome single-nucleotide-polymorphism diversity for the identification of geographic population structure and genetic ancestry. Am. J. Hum. Genet. 2006;78:680–690. doi: 10.1086/501531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kidd KK, Pakstis AJ, Speed WC, Grigorenko EL, Kajuna SL, Karoma NJ, Kungulilo S, Kim JJ, Lu RB, Odunsi A, et al. Developing a SNP panel for forensic identification of individuals. Forensic Sci. Int. 2006;164:20–32. doi: 10.1016/j.forsciint.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 13.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, Tchinda J, Tomlins SA, Hofer MD, Pienta KG, et al. TMPRSS2:ERG fusion associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 14.Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 16.Monks A, Scudiero DA, Johnson GS, Paull KD, Sausville EA. The NCI anti-cancer drug screen: a smart screen to identify effectors of novel targets. Anticancer Drug Des. 1997;12:533–541. [PubMed] [Google Scholar]
- 17.Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat. Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 18.Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, Kohn KW, Reinhold WC, Myers TG, Andrews DT, et al. A gene expression database for the molecular pharmacology of cancer. Nat. Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 19.Bright RK, Vocke CD, Emmert-Buck MR, Duray PH, Solomon D, Fetsch P, Rhim JS, Linehan WM, Topalian SL. Generation and genetic characterization of immortal human prostate epithelial cell lines derived from primary cancer specimens. Cancer Res. 1997;57:995–1002. [PubMed] [Google Scholar]
- 20.Macoska JA, Paris P, Collins C, Andaya A, Beheshti B, Chaib H, Kant R, Begley L, MacDonald JW, Squire JA, et al. Evolution of 8p loss in transformed human prostate epithelial cells. Cancer Genet. Cytogenet. 2004;154:36–43. doi: 10.1016/j.cancergencyto.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Begley L, Keeney D, Beheshti B, Squire JA, Kant R, Chaib H, MacDonald JW, Rhim J, Macoska JA. Concordant copy number and transcriptional activity of genes mapping to derivative chromosomes 8 during cellular immortalization in vitro. Genes Chromosomes Cancer. 2006;45:136–146. doi: 10.1002/gcc.20274. [DOI] [PubMed] [Google Scholar]
- 22.Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Koster H. Chip-based genotyping by mass spectrometry. Proc. Natl Acad. Sci. USA. 1999;96:10016–10020. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Team RDC. R: A Language and Environment for Statistical Computing. Vienna, Austria: 2006. [Google Scholar]
- 24.Hurnanen D, Chan HM, Kubow S. The protective effect of metallothionein against lipid peroxidation caused by retinoic acid in human breast cancer cells. J. Pharmacol. Exp. Ther. 1997;283:1520–1528. [PubMed] [Google Scholar]
- 25.Elstner E, Linker-Israeli M, Said J, Umiel T, de Vos S, Shintaku IP, Heber D, Binderup L, Uskokovic M, Koeffler HP. 20-epi-vitamin D3 analogues: a novel class of potent inhibitors of proliferation and inducers of differentiation of human breast cancer cell lines. Cancer Res. 1995;55:2822–2830. [PubMed] [Google Scholar]
- 26.Macoska JA, Beheshti B, Rhim JS, Hukku B, Lehr J, Pienta KJ, Squire JA. Genetic characterization of immortalized human prostate epithelial cell cultures. Evidence for structural rearrangements of chromosome 8 and i(8q) chromosome formation in primary tumor-derived cells. Cancer Genet. Cytogenet. 2000;120:50–57. doi: 10.1016/s0165-4608(99)00248-4. [DOI] [PubMed] [Google Scholar]
- 27.Steenbergen RD, Hermsen MA, Walboomers JM, Joenje H, Arwert F, Meijer CJ, Snijders PJ. Integrated human papillomavirus type 16 and loss of Heterozygosity at 11q22 and 18q21 in an oral carcinoma and its derivative cell line. Cancer Res. 1995;55:5465–5471. [PubMed] [Google Scholar]
- 28.van Bokhoven A, Varella-Garcia M, Korch C, Hessels D, Miller GJ. Widely used prostate carcinoma cell lines share common origins. Prostate. 2001;47:36–51. doi: 10.1002/pros.1045. [DOI] [PubMed] [Google Scholar]
- 29.Consortium IH. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Z, Owen AB, Altman RB. Genetics. Genomic research and human subject privacy. Science. 2004;305:183. doi: 10.1126/science.1095019. [DOI] [PubMed] [Google Scholar]
- 31.Gaustadnes M, Orntoft TF, Jensen JL, Torring N. Validation of the use of DNA pools and primer extension in association studies of sporadic colorectal cancer for selection of candidate SNPs. Hum. Mutat. 2006;27:187–194. doi: 10.1002/humu.20248. [DOI] [PubMed] [Google Scholar]