


Abstract
The search for feature enrichment is a widely used method to characterize a set of genes. While several tools have been designed for nominal features such as Gene Ontology annotations or KEGG Pathways, very little has been proposed to tackle numerical features such as the chromosomal positions of genes. For instance, microarray studies typically generate gene lists that are differentially expressed in the sample subgroups under investigation, and when studying diseases caused by genome alterations, it is of great interest to delineate the chromosomal regions that are significantly enriched in these lists. In this article, we present a positional gene enrichment analysis method (PGE) for the identification of chromosomal regions that are significantly enriched in a given set of genes. The strength of our method relies on an original query optimization approach that allows to virtually consider all the possible chromosomal regions for enrichment, and on the multiple testing correction which discriminates truly enriched regions versus those that can occur by chance. We have developed a Web tool implementing this method applied to the human genome (http://www.esat.kuleuven.be/~bioiuser/pge). We validated PGE on published lists of differentially expressed genes. These analyses showed significant overrepresentation of known aberrant chromosomal regions.
INTRODUCTION
Microarrays are powerful tools to study gene expression patterns on a genome wide scale. From the raw microarray data, various gene lists are sifted out based on their differential expression in the subgroups under investigation. These gene lists provide the foundation from which to begin the exploration of the underlying cellular biology resulting in the observed phenotype. A data-mining approach that is increasingly used for study of such gene lists is the search for enriched terms associated with the individual genes. Instead of focusing on the actual genes, such analyses try to summarize the information using annotated and structured information, such as hierarchically organized Gene Ontology (GO) annotation terms and KEGG cellular pathways. For enrichment analysis of the latter two gene classifications, many software tools are available (1–3).
Another—less frequently explored—gene characteristic for the study of microarray gene lists is the chromosomal position of the genes, especially when studying diseases caused by genome alterations, such as cancer. Exploration of the relationship between gene copy number alterations and gene expression in breast tumors for instance revealed that a high percentage of amplified genes were over expressed (4–6). Other studies showed systematic up-regulation of many genes on chromosome 21 in Down syndrome patients, harboring an extra copy of this chromosome in all their cells (7). In acute myeloid leukemia DNA gains and losses caused by multiple chromosome rearrangements were shown to result in altered gene expression in a gene-dosage-dependent manner (8). Comparable to the GO and KEGG pathway analysis tools, it would be of great interest to explore the overrepresentation of chromosomal regions in the generated microarray gene lists. Currently, various tools exist that identify entire chromosomes that are overrepresented in the gene lists (1,9,10). However, the resolution is not sufficient enough to pinpoint critical dosage sensitive regions. Other tools allow the identification of overrepresented regions along the chromosomes in higher detail, such as expression imbalance maps (11), ChARM (12), MACAT (13) and DIGMAP (14). The downside is that these methods need the microarray data as input, which is not always possible (e.g. raw data not made available) or more importantly because it restricts the search of enriched chromosomal region to gene expression and prevents its application to a given set of genes of interest. More recently, ChroCoLoc was developed and accepts a given list of genes as input (15). However, the resolution is limited to chromosomal bands i.e. only a fixed set of chromosomal regions are tested for enrichment.
Here, we present an innovative algorithm and accompanying software—accessible through an online Web interface—for positional gene enrichment (PGE) analysis.
METHODS
Statistics
To test if a chromosomal region is enriched in a query set of genes of interest (for example differentially expressed genes), we apply the hypergeometric distribution as follows: Let g be the total number of genes considered (genome), t the number of genes in the region (target set), q the number of genes of interest (query set) and c the number of genes of interest in the region (genes common to the query and target set), then 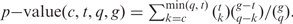 This corresponds to the probability of having at least the observed number of genes of interest in that region. To correct for multiple testing (a large number of regions are tested), adjusted P-values are calculated using the minimum P-values cumulative distribution function. This function provides the probability of obtaining a P-value at least as good (lower or equal) by chance i.e. by submitting a random set of the same size. Unfortunately, it is practically impossible to model this distribution, so we approximate it by sampling to obtain an empirical function as described in (3) and (16).
This corresponds to the probability of having at least the observed number of genes of interest in that region. To correct for multiple testing (a large number of regions are tested), adjusted P-values are calculated using the minimum P-values cumulative distribution function. This function provides the probability of obtaining a P-value at least as good (lower or equal) by chance i.e. by submitting a random set of the same size. Unfortunately, it is practically impossible to model this distribution, so we approximate it by sampling to obtain an empirical function as described in (3) and (16).
Identification of pertinent chromosomal regions to test
To obtain the highest possible resolution i.e. base pairs, our method virtually tests all the sets of genes that are located in all possible windows of all possible widths. Obviously, testing all window positions and widths is inefficient and yields a lot of redundancy in the computations. For example, when shifting the window by one nucleotide, the resulting window will often be on the same genes. A simple observation overcomes this problem: one will obtain the same results if, instead of considering nucleotides, we consider genes ordered on the chromosome. Still, the number of computations to perform can be very large as there are  windows (sets of adjacent genes) on a chromosome with n genes (Figure 1).
windows (sets of adjacent genes) on a chromosome with n genes (Figure 1).
Figure 1.

Sets of genes that are adjacent on the chromosome. Genes are ordered on the chromosome by their start position (in base pairs). Each pair of genes defines an interval i.e. a set of adjacent genes.
The other and most important problem with this setup is that a lot of redundancy in terms of overlapping enriched regions is obtained. For example, a region R1 of 100 genes containing 90 genes of interest. This is very unlikely (i.e. significant) and will be reported as an enriched region. If we now consider a slightly larger region R2 of 101 genes that includes R1 and that also contains 90 genes of interest, then R2 will also be reported as enriched. This is statistically true, but reporting R2 is redundant with R1 because it is merely the same region to which we add one gene not of interest. Hence, R2 should not be part of the results.
To avoid redundancy and identify the pertinent regions to consider, a formal definition is provided in (17) for the pertinence of a target set (here a chromosomal region) comparing it to a given query set (genes of interest). From this definition, three rules are derived which are better suited for the design of an efficient algorithm. These rules are illustrated in Figure 1 with the genes of interest (genes of rank 2, 3, 5 and 7 on the chromosome) and the pertinent regions ([2,3], [2,5], [2,7] and [5,7]) represented by black-filled nodes in the implicit lattice. Informally, the pertinence definition allows to consider only regions bounded by genes of interest, and more specifically, the largest ones (in the example in Figure 1, the region [3,5] is not pertinent because it is included in the larger pertinent region [2,5]). Unfortunately in practice, those three rules do not reduce as much redundancy as it might be desirable. Figure 2 illustrates a concrete example of this problem (biological meaning addressed in the discussion section). In this figure, the whole q arm of chromosome 21 is enriched and the largest region has the best P-value. As the whole q arm is enriched, one should be tempted to report only this region. However, there might be smaller regions that are worth considering. In the following, we extend the pertinence rules to overcome this issue in the particular context of enriched genomic regions. These extended rules are based on two main observations: first, the use of the P-value allows the selection of statistically significant regions but this measure is biased towards large regions. Second, similar to the a priori algorithm (18) that makes use of the anti-monotonic property of the support of an item-set to prune the search space, if a region shows less than expected genes of interest (based on the ratio of the number of genes of interest over the total number of genes) then all the regions including this region should not be considered for enrichment.
Figure 2.

Filtering redundant chromosome regions significantly enriched in genes differentially expressed in tissues of Down syndrome patients on chromosome 21: (A) regions are displayed from left to right by increasing P-value significance (decreasing P-values) and are plotted as –10 log P-value; (B) the same regions plotted by the percentage of genes of interest; (C) enriched regions are filtered for redundancy and plotted by P-value: see rules 4 and 5 in text; (D) the same regions plotted by the percentage of enrichment.
Our first observation is that the P-value allows to assess the statistical significance of the enrichment but it does not reflect the coverage of the region by genes of interest. For example in Figure 2, we see that the q arm has an enrichment of ∼20% in genes of interest, and there is also a smaller region that has almost 50% enrichment, thus it might be interesting to consider this smaller more specific region for investigation even if it is statistically less significant. More generally, these two measures are biased towards opposite directions: small regions tend to have higher percentages of genes of interest while large regions tend to have better P-values. To improve the relevance of the enriched regions reported by our method while reducing the redundancy, we propose two additional rules (Rule 4 and Rule 5) that result in a balance between coverage and statistical significance.
Our second observation is related to improving the efficiency of the resulting algorithm. The P-value significance threshold is not anti-monotonic i.e. if a given region is not statistically significant, there might be a larger encompassing region that is significant. Thus, regions including non-significant regions have to be tested. However, although the percentage of genes of interest is also not anti-monotonic, it can be used to prune the search space: if a region contains less genes of interest than expected (for example the genes of interest represent 2% of the genome and a region contains only 1% of genes of interest) then any region encompassing this region should not be considered in the search. Then, given q the number of genes of interest, a region spanning over t genes is expected to contain on average q/g*t genes of interest (with g the total number of genes). Hence, a region is pertinent if it does not contain any sub-region made of g/q adjacent genes that are not of interest.
Finally, a region is pertinent if the following rules hold:
Rule 1: it contains at least two genes of interest,
Rule 2: there is no smaller region containing the same genes of interest,
Rule 3: there is no bigger region with more genes of interest and the same genes not of interest,
Rule 4: there is no larger encompassing region with a higher percentage of genes of interest,
Rule 5: there is no smaller encompassed region with a better P-value,
Rule 6: it does not contain any region having less than expected genes of interest.
RESULTS
Implementation of the PGE algorithm as a web interface
The PGE method was implemented as a Perl script that is publicly accessible as a web interface (http://homes.esat.kuleuven.be/~bioiuser/pge/). Currently, users can choose to submit lists of Affymetrix probeset ids or Ensembl ids. Probeset ids are mapped either to gene symbols or Ensembl ids by using the Affymetrix human genome array plate set annotations file provided on the Affymetrix Web site. As users might be specifically interested to check the enrichment in a specific chromosome, they have the possibility to only calculate enrichment in the chromosome of interest. To build the empirical minimum P-values cumulative distribution function for P-values significance assessment, we perform 500 simulations with random query sets of the same size as proposed in (3). Because of the computational cost of these simulations, this correction method is available only for gene lists of up to 500 genes; otherwise the False Discovery Rate (19) is applied. The threshold for adjusted P-value significance is set to 0.05. The tool visualizes the enriched chromosomal regions along the chromosomes, based on P-value or percentage of enrichment. A mouse-over pop-up window gives further information on the Ensembl ids or gene symbols that are present in the enriched chromosomal region. Clicking on a region, redirects to the Ensembl genome browser including a PGE track. In addition, enriched regions are provided in BED format to enable the upload of results to other genome browsers.
Validation and application of PGE on published gene lists
We applied the PGE analysis on published gene lists from microarray gene expression studies in order to validate the algorithm. A first test was performed on a list of 407 genes obtained by comparing genetic subtypes of B-cell chronic lymphocytic leukemia (CLL) (20). In B-CLL, the most frequent losses of genomic material are deletions of chromosome bands 13q14, 11q22–23, 17p13 and 6q21, and the most frequent gains affect 12q13, 8q24 and 3q26. In this study, the authors identified genes that are differentially expressed based on microarray gene expression data in different B-CLL subgroups (considering following genetic parameters: 17p13, 11q22–q23, 13q14 and 6q21 deletion, as well as trisomy 12q13) and they visually noticed that a large number of the differentially expressed genes mapped in the chromosomal regions affected by the respective genomic losses or gains. To confirm this correlation, we submitted the set of differentially expressed genes to our PGE tool which reported significantly enriched regions in agreement with the most frequent genomic alterations (Table 1 and Figure 3).
Table 1.
Chromosome regions significantly enriched in genes differentially expressed in subtypes of B-CLL
| Chr. | Band(s) | Coordinates | Genes of interest/genes in the region |
|---|---|---|---|
| 6 | p21.32–22.2 | 26,163,912; 33,851,518 | 13/58 |
| 6 | p22.1 | 27,208,799; 27,941,634 | 7/15 |
| 6 | p22.1 | 27,883,200; 27,941,634 | 5/7 |
| 6 | q14.3–23.2 | 86,216,527; 132,690,949 | 12/77 |
| 11 | q14.1–24.3 | 77,603,590; 127,897,218 | 18/124 |
| 11 | q23.1–23.3 | 111,117,019; 117,775,136 | 8/28 |
| 12 | p13.31–24.33 | 7,233,850; 131,915,071 | 64/408 |
| 12 | p11.21–q13.11 | 31,117,786; 44,641,909 | 5/10 |
| 12 | q23.3 | 104,025,639; 106,630,469 | 4/5 |
| 12 | q24.31 | 120,230,432; 121,194,727 | 4/6 |
| 17 | p13.1–13.3 | 594,403; 8,006,662 | 19/90 |
| 17 | p13.1 | 7,084,456; 8,006,662 | 11/25 |
| 17 | p13.2 | 3,746,634; 4,742,127 | 5/11 |
Figure 3.

PGE Chromosome view of regions significantly enriched in genes differentially expressed in subtypes of B-CLL (Table 1).
Using the PGE algorithm we also noticed a high degree of correlation between the chromosomal localization of differentially expressed genes and the respective genomic aberrations. We found enrichment of genes on 17p13, entire chromosome 12, 11q23, 6q14.3–q23.2 and 6p21.32–p22.2.
For chromosome 6, we find gene enrichment for the region 6q14.3–q23.2 which includes the frequently deleted chromosomal band 6q21, and enrichment in the region 6p21.32–p22.2. From the figure, it appears that some genes cluster on band q21, but this enrichment is not statistically significant which means that such a cluster could arise by chance. The authors suggest that genes located outside of the gained or lost regions might be downstream effectors of the genes directly affected by the loss or gain and may contribute to the disease phenotype. The region 6p21–p22 contains genes that are members of the HIST1 major histone gene locus. This HIST cluster enrichment might be due to the fact that Affymetrix probeset-ids for the different HIST genes are not specific enough, or might only indicate that when HIST genes are expressed, the whole cluster is transcribed.
With our tool, we see that the loss of 13q14 do not lead to clusters of genes differentially expressed in that region. By correcting the P-values with the False Discovery Rate (19), this appeared to be significantly enriched (data not shown). This highlights the importance of the multiple testing adjustment method applied. In this case, the minimum P-values distribution function shows that the number of differentially expressed genes observed on 13q14 could have occurred by chance by selecting 407 genes at random in the human genome.
For a second test, we mapped the genes that are higher expressed in neuroblastoma tumors with amplification of the proto-oncogene MYCN, located on cytoband 2p24, compared to normal neuroblasts (21). A region with high significance for overrepresentation is found on 2p. Within this region a smaller significant region with a higher enrichment percentage is found at band 2p24 (from 5.8 Mb to 24.1 Mb), containing the MYCN, DDX1, NAG and VSNL1 genes (Figure 4). DDX1 and NAG are known to be frequently co-amplified and over expressed in MYCN amplified neuroblastomas. PGE on the same data set also shows an overrepresentation of genes on 17q which is in concordance with the finding that neuroblastoma tumors with MYCN amplification almost always present with gain of the long arm of chromosome 17.
Figure 4.

Regions significantly enriched in genes differentially expressed in neuroblastoma tumors.
As a third example, we mapped the genes that are higher expressed in tissues from Down syndrome patients compared to normal samples (22) and found a significant overrepresentation of genes on chromosome 21 (Figure 2). A smaller region with very high significance emerged that contains the DSCR2 gene that is located in the minimal region for Down syndrome and is a candidate to be involved in the dosage effect of trisomy 21.
DISCUSSION
In this article, we present a new efficient method for the high-resolution identification of chromosomal regions that are overrepresented in custom gene lists together with its application to the human genome. In contrast to many other methods, PGE does not require any user analysis parameter, is user-friendly and performs rapid calculations (few seconds for gene lists of 100–500 genes). The unique approach of this algorithm allows to exhaustively evaluate the overrepresentation rate at all resolution levels simultaneously i.e. from pairs of genes to entire chromosomes. The simulations performed for assessing the significance of enriched regions are essential because they allow discriminating which regions are truly enriched from those that could occur by chance. Application of the PGE analysis on published differentially expressed gene lists showed an overrepresentation of aberrant chromosomal regions, demonstrating the validity of the tool. Moreover, within these regions, smaller regions with significant overrepresentation (lower significance but higher enrichment) might highlight specific genes that are of interest in the aberrant regions, and which deserve further study.
In the near future, we will extend the tool with additional identifier types (RefSeq, HGNC, …) and species (mouse, fly, worm, yeast, …). The software is generic in the sense that only a reference data set file mapping identifiers to chromosome locations is needed to propose other identifiers or species.
We will also adapt and implement our method in the context of circular genomes. Actually, very little modifications will be needed: either Rule 6 allows removing at least one region that cannot be enriched and we are left with a linear chromosome, or we have to consider that a pair of (query) genes defines two regions. In the latter case, the worst case time complexity of our method is still in O(q2) with q the number of genes of interest.
In this article, we focused on gene expression correlated with genomic alteration. This method and tool will also be valuable to test other hypotheses such as tissue-specific chromosomal region accessibility and expression with sets of genes corresponding to EST expressed in different tissues, and genes participating in the same pathways or biological processes.
ACKNOWLEDGEMENTS
The authors are grateful to the reviewers for their helpful comments and suggestions. This text presents research results of the Belgian program of Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming. K.D.P. and J.V. are post-doctoral researchers with a grant from the Fund for Scientific Research Flanders (FWO). R.B. is a post-doctoral researcher with a grant from the research Council Katholieke Universiteit Leuven, Center of Excellence EF/05/007 SymBioSys. This work was supported by the Fund for Scientific Research Flanders (‘Krediet aan Navorsers’ J.V. 1.5.243.05 and K.D.P. 1.5.117.06, FWO-grant G.0185.04) and UGent (GOA-grant 12051203, BOF grant 011F1200 and 011B4300), Kinderkankerfonds and Stichting tegen Kanker. Funding to pay the Open Access publication charges for this article was provided by the Fund for Scientific Research Flanders (FWO).
Conflict of interest statement. None declared.
REFERENCES
- 1.Khatri P, Draghici S, Ostermeier GC, Krawetz SA. Profiling gene expression using onto-express. Genomics. 2002;79:266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 2.Zhang B, Schmoyer D, Kirov S, Snoddy J. GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics. 2004;5:16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barriot R, Poix J, Groppi A, Barre A, Goffard N, Sherman D, Dutour I, de Daruvar A. New strategy for the representation and the integration of biomolecular knowledge at a cellular scale. Nucleic Acids Res. 2004;32:3581–3589. doi: 10.1093/nar/gkh681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyman E, Kauraniemi P, Hautaniemi S, Wolf M, Mousses S, Rozenblum E, Ringner M, Sauter G, Monni O, Elkahloun A, et al. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 2002;62:6240–6245. [PubMed] [Google Scholar]
- 5.Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl Acad. Sci. USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buness A, Kuner R, Ruschhaupt M, Poustka A, Sultmann H, Tresch A. Identification of aberrant chromosomal regions from gene expression microarray studies applied to human breast cancer. Bioinformatics. 2007;23:2273–2280. doi: 10.1093/bioinformatics/btm340. [DOI] [PubMed] [Google Scholar]
- 7.Kahlem P. Gene-dosage effect on chromosome 21 transcriptome in trisomy 21: implication in Down syndrome cognitive disorders. Behav Genet. 2006;36:416–28. doi: 10.1007/s10519-006-9053-z. [DOI] [PubMed] [Google Scholar]
- 8.Lindvall C, Furge K, Bjorkholm M, Guo X, Haab B, Blennow E, Nordenskjold M, Teh BT. Combined genetic and transcriptional profiling of acute myeloid leukemia with normal and complex karyotypes. Haematologica. 2004;89:1072–1081. [PubMed] [Google Scholar]
- 9.Hosack DA, Dennis G., Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kano M, Nishimura K, Ishikawa S, Tsutsumi S, Hirota K, Hirose M, Aburatani H. Expression imbalance map: a new visualization method for detection of mRNA expression imbalance regions. Physiol. Genomics. 2003;13:31–46. doi: 10.1152/physiolgenomics.00116.2002. [DOI] [PubMed] [Google Scholar]
- 12.Myers CL, Dunham MJ, Kung SY, Troyanskaya OG. Accurate detection of aneuploidies in array CGH and gene expression microarray data. Bioinformatics (Oxford, England) 2004;20:3533–3543. doi: 10.1093/bioinformatics/bth440. [DOI] [PubMed] [Google Scholar]
- 13.Toedling J, Schmeier S, Heinig M, Georgi B, Roepcke S. MACAT–microarray chromosome analysis tool. Bioinformatics. 2005;21:2112–2113. doi: 10.1093/bioinformatics/bti183. [DOI] [PubMed] [Google Scholar]
- 14.Yi Y, Mirosevich J, Shyr Y, Matusik R, George AL., Jr Coupled analysis of gene expression and chromosomal location. Genomics. 2005;85:401–412. doi: 10.1016/j.ygeno.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Blake J, Schwager C, Kapushesky M, Brazma A. ChroCoLoc: an application for calculating the probability of co-localization of microarray gene expression. Bioinformatics. 2006;22:765–767. doi: 10.1093/bioinformatics/btk022. [DOI] [PubMed] [Google Scholar]
- 16.Dufour J-M Technical Report, C.R.D.E., Canada: Université de Montréal, Montreal; 1995. Monte Carlo Tests with Nuisance Parameters: A General Approach to Finite-Sample Inference and Nonstandard Asymptotics in Econometrics. [Google Scholar]
- 17.Barriot R, Sherman DJ, Dutour I. How to decide which are the most pertinent overly-represented features during gene set enrichment analysis. BMC Bioinformatics. 2007;8:332. doi: 10.1186/1471-2105-8-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agrawal A, Imielinski T, Swami A. Proceedings of the ACM-SIGMOD 1993 Int'l Conference on Management of Data. Washington D.C: 1993. Mining Associations between Sets of Items in Massive Databases. [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statisit. Soc. 1995;B 57:289–300. [Google Scholar]
- 20.Haslinger C, Schweifer N, Stilgenbauer S, Dohner H, Lichter P, Kraut N, Stratowa C, Abseher R. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. J. Clin. Oncol. 2004;22:3937–3949. doi: 10.1200/JCO.2004.12.133. [DOI] [PubMed] [Google Scholar]
- 21.De Preter K, Vandesompele J, Heimann P, Yigit N, Beckman S, Schramm A, Eggert A, Stallings RL, Benoit Y, Renard M, et al. Human fetal neuroblast and neuroblastoma transcriptome analysis confirms neuroblast origin and highlights neuroblastoma candidate genes. Genome Biol. 2006;7:R84. doi: 10.1186/gb-2006-7-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao R, Zielke CL, Zielke HR, Pevsner J. Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain. Genomics. 2003;81:457–467. doi: 10.1016/s0888-7543(03)00035-1. [DOI] [PubMed] [Google Scholar]