

Abstract
Glutarylamidase is an important enzyme employed in the commercial production of 7-aminocephalosporanic acid, a starting compound in the synthesis of cephalosporin antibiotics. 7-aminocephalosporanic acid is obtained from cephalosporin C, a natural antibiotic, either chemically or by a two-step enzymatic process utilizing the enzymes D-amino acid oxidase and glutarylamidase. We have investigated possibilities for redesigning glutarylamidase for the production of 7-aminocephalosporanic acid from cephalosporin C in a single enzymatic step. These studies are based on the structures of glutarylamidase, which we have solved with bound phosphate and ethylene glycol to 2.5 Å resolution and with bound glycerol to 2.4 Å. The phosphate binds near the catalytic serine in a way that mimics the hemiacetal that develops during catalysis, while the glycerol occupies the side-chain binding pocket. Our structures show that the enzyme is not only structurally similar to penicillin G acylase but also employs essentially the same mechanism in which the α-amino group of the catalytic serine acts as a base. A subtle difference is the presence of two catalytic dyads, His B23/Glu B455 and His B23/Ser B1, that are not seen in penicillin G acylase. In contrast to classical serine proteases, the central histidine of these dyads interacts indirectly with the Oγ through a hydrogen bond relay network involving the α-amino group of the serine and a bound water molecule. A plausible model of the enzyme–substrate complex is proposed that leads to the prediction of mutants of glutarylamidase that should enable the enzyme to deacylate cephalosporin C into 7-aminocephalosporanic acid.
Keywords: Cephalosporin acylase, glutaryl acylase, cephalosporin C, catalytic triad, Ntn-hydrolase, X-ray structure
Glutarylamidase (E.C. 3.5.1.11; synonyms cephalosporin acylase, glutaryl acylase) deacylates glutaryl-7-aminocephalosporanic acid (Gl-7-ACA) to glutaric acid and 7-aminocephalosporanic acid (7-ACA) (Aramori et al. 1991). Catalytically competent glutarylamidase (GA) from Pseudomonas sp. is formed by a two-step autocatalytic process from the inactive precursor. In the first step the precursor polypeptide chain is cleaved into a short (residues A1–A170) and a long chain (residues B1–B522). In the second step the spacer peptide A161–A170 in chain A is removed, catalyzed by the N-terminal Ser B1 of chain B (Lee and Park 1998). The resulting enzyme forms an A2B2 heterotetramer complex from two A- and B-chains (Ichikawa et al. 1981; Matsuda and Komatsu 1985), or A1B1 heterodimers depending on the species. The N-terminal serine in the B-chain is also an essential residue of the mature enzyme that acts as a nucleophile in catalysis; GA is a member of the serine-protease family (Nobbs et al. 1994; Ishii et al. 1995). The exact role of GA in the cell is still unknown.
The recently determined structure of GA (Kim et al. 2000) shows extensive similarity with the structure of penicillin G acylase (PGA) (Duggleby et al. 1995). PGA is a founding member of the Ntn (N-terminal nucleophile)-hydrolase superfamily (Brannigan et al. 1995). Like GA, members of this superfamily consist of two different amino acid chains generated by post-translational processing from inactive precursors (Brannigan et al. 1995). Ntn-hydrolases share a common fold in which the N-terminal nucleophile and other catalytic groups occupy equivalent sites. Serine proteases differ in details of proton abstraction from the catalytic Ser-hydroxyl group, involving either the classical catalytic triad, consisting of Ser, His, and Asp, or hydroxyl/ɛ-amine or hydroxyl/α-amine catalytic dyads (Paetzel and Dalbey 1997). It has been suggested that the catalytic triad should be regarded as two dyads, Ser/His and His/Asp dyad (Liao et al. 1992). PGA employs the α-amino group of the catalytic serine for proton abstraction from the Ser-hydroxyl (Duggleby et al. 1995).
The two serine proteases PGA and GA share a common fold, but they convert different substrates (Scheme 1 ▶). PGA catalyzes the hydrolysis of the antibiotic penicillin G into phenylacetic acid and 6-aminopenicillanic acid (6-APA), whereas GA deacylates Gl-7-ACA to 7-ACA. Another GA from the Pseudomonas sp. strain N176 (N176) converts in addition cephalosporin C (CPC), although at low efficiency (Ishii et al. 1995). CPC is the natural form of the antibiotic cephalosporin, which is produced by the fungus Cephalosporin acremonium. The substrates shown in Scheme 1 ▶ are all cleaved at the amide bond linking the β-lactam core with the side chain. In penicillin G, the side chain consists of a hydrophobic phenyl moiety, while the side chains of Gl-7-ACA and CPC include charged groups (Scheme 1 ▶). Substrate specificity of the enzymes is associated with recognition of the side chain and not of the β-lactam core, as shown for the case of PGA (Huang et al. 1963).
Scheme 1.

GA is employed in the industrial production of semisynthetic cephalosporin C antibiotics like Claforan™ from 7-ACA, the end product of the enzymatic reactions. Gl-7-ACA, a substrate of GA, is obtained from CPC by an additional enzymatic step using D-amino acid oxidase. An enzyme that is catalytically competent to produce 7-ACA directly from CPC would therefore be of great commercial interest (Ishii et al. 1995; McDonough et al. 1999; Kim et al. 2000). So far, none of the numerous attempts to modify PGA or GA for this purpose have been successful.
We describe here a structure-based approach to achieve this goal that is guided by the structure of GA and comparison with the related enzymes PGA and N176. We have solved the structure of GA with a phosphate bound near the catalytic serine and with glycerol in the side-chain binding pocket, and derive the putative structure of Gl-7-ACA in complex with GA. This allowed us to pinpoint the required modifications that should enable the new enzyme to convert CPC to 7-ACA.
Results and Discussion
Structure determination
We have obtained monoclinic crystals of glutarylamidase (GA) from Pseudomonas sp. and of selenomethionine-GA (Se–GA). The enzyme is an A2B2 heterotetramer in solution; the crystals contain two A1B1 heterodimers in the asymmetric unit, related by a pure translation. We used the multiwavelength anomalous dispersion (MAD) method to solve the Se–GA structure at 2.5 Å resolution. The GA structure at 2.4 Å was subsequently obtained by molecular replacement.
In the active center the GA structure contains a glycerol and a water, whereas an ethylene glycol and a phosphate are found in the Se–GA structure. Otherwise, both structures are very similar.
Overall structure
The A2B2 heterotetramer of GA (Fig. 1A ▶) consists of two A1B1 heterodimers, each built from two polypeptide chains (A: residues Ala A1–Glu A160; B: residues Ser B1–Pro B522). The two chains fold into a compact structure as one might expect for a protein derived from a single-chain precursor. The two heterodimers, related by a crystallographic twofold axis, form a tight contact (buried surface area 1862 Å2) that involves only residues of the B-chains (Fig. 1A ▶). The symmetry axis is located between Tyr B380 (heterodimer 1) and Tyr B380 (heterodimer 2). The tyrosine rings are oriented antiparallel, and their hydroxyl groups interact, mediated by a bridging water, with the carbonyl group of Ser B377 of the other heterodimer (Fig. 1B ▶).
Fig. 1.
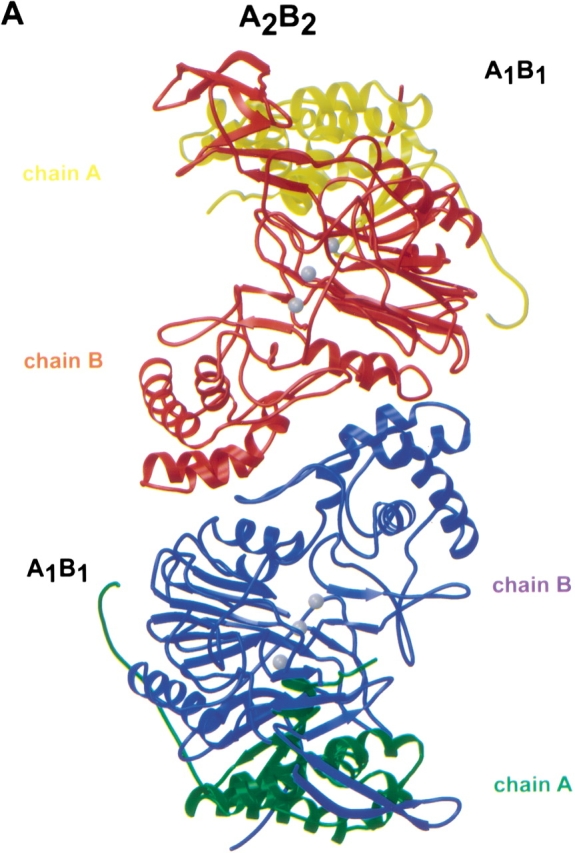

A2B2 heterotetramer of GA. (A) Ribbon representation of the A2B2 heterotetramer of GA. The A1B1 dimers are related by a crystallographic twofold axis. The three gray balls in the two active sites mark ligand electron density. (B) Contact region between two A1B1 dimers that form the heterotetramer of GA. The crystallographic twofold axis relating the two heterodimers is approximately perpendicular to the drawing plane. Only residues of the B-chain are involved in the contact. Water molecules are shown as gray balls. Figures were created with the programs Molscript (Kraulis 1991) and Raster3D (Meritt and Bacon 1997).
The structure of the A1B1 heterodimer (Fig. 2A ▶) appears to be very similar to the structure of cephalosporin acylase reported by Kim et al. (Fig. 1c ▶ of Kim et al. 2000); the atomic coordinates of the latter structure were not released at the time of this writing. The heterodimer forms one large and two small domains. One of the small domains (B301–B452) contains mainly β-sheets, while the other (A61–A86, A151–A160, B75–B142, B199–B221) is helical, and involved in A2B2 heterotetramer contacts (Fig. 1A ▶). The central core of the large domain comprises four layers of αββα ( Fig. 2A ▶) that display the characteristic structural motif of the Ntn-hydrolase superfamily (Brannigan et al. 1995).
Fig. 2.
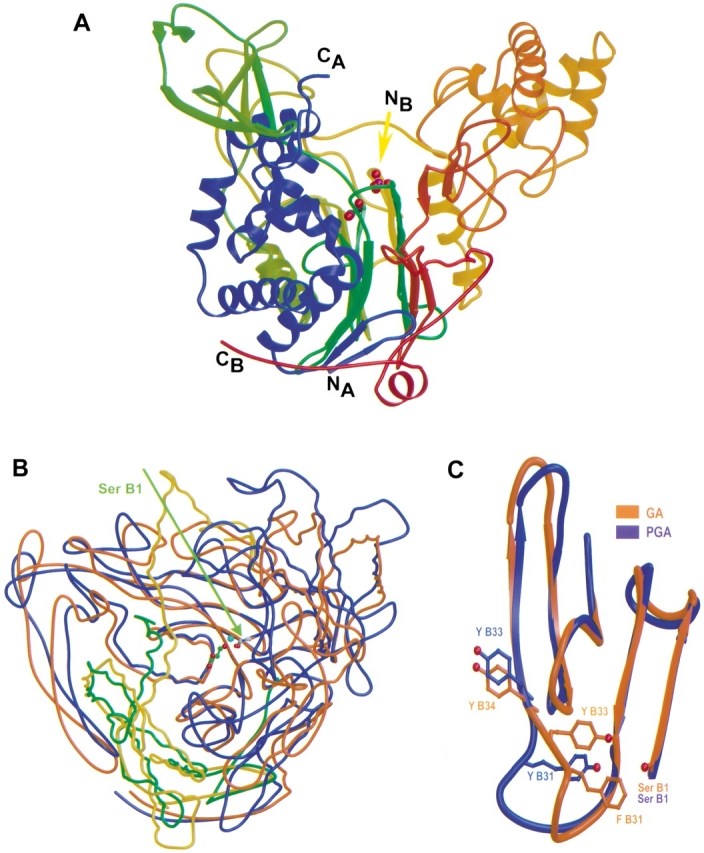
A1B1 heterodimer. (A) Ribbon representation of the A1B1 heterodimer. N- and C-termini of the A- and B-chain are indicated (chain A: blue; chain B: rainbow). The Se–GA structure is shown with ethylene glycol (red and green balls) in the side-chain binding pocket and with bound phospate (red and magenta balls) near Ser B1(NB). (B) Superimposed structures of GA (green, brown) and PGA (yellow, blue). The catalytic residue Ser B1 is indicated. Glycerol (red and green balls) and water W1 (blue ball) are shown in the active site. (C) Comparison of GA and PGA in the side-chain binding pocket of the active site. Figures were created with the programs Molscript (Kraulis 1991) and Raster3D (Meritt and Bacon 1997).
We have identified 401 equivalent residues in GA and in PGA (PMSF-PGA, Protein Data Bank accession code: 1pnm; Duggleby et al. 1995), a member of this superfamily, and found that their Cα coordinates can be superimposed with a r.m.s.d. of 1.9 Å (Fig. 2B ▶), which is similar to the results of Kim et al. (2000).
Structure of the active center
According to the above alignment the structural similarity between GA and PGA includes the active site centered at the catalytic residue Ser B1 (Fig. 2B ▶). As shown in Figure 2A ▶, Ser B1 is located at the top of the flat β-sheet, while the substrate binding pocket extends into the more twisted β-sheet. In the neighborhood of the catalytic residue Ser B1 we found significant electron density (4–5 σ) in both of our structures that could not be explained by protein atoms.
In the GA structure this density can be explained by a glycerol and a water molecule (W1) (Fig. 3A ▶). In both crystallographically independent copies, W1 occupies a well-defined position, as indicated by its low temperature factor of 21 Å2. Glycerol is somewhat more mobile, with an average temperature factor of 49 Å2, but shows clear density in the annealed omit map. Glycerol interacts with Ser B1, Tyr A150, Tyr B33, Gln B50, Arg B57, Val B70, and W1, which bridges the hydroxyl group and the amino group of Ser B1 (Fig. 4A ▶).
Fig. 3.

Stereoview of the active site with bound ligands and superimposed omit density contoured at 1.2 σ. (A) GA structure with bound glycerol. (B) Se–GA structure with bound phosphate and ethylene glycol.
Fig. 4.
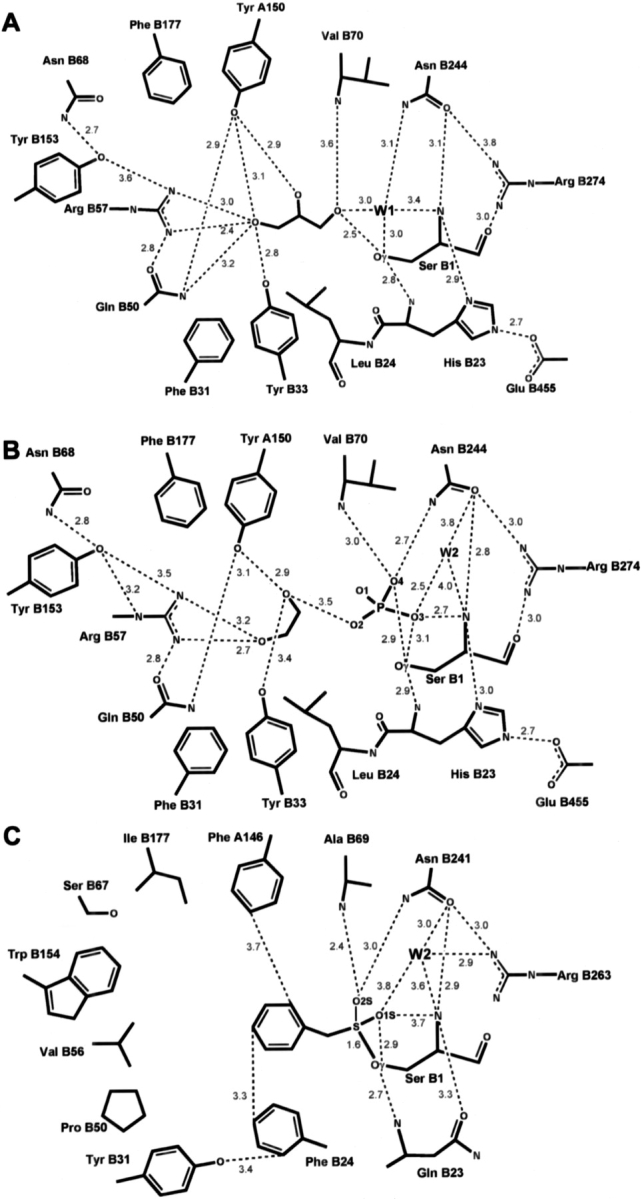
Schematic drawing of the active site. Distances are in Å. (A) GA with bound glycerol. (B) Se–GA with bound phosphate and ethylene glycol. (C) PGA with bound affinity label PMSF, derived from coordinate data set 1pnm of Duggleby et al (1995).
In the Se–GA structure, density for an ethylene glycol molecule and a phosphate group was found instead of a glycerol and a water molecule (Fig. 3B ▶). Refined temperature factors of the ligand atoms are 30 Å2 and 40 Å2, respectively. Ethylene glycol interacts with Tyr A150, Tyr B33, and Arg B57. Phosphate oxygen O2 interacts with ethylene glycol and the O3 atom forms hydrogen bonds with the Ser B1 hydroxyl and backbone-amino group, while O4 interacts with the hydroxyl-group of Ser B1 and with the amino groups of residues Val B70 and Asn B244 (Fig. 4B ▶).
In both structures the catalytic residue Ser B1 is fixed by numerous interactions with the neighboring residues His B23, Asn B244, and Arg B274 (Figs. 4A,B ▶). Hydrogen bonds are formed between Ser B1 Oγ and the main-chain amino group of His B23, between the α-amino-group of Ser B1 with OD1 of Asn B244 and NE2 of His B23, and between the main-chain carbonyl group of Ser B1 and the side chain of Arg B274. The Asn B244 side chain is fixed by the guanidino group of Arg B274.
Mechanism
In our structures Glu B455 is buried in the protein, and there is no other charged residue to compensate for this negative charge. Glu B455 interacts with the imidazole of His B23 so that the three residues, Ser B1, His B23, and Glu B455, form a cascade of two dyads (Figs. 4A,B ▶), reminiscent of the catalytic dyads of serine proteases (Liao et al. 1992). However, the hydrogen bond between Ser B1 and His B23 involves the amino group of Ser and not the hydroxyl group, as found in serine proteases. This points to the importance of the backbone amino group of Ser B1, indicating that the catalytic mechanism of GA could be similar to PGA (Duggleby et al. 1995). Indeed, we observed a close structural similarity between Se–GA and the PMSF–PGA complex that includes the active site residues around the catalytic Ser B1. If we superimpose equivalent main-chain atoms and Cβ-atoms (Kabsch 1978), we obtain a r.m.s.d. of 0.5 Å for 69 atoms (GA: B1–B3, B22–B24, B68–B71; B242–B245; PGA: B1–B3, B22–B24, B67–B70, B239–B242).
The mechanism proposed by Duggleby et al. (1995) for PGA assigns the Ser B1 α-amino group as a general base. This amino group subtracts a proton from water W1, which bridges the amino and hydroxyl groups of Ser B1. The resulting OH− attracts the proton of the Ser B1 hydroxyl group. The negatively charged Ser B1 Oγ attacks the substrate's carbonyl carbon, thereby forming a negatively charged intermediate stabilized by the oxyanion hole. Collapse of this tetrahedral intermediate leads to a seryl acyl enzyme and release of free 6-aminopenicillanic acid (6-APA). Attack of the acyl enzyme by water W2 leads to the formation of a second tetrahedral intermediate that is stabilized by the same interactions as the first one. Finally, this intermediate collapses, releasing free phenylacetic acid.
The interactions made by the tetrahedral intermediates developing during the catalytic cycle have been deduced from the structure of the PMSF–PGA complex in which the sulphonyl group is thought to model the hemiacetal (Duggleby et al. 1995). The same interactions occur in our Se–GA structure if we identify O3, O4 of the phosphate group and water W2 with O1S, O2S of PMSF and W2 in the PMSF–PGA complex, respectively (Figs. 4B,C ▶). In Se–GA, the O4 atom interacts with nitrogens of the main chain of Val B70 and the side chain of Asn B244. These residues form together with His B23 the oxyanion hole, a structural motif that is characteristic of serine proteases (Stroud 1974). The corresponding residues in PGA are Ala B69, Asn B241, and Gln B23, respectively.
We conclude that the network of hydrogen bonds seen in our structures closely matches the network in the active site of the PGA structures, which is a strong indication that both enzymes adopt the same catalytic mechanism. The Ser B1 α-amino group in PGA and GA can only work as a base if this group is uncharged. This is likely to be true for both enzymes at their pH optimum (PGA: 8.2 [Cole 1969], GA: 7.8) if the pKa of the free α-amino group assumes a value about 8, as is usually found in proteins (Stryer 1988). In GA, the uncharged state of the α-amino group is maintained by His B23 and Glu B455.
One structural difference remains, however. In PGA, the side-chain oxygen of Gln B23 forms a hydrogen bond with the α-amino group of Ser B1 (Fig. 4C ▶), whereas the role of the oxygen is assumed by the nitrogen NE1 of His B23 in GA (Fig. 4B ▶). As mentioned above, this interaction is stabilized by Glu B455. At the pH optimum of GA the His and Glu side chains (pKa 6; pKa 4.25, Stryer) are unprotonated. The unprotonated side of the histidine residue (NE1) is oriented towards the α-amino group, while the histidine NE2 forms a hydrogen bond with the acidic residue. The strength of a hydrogen bond depends on the pK values of the heteroatoms sharing the proton: two similar pK result in a short, strong hydrogen bond with almost no barrier between the two energy minima of the shared proton (Cleland 2000). Transient positive charges develop during the catalytic cycle at the α-amino group or at the His side chain. It is then likely that in GA the hydrogen bond between His B23 and Glu B455 (distance ≈2.7 Å) changes to a low-barrier hydrogen bond (distance ≈2.5 Å), because the pK of the now charged histidine matches that of the Glu side chains. The proton could then readily move between the heteroatoms forming the low-barrier hydrogen bond. The energy released upon formation of the low barrier hydrogen bond is used to ease proton transfer during the catalytic cycle by lowering the activation energies.
Taken together, it appears that GA has efficiently combined reaction principles from the PGA mechanism and from the classical catalytic triad.
Side-chain binding pocket
GA deacylates Gl-7-ACA to 7-ACA, whereas PGA catalyzes the hydrolysis of penicillin G into phenylacetic acid and 6-APA. PGA specifically recognizes the side-chain phenyl moiety and not the β-lactam core of the substrate Penicillin G (Huang et al. 1963). In our GA structure glycerol is located at a position in the active center that corresponds to phenyl acetic acid in the structure of the complex with PGA (1pnl; Duggleby et al. 1995).
The side-chain pocket in PGA consists of 20 residues (A142–A146, B48, B49, B52–B57, B153–B155, B175–B178), which could be aligned with residues (A146–A150, B49, B50, B53–B58, B152–B154, B175–B178) in our GA structures (r.m.s.d. = 1.4 Å for 99 equivalent pairs of main-chain and Cβ atoms). Most of these residues reside on the twisted β-sheet, except for the GA residues Tyr A150 and Met A146 located on the C-terminal helix of chain A, and TyrB33, which is at a branching point where the polypeptide chains follow different courses in the two enzymes (Fig. 2C ▶).
Only hydrophobic amino acids line the side-chain pocket in PGA, while GA possesses Arg B57, which binds glycerol as observed in our GA structure (Figs. 4A ▶, 5A,B). The hydroxyl group at the other end of glycerol can only marginally interact with the oxyanion hole because the molecule is too short. The glutaryl side chain of the Gl-7-ACA substrate is just long enough to allow simultaneous interactions with Arg B57 and the oxyanion hole (Fig. 5C ▶).
Fig. 5.

Views of the active site. (A) PGA with bound phenyl acetic acid according to the coordinate data set 1pnl of Duggleby et al. (1995). (B) GA with bound glycerol. (C) Putative GA structure in complex with Gl-7-ACA. (D) Putative N176 structure in complex with CPC. (E) CPC in the GA structure, modeled according to D shows unfavorable interactions. Mutations of GA residues in the side-chain pocket as shown in (F) are expected to lead to a modified enzyme that could accept CPC as a substrate. (F) Suggested residues for binding the carboxylate and the α-amino group of CPC are shown in blue (one of R B57; Q B50 R; L B24 R) and red (one of Y A150 E; F B177 H and T B176 D), respectively.
Putative structure of the enzyme–substrate complex
Our results described so far allow a stereochemical modeling of the substrate Gl-7-ACA in complex with GA. Figure 5C ▶ shows a model of the first tetrahedral intermediate. The carbonyl carbon is bound to Ser B1 Oγ, and the carbonyl oxygen fits into the oxyanion hole. The carboxylate group of the glutaryl side-chain interacts with Arg B57, Tyr B33, and Tyr A150, similar to the interactions seen for glycerol. The glutaryl side chain of Gl-7-ACA in GA and the side-chain phenyl moiety in PGA are then orientated in the same manner, while the β-lactam core makes no interactions with the protein.
Comparison with glutarylamidase from the Pseudomonas strain N176
Glutarylamidase from the Pseudomonas sp. strain N176 (N176) hydrolyzes Gl-7-ACA and also CPC, although the latter only at low efficiency (Ishii et al. 1995). GA and N176 share 23% sequence identity and 50% sequence similarity. To predict the structure of N176 we have aligned its sequence with the GA and PGA structures as shown in Figure 6 ▶. The alignment suggests that the three enzymes adopt the same catalytic mechanism, all using Ser B1 as catalytic residue and three NH groups forming the oxyanion hole. One of these NH groups is provided by the side chain of a conserved asparagine residue, which is fixed by a conserved arginine. It is possible that the catalytic dyads seen in GA are also present in N176. Both proteins use a histidine that is either linked to Glu B455 in GA or to Asp B21 in N176 (Fig. 5D,E ▶).
Fig. 6.

Portion of sequence alignment of GA, N176, and PGA covering the active site.
In the side-chain binding pocket, the loop regions of the polypeptide chains between B26 and B33 in GA and between B26 and B32 in PGA follow different courses, as mentioned above (Fig. 2C ▶). At the C-terminal end of this loop we have aligned N176 Tyr B32 with Tyr B33 in GA and Tyr B31 in PGA. The side chain of Tyr B32 would then point towards Ser B1, consistent with mutational studies of N176 (Ishii et al. 1995) that show that Tyr B32 could interact with Ser B1, and that the mutation Tyr B32 Glu abolishes enzymatic activity (Figs 5D, 7 ▶ ▶). Our alignment nicely explains the loss of catalytic activity when this residue is mutated to the negatively charged Glu residue. The two neighboring residues of Tyr B32 in N176, namely Ala B33 and Met B31, correspond to Tyr B34 and Phe B31 in GA. Studies of the mutants Ala B33 Tyr and Met B31 Phe show moderately increased activities for catalysis of CPC with respect to the wild type, and essentially no change of activity for the substrate Gl-7-ACA (Ishii et al. 1995).
Fig. 7.

Equivalent residues of glutarylamidase from Pseudomonas sp. (GA, black), glutarylamidase from Pseudomonas sp. strain N176 (N176, orange), and penicillin G acylase (PGA, green) in the active site. GA residues suggested for mutation are colored magenta. Tyr A150 in GA aligns with Phe A146 in PGA, whereas the corresponding residue in N176 is ambiguous. Also, no residues corresponding to Glu B455 in GA are found in the other structures.
The considerations described above lead us to suggest the putative structure of the active site of N176 in complex with CPC (Fig. 5D ▶). The carboxylate group of CPC interacts with Arg B24, whereas the carboxylate group of Gl-7-ACA is bound by Arg B57 in GA, as mentioned above (Fig. 7 ▶). In PGA, the side chain of Penicillin G is purely hydrophobic, and as a consequence, there is no positively charged residue in the pocket. In N176, His B178 interacts with the α-amino adipyl moiety of CPC. This interaction is stabilized through the negatively charged side chain of Asp B177 that points towards the histidine side chain. The α-amino adipyl moiety, together with the His and Asp residues, structurally resembles the catalytic dyads described above. For the formation of the dyads it is necessary that the side chains of the adjacent Asp and His residues point into the same direction, which leads to backbone torsion angles outside of the allowed region of the Ramachandran plot. It has been noted that steric strain in the polypeptide backbone, when it occurs, is often found in functional regions of the protein (Herzberg and Moult 1991).
Implications for CPC converting mutants of GA
An enzyme that is catalytically competent to produce 7-ACA directly from cephalosporin C (CPC) would be of great interest for the industrial production of semisynthetic cephalosporin antibiotics (Ishii et al. 1995; McDonough et al. 1999; Kim et al. 2000). Our approach to achieving this goal is based on the putative structures of Gl-7-ACA in complex with GA (Fig. 5C ▶) and of CPC in complex with N176 (Fig. 5D ▶). We attempt to pinpoint the modifications required in GA if its substrate is to be CPC.
Instead of the carboxylate moiety at the end of the Gl-7-ACA side chain, CPC carries a carboxylate and an amino group (Scheme 1 ▶, Fig. 5D,E ▶). Replacing Gl-7-ACA in our model structure by CPC would lead to no interactions of the α-amino adipyl moiety. This is consistent with the finding that GA does not accept CPC as a substrate.
For binding of the α-amino adipyl moiety, Figures 5D ▶–F and 7 suggest that the GA mutant Tyr A150 Glu could lead to CPC binding. The carboxylate group of CPC could interact with Arg B57 (Fig. 5F ▶). Considering the putative structure of CPC in complex with N176 (Fig. 5D ▶), binding of the α-amino adipyl moiety would require the mutation Phe B177 His and perhaps Thr B176 Asp to match the triad seen in N176. In addition, we expect that the mutants Leu B24 Arg or Gln B50 Arg would lead to more favorable interactions with the carboxylate group.
Summarizing our analysis, a modified GA that accepts CPC as a substrate should include a histidine or a glutamate for binding the α-amino adipyl moiety and exactly one positively charged residue for binding the carboxylate group. This leads to the specific suggestions for modifying the sequence of GA that are summarized in Table 1. The additional mutations Y A150 A and Y B153 L, which enlarge the side-chain binding pocket, may be needed to accommodate the larger substrate CPC. Preparation and biochemical characterization of these GA mutants are underway.
Table 1.
Suggestions for mutations of GA
| Interacting residue(s) for the α-amino adipyl group | Only one positively charged residue for the carboxylate group | To stabilize or to enlarge the volume | |||
| Tyr A150 Glu | Arg B57 existing | ||||
| Tyr A150 Glu | Arg B57 existing | Phe B177 His | |||
| Tyr A150 Glu | Leu B24 Arg | Arg B57 His | |||
| Tyr A150 Glu | Leu B24 Arg | Arg B57 His | Phe B177 His | ||
| Tyr A150 Glu | Gln B50 Arg | Arg B57 His | |||
| Tyr A150 Glu | Gln B50 Arg | Arg B57 His | Phe B177 His | ||
| Phe B177 His | Thr B176 Asp | Arg B57 existing | |||
| Phe B177 His | Thr B176 Asp | Arg B57 existing | Tyr A150 A | ||
| Phe B177 His | Thr B176 Asp | Leu B24 Arg | Arg B57 His | ||
| Phe B177 His | Thr B176 Asp | Leu B24 Arg | Arg B57 His | Tyr A150 A | |
| Phe B177 His | Thr B176 Asp | Gln B50 Arg | Arg B57 His | ||
| Phe B177 His | Thr B176 Asp | Gln B50 Arg | Arg B57 His | Tyr A150 A | |
| Phe B177 His | Leu B24 Arg | Arg B57 His | |||
| Phe B177 His | Val B70 His | Leu B24 Arg | Arg B57 His | Gln B50 A | |
| Phe B177 His | Val B70 His | Leu B24 Arg | Arg B57 His | Gln B50 A | Tyr B153 Leu |
Materials and methods
Protein preparation
The glutarylamidase (GA) gene of Pseudomonas sp. was cloned and overexpressed in Escherichia coli using a noninduced, continuous expression system yielding up to 2.5 g of active enzyme per liter in production scale (Aretz et al. 1998).
Gel chromatography of GA using superose 12 in the presence of a 50-mM potassium phosphate buffer at pH 7.0 revealed a molecular weight of 140,000.
The pH profile of GA activity was determined at 37°C with 25 mM Glutaryl-7-ACA in the presence of 100-mM sodium phosphate buffers (pH 6.0 to 8.0). Glutarylamidase was added in appropriate dilution to get a linear velocity. The reaction was stopped after 10 min by addition of acid and aldehyde according to Shibuja et al. (1981). The resulting Schiff base was determined photometrically. Conversion to 7-ACA was about 8%. The pH profile displayed a broad optimum between pH 7 to 8 and a significant increase of specific activity between pH 6 and 7.
Crystallization
For bulk industrial production the enzyme was crystallized in the last purification step as described in the patent (Bayer 1996). To obtain X-ray grade crystals, small crude crystals were dissolved at 20–30 mg/mL in 0.5 M potassium phosphate buffer at pH 7.5 in the presence of 5 mM DTT. Crystals of GA were grown at 18°C by the hanging drop method using potassium phosphate (1.5–2.0 M, pH 7.0–9.0) as the precipitating agent. Diamond-shaped crystals of the size 400 × 300 × 40 μm appeared after 2–3 days. The crystals were monoclinic and contained two A1B1 heterodimers in the asymmetric unit, leaving a solvent content of 58%. Preparation of selenomethionyl-GA (Se–GA) was carried out as described (Budisa et al. 1995), applying the same continuous expression system in 10 L lab-scale fermentation (Aretz et al. 1998). Se–GA crystals were obtained under the same conditions as above. For cryoprotection, the GA and Se–GA crystals were transfered to crystallization buffer containing 10% (w/v) glycerol and 10% ethylene glycol, respectively.
Data collection
All diffraction data were recorded by the rotation method and processed by XDS (Kabsch 1988), which includes routines for space group determination, scaling, and conversion to reduced structure factor amplitudes (Kabsch 1993). Data set GA (Table 2) was collected from a single crystal at 100 K in the laboratory (X-rays: Cu Kα; generator: Nonius Fr571, operated at 45 kV/80 mA; detector: Mar300 image plate, xrayresearch; rotation/image: 0.5°). Multiwavelength anomalous dispersion (MAD) data were collected from the same selenomethionyl crystal (Se–GA) at 100 K using synchrotron radiation (ESRF beamline BM-14). Data sets Se–GApk, Se–GAip, and Se–GArm (Table 2) were obtained at wavelengths at the peak anomalous signal, at the inflection point, and remote from the absorption edge, respectively (detector: MAR345, xrayresearch; rotation/image: 1°).
Table 2.
Data collection statistics
| Data set | Space group | a (Å) | b(Å) | c(Å) | α(°) | (β)° | γ(°) |
| Se–GA | C2 | 228.37 | 69.91 | 113.55 | 90.00 | 97.57 | 90.00 |
| GA | C2 | 230.29 | 70.44 | 114.80 | 90.00 | 97.48 | 90.00 |
| Data set | Se–GAm | Se–GApk | Se–GAip | GA |
| a Values within parentheses are for data in the last resolution shell. | ||||
| bRsym = 100 Σ|fh − 〈I〉|/Σlh | ||||
| cRmeas and Rmrgd-F as defined by Diederichs and Karplus (1997) are quality measures of the individual intensity observations and of the reduced structure factor amplitudes, respectively. | ||||
| d Phasing power = sqrt {〈Fh|2〉/〈|E2|〉}, where Fh is the heavy-atom structure factor and E the lack-of-closure error. Reflections between 20 and 3 Å resolution were used. | ||||
| e Overall figure-of-merit for reflections between 20 and 3 Å resolution were used for computing the initial map. | ||||
| λ (Å) | 0.8854 | 0.9776 | 0.9779 | 1.5418 |
| Resolution range (Å) | 20–2.5 | 20–2.5 | 20–2.5 | 15–2.4 |
| Last shell (Å)a | 2.6–2.5 | 2.6–2.5 | 2.6–2.5 | 2.5–2.4 |
| Completeness (%) | 98.6 (95.9) | 95.8 (70.2) | 96.1 (72.1) | 93.7 (92.7) |
| Mean redundancy | 2.0 (1.85) | 1.98 (1.47) | 2.18 (1.56) | 1.91 (1.92) |
| Rsym (%)b | 4.0 (8.2) | 4.7 (9.7) | 4.9 (10.5) | 9.3 (26.2) |
| Rmeas (%)c | 5.4 (11.1) | 6.3 (13.2) | 6.5 (13.9) | 12.5 (35.5) |
| Rmrgd-F (%)c | 6.6 (13.6) | 7.6 (15.9) | 7.7 (15.9) | 17.0 (38.2) |
| f`/f` (electrons) | −3.00/3.48 | −9.80/6.47 | −13.38/4.62 | |
| Dispersive phasing powerd | — | 1.24 | 1.82 | |
| Anomalous phasing powerd | 1.22 | 1.93 | 2.13 | |
| Figure-of-merite | 0.64 | |||
Phasing
Reflection phasing and structure refinements were carried out with CNS (Brünger et al. 1998). Twenty-one out of the 24 selenium atoms expected from the number of methionines in the asymmetric unit of the GA crystals were initially located, all of which turned out to be correct. The remaining three sites appeared after the first round of phase refinement. The figure of merit of the phased reflections was 0.64- to 3-Å resolution. The initial map revealed clear solvent boundaries only for the correct choice of the enantiomorph of the anomalous scatterer. The 24 positions of the selenium atoms were found to comprise two sets of 12 equivalent pairs that could be superimposed with a rms of 0.17 Å, which confirmed the correctness of our analysis. Moreover, the translation/rotation operator relating the two sets and inspection of the initial map revealed that the crystals contain two A1B1 heterodimers in the asymmetric unit that are related by a pure translation. The starting map at 2.5 Å resolution used for model building was obtained by combining MAD phase information with density modification involving solvent flipping (Abrahams and Leslie 1996). This map (figure of merit 0.83) showed very clear density for all but the seven N-terminal residues of the A-chains.
Model building and refinement
An initial atomic model of Se–GA was fitted to this map using the interactive graphics program O (Jones et al. 1991). Refinement of the model was completed after a few rounds of model correction, thereby imposing restraints on the two independent A1B1 heterodimers to obey noncrystallographic symmetry (Table 3). The final model of the A2B2 heterotetramer comprises residues Gln 8 to Glu 160 of the A chains, residues Ser 1 to Pro 522 of the B chains, one phosphate, and one ethylene glycol in each of the two active sites, and 710 water molecules in the asymmetric unit. The model displays good stereochemistry, as verified by Procheck (Laskowski et al. 1993). Except for residue Phe B 177, all backbone torsion angles are within the allowed region of the Ramachandran plot, and four prolines (P A132, P B253, P B379, P B466) assume a cis-peptide conformation, as has been noted previously by Kim et al. (2000). The GA structure was solved by molecular replacement using the Se–GA structure as a starting model. The final model contains the same number of protein atoms as the Se–GA structure, one glycerol molecule in each of the two active sites, and 706 water molecules (Table 3).
Table 3.
Model and refinement statistics
| Data set | Se–GA | GA |
| Resolution range (Å): | 20.0–2.5 | 15.0–2.4 |
| No. of reflections used in refinement.a | 118,686 | 67,222 |
| No. of reflections used for Rfree:b | 3585 (3%) | 3362 (5%) |
| Rcryst (%)a | 20.7 | 18.0 |
| Rfree (%)b | 22.2 | 21.9 |
| Content of the model | ||
| Protein atoms (non-hydrogen): | 10,650 | 10,650 |
| Solvent molecules: | 710 | 706 |
| Heterogen atoms: | 18 | 12 |
| Rms deviation | ||
| Bond distances (Å) | 0.007 | 0.006 |
| Bond angles (°) | 1.4 | 1.3 |
| Average B-factors (Å2) | 14.1 | 19.6 |
| Estimated coordinate error (Å)c | 0.3 | 0.3 |
aRcryst = 100 Σ∥Fobs| − |Fcalc∥/Σ|Fobs| calculated for all observed data. No sigma cutoff was used.
bRfree = 100 Σ ∥Fobs| − |Fcalc∥/Σ|Fobs| calculated for a specified number of randomly chosen reflections that were excluded from the refinement. No sigma cutoff was used.
c Crossvalidated estimated coordinate error from Luzatti plot.
Coordinates and measured reflection amplitudes have been deposited in the Brookhaven Protein Data Bank (PDB, accession code 1gk0 and 1gk1).
Acknowledgments
We gratefully acknowledge the dedicated technical assistance of Wilhelm Klingenberg, Anke Schickel, Christiane Metz-Weidmann, and Oliver Bronk. We thank Andreas Becker, Susanne Eschenburg, and John Wray for critical discussions and reading of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.27502.
References
- Abrahams, J.P. and Leslie, A.G.W. 1996. Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta Crystallogr. D 52 30–42. [DOI] [PubMed] [Google Scholar]
- Aramori, I., Fukagawa, M., Tsumura, M., Iwami, M., Isogai, T., Ono, H., Ishitani, Y., Kojo, H., Kohsaka, M., Ueda, Y., and Imanaka, H. 1991. Cloning and nucleotide sequencing of new Glutaryl 7-ACA and cephalosporin C acylase genes from Pseudomonas strain. J. Ferment. Bioeng. 72 232–243. [Google Scholar]
- Aretz, W., Holst, U., and Koller, K.-P. 1998. Induction-free process for the recombinant preparation of glutarylamidase. U.S. Patent No. 5766881.
- Bayer, T. 1996. Enzymatische Herstellung von 7-Aminocephalosporansäure. Dechema Jahrestagungen 1 230–233. [Google Scholar]
- Brannigan, J.A., Dodson, G., Duggleby, H.J., Moody, P.C.E., Smith, J.L., Tomchick, D.R., and Murzin, A.G. 1995. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 378 416–419. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography & NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. D 54 : 905–921. [DOI] [PubMed] [Google Scholar]
- Budisa, N., Steibe, B., Demange, P., Eckerskorn, C., Kellermann, J., and Huber, R. 1995. High-level incorporation of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur. J. Biochem. 230 788–796. [DOI] [PubMed] [Google Scholar]
- Cleland, W.W. 2000. Low-barrier hydrogen bonds and enzymatic catalysis. Arch. Biochem. Biophys. 382 1–5. [DOI] [PubMed] [Google Scholar]
- Cole, M. 1969. Hydrolysis of penicillins and related compounds by the cell-bound penicillin acylase of E. coli. Biochem. J. 115 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs, K. and Karplus, P.A. 1997. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4 269–275. [DOI] [PubMed] [Google Scholar]
- Duggleby, H.J., Tolley, S.P., Hill, C.P., Dodson, E.J., Dodson, G., and Moody, P.C.E. 1995. Penicillin acylase has a single-amino-acid catalytic center. Nature 373 264–268. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graphics 15 132–134. [DOI] [PubMed] [Google Scholar]
- Herzberg, O. and Mould, J. 1991. Analysis of the steric strain in the polypeptide backbone of protein molecules. Proteins 11 223–229. [DOI] [PubMed] [Google Scholar]
- Huang, H.T., Seto, T.A., and Shull, G.M. 1963. Distribution and substrate specificity of benzylpenicillin acylase. Appl. Microbiol. 11 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa, S., Shibuya, Y., Matsumoto, K., Fujii, T., Komatsu, K., and Kodaira, R. 1981. Purification and properties of 7β-(4-carboxybutanamido-)cephalosporanic acid acylase produced by mutants derived from Pseudomonas. Agric. Biol. Chem. 45 2231–2236. [Google Scholar]
- Ishii, Y., Saito, Y., Fujimura, T., Sasaki, H., Noguchi, Y., Yamada, H., Niwa, M., and Shimomura, K. 1995. High-level production, chemical modification and site-directed mutagenesis of a cephalosporin C acylase from Pseudomonas strain N176. Eur. J. Biochem. 230 773–778. [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods of building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. 1978. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A34 827–828. [Google Scholar]
- ———. 1988. Evaluation of single-crystal X-ray diffraction data from a position-sensitive detector. J. Appl. Crystallogr. 21 916–924. [Google Scholar]
- ———. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26 795–800. [Google Scholar]
- Kim, Y., Yoon, K.-H., Khang, Y., Turley, S., and Hol, W.G.J. 2000. The 2.0 Å crystal structure of cephalosporin acylase. Structure 8 1059–1068. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Lee, Y.S. and Park, S.S. 1998. Two-step autocatalytic processing of the glutaryl 7-aminocephalosporanic acid acylase from Pseudomonas sp. strain GK16. J. Bacteriol. 180 4576–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, D.I., Breddam, K., Sweet, R.M., Bullock, T., and Remington, S.J. 1992. Refined atomic model of wheat serine carboxypeptidase II at 2.2 Å resolution. Biochemistry 31 9796–9812. [DOI] [PubMed] [Google Scholar]
- Matsuda, A. and Komatsu, K. 1985. Molecular cloning and structure of the gene for 7β-(4-carboxybutanamido)cephalosporanic acid acylase from a Pseudomonas strain. J. Bacteriol. 163 1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough, M.A., Klei, H.E., and Kelly, J.A. 1999. Crystal structure of penicillin G acylase from the Bro1 mutant strain of Providencia rettgeri. Protein Sci. 8 1971–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Nobbs, T.J., Ishii, Y., Fujimura, T., Saito, J., and Niwa, M. 1994. Chemical modification and site-directed mutagenesis of tyrosines residues in Cephalosporin C acylase from strain N176. J. Ferment. Bioeng. 77 604–609. [Google Scholar]
- Paetzel, M. and Dalbey, R.E. 1997. Catalytic hydroxyl/amine dyads within serine proteases. Trends Biosci. 22 28–31. [DOI] [PubMed] [Google Scholar]
- Shibuya, Y., Matsumoto, K., and Fujii, T. 1981. Isolation and properties of 7β(4-carboxybutanamido)-cephalosporanic acid acylase producing bacteria. Agric. Biol. Chem. 45 1561–1567. [Google Scholar]
- Stroud, R.M. 1974. A family of protein-cutting proteins. Sci. Am. 231 74–88. [DOI] [PubMed] [Google Scholar]
- Stryer, L. 1988. Biochemistry, 3rd ed., pp. 21, 42. New York: W.H. Freeman and Company.