

Abstract
The cleavage pattern of oligocytidylic acid substrates by bovine pancreatic ribonuclease A (RNase A) was studied by means of reversed-phase HPLC. Oligocytidylic acids, ranging from dinucleotides to heptanucleotides, were obtained by RNase A digestion of poly(C). They were identified by MALDI-TOF mass spectrometry; it was confirmed that all of them corresponded to the general structure (Cp)nC>p, in which C>p indicates a 2`,3`-cyclic phosphate. This is a confirmation of the proposed mechanism for RNase A, wherein the so-called hydrolytic (or second) step is in fact a special case of the reverse of transphosphorylation (first step). The patterns of cleavage for the oligonucleotide substrates show that the native enzyme has no special preference for endonucleolytic or exonucleolytic cleavage, whereas a mutant of the enzyme (K7Q/R10Q-RNase A) lacking p2 (a phosphate binding subsite adjacent, on the 3` side, to the main phosphate binding site p1) shows a clear exonucleolytic pattern; a mutant (K66Q-RNase A) lacking p0 (a phosphate binding subsite adjacent, on the 5` side, to the main phosphate binding site p1) shows a more endonucleolytic pattern. This indicates the important role played by the subsites on the preference for the bond cleaved. Molecular modeling shows that, in the case of the p2 mutant, the amide group of glutamine can form a hydrogen bond with the 2`,3`-cyclic terminal phosphate, whereas the distance to a 3`,5`-phosphodiester bond is too long to form such a hydrogen bond. This could explain the preference for exonucleolytic cleavage shown by the p2 mutant.
Keywords: Ribonuclease A, subsites, kinetics, oligocytidylic acids
Bovine pancreatic ribonuclease (RNase A, EC 3.1.27.5) has been studied extensively (e.g., see D'Alessio and Riordan 1997; Raines 1998). However, fine kinetic analysis of its specificity has always been hampered by the enormous complexity of its substrate RNA. In fact, the efficiency of breakdown of the different bonds in RNA depends not only on the sequence and number of nucleotides but also on the structure of the substrate. Thus, RNase A cuts only single-stranded RNA; also, the nucleotide on the 3` position of the phosphodiester bond broken must be a pyrimidine, with cytidine being preferred over uridine. The base in the nucleotide on the 5` position of the broken bond is also important in defining its efficiency, A>G>C>U (Witzel and Barnard 1962). The length of the substrate is another factor that affects the rate of transphosphorylation. Thus, in the case of homo-oligonucleotides of uridine, the trinucleotide is split faster than the dinucleotide (Irie et al. 1984). For cytidine oligonucleotides, the trinucleotide is cut faster than the dinucleotide and also faster than both the tetra and the pentanucleotide; however, the polynucleotide polycytidylic acid (poly(C)) is cut much faster than any of the oligonucleotides and the reaction approaches a diffusion-limited reaction rate (Moussaoui et al. 1998).
The cleavage of poly(C) by RNase A follows an endonucleolytic pattern showing a preference for the longer substrate molecules and for the release of fragments containing 6–8 nucleotide units (Moussaoui et al. 1996). From these results and previous structural and kinetics studies, a model for the cleavage of the RNA chain based on the complementary binding between the multi-subsite structure of RNase A and the phosphates of the polynucleotide was proposed. The model indicated that, in addition to the phosphate group that binds to the active site (named p1), there are two adjacent phosphate binding sites, one of them towards the 5` end of the substrate, p0 (Lys 66), and the other towards the 3` end, p2 (Lys 7 and Arg 10) (Parés et al. 1980, 1991; De Llorens et al. 1989; Boix et al. 1994; Nogués et al. 1995; Fisher et al. 1998a ,1998b). Other binding sites, such as p-1 that includes Arg 85 (Fontecilla-Camps et al. 1994; Fisher et al. 1998a), and other sites located at the surface of the protein are also considered (Fig. 1 ▶). The cleavage pattern must depend on the subsite structure of the enzyme, because, in a mutant lacking the p2 subsite (K7Q/R10Q-RNase A mutant), a more exonucleolytic pattern with poly(C) as substrate was observed (Moussaoui et al. 1996). Other RNases, such as eosinophil cationic protein (ECP; also known as RNase-3) that seems to lack a p2 site, also show a preference for external bonds (Boix et al. 1999). To gain greater insight of the size preference of the enzyme and the role of the non-catalytic phosphate binding sites, we analyzed the pattern of cleavage of oligocytidylic acids of different lengths (from tetra- to hexanucleotide) by reversed-phase HPLC as a function of time. We used the wild-type enzyme and the mutants that show specific deletion of the electrostatic interaction in the p2 (K7Q/R10Q-RNase A) and p0 (K66Q-RNase A) sites. The results indicate that in native RNase A the increase in the length of the substrate favors slightly the endonucleolytic activity of RNAse A, whereas the mutant lacking p2 shows a clear exonucleolytic pattern. The mutant lacking p0 is more endonucleolytic than the native enzyme.
Fig. 1.
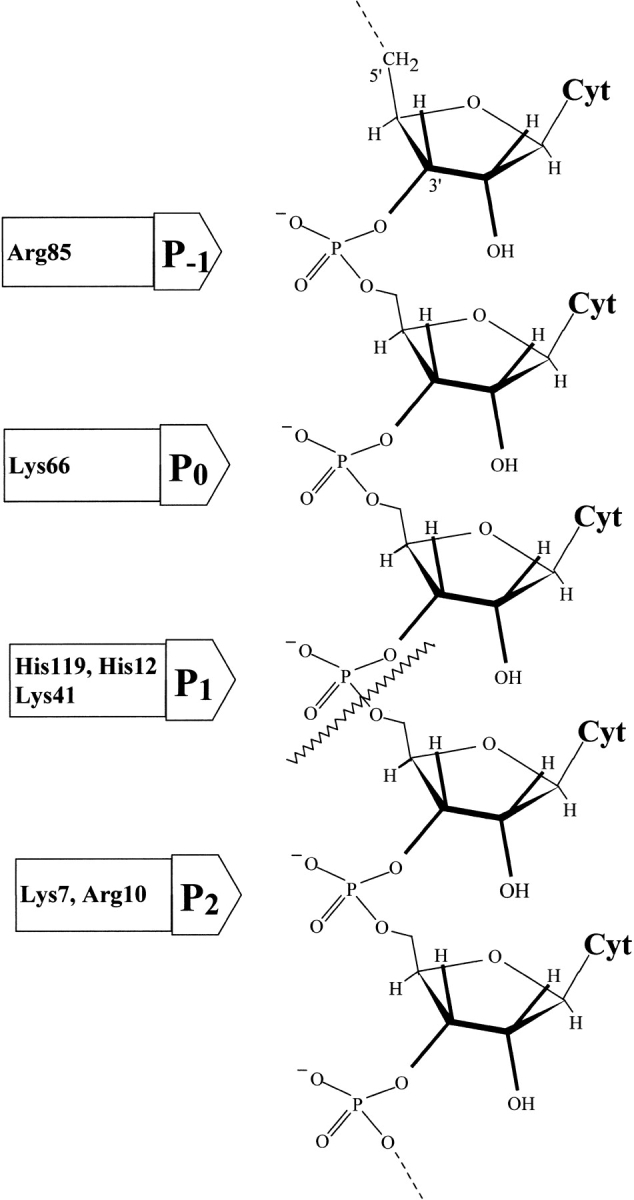
Structure of a poly(C) chain showing the ionic interactions between the negatively charged phosphates and the corresponding binding subsites in the enzyme. The residues identified as pertaining to each subsite are indicated. The scissile bond (wavy line) is placed in p1.
Results
Preparation and characterization of the oligocytidylic acid substrates
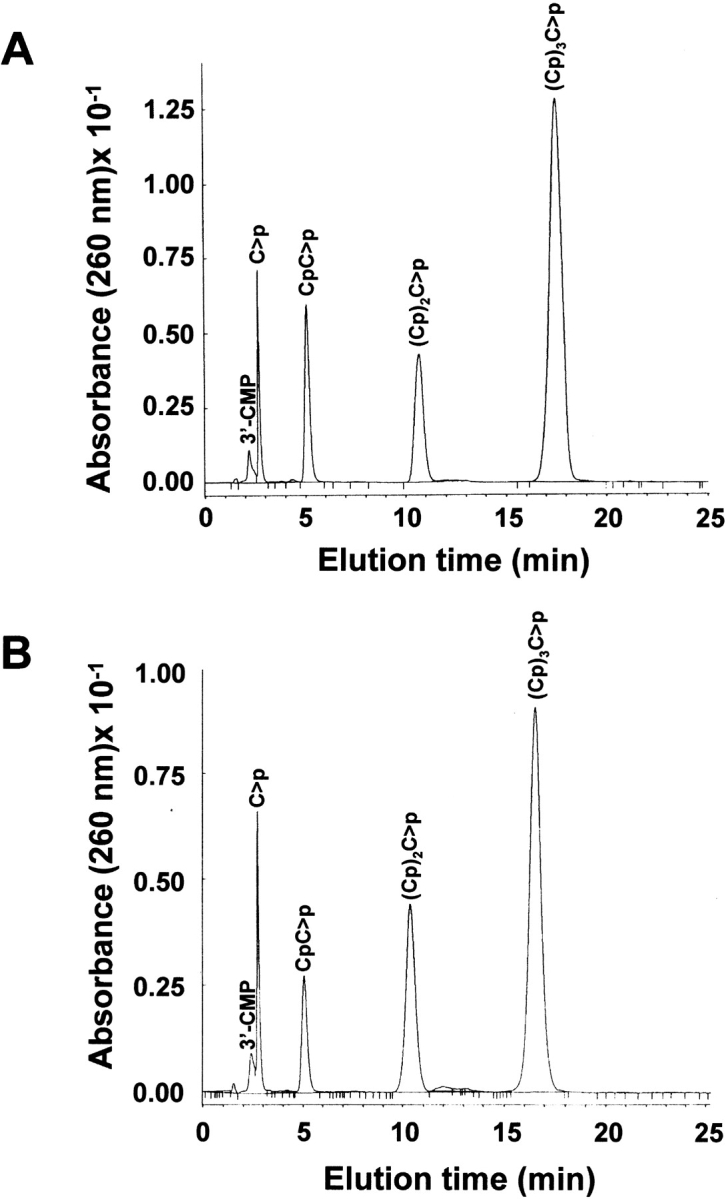
Oligocytidylic acids of the type (Cp)nC>p, in which C>p indicates a 2`,3`-cyclic-phosphate and n varies from 3 to 5 were prepared as described by Moussaoui et al. (1996) and Nogués and Cuchillo (2001). Figure 2 ▶ shows a typical chromatogram of the products of poly(C) breakdown by RNase A. Owing to the preparative nature of the run, the height of the peaks from the dinucleotide, CpC>p, to the hexanucleotide, (Cp)5C>p, all had the same maximum because of saturation of the detection device. The identity and homogeneity of the peaks was corroborated by means of MALDI-TOF MS (matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry). The technique confirmed the correctness of the previous assignments and the molecular mass obtained (Table 1) indicated that the terminal nucleotide had a 2`,3`-cyclic phosphate in all cases. Only in the case of the mononucleotide (the peak with a retention time ∼3 min) were no clear mass spectrometry results obtained.
Fig. 2.

Preparation of oligocytidylic acids. Elution profile on a reversed-phase HPLC column (Nova-Pak C18 column) of oligocytidylic acids (Cp)nC>p (n =0–7) obtained from a preparative poly(C) digestion. The elution conditions are described in Materials and Methods.
Table 1.
Molecular mass of oligocytidylic acids obtained from poly(C) cleavage by ribonuclease A and comparison with the theoretical values
| Theoretical value | Experimental valuea | |||
| CpCp | 628 | CpC>p | 610 | 610.9 |
| (Cp)2Cp | 933 | (Cp)2C>p | 915 | 915.6 |
| (Cp)3Cp | 1238 | (Cp)3C>p | 1220 | 1221.0 |
| (Cp)4Cp | 1543 | (Cp)4C>p | 1525 | 1527.1 |
| (Cp)5Cp | 1848 | (Cp)5C>p | 1830 | 1834.4 |
| (Cp)6Cp | 2153 | (Cp)6C>p | 2135 | 2135.6 |
| (Cp)7Cp | 2458 | (Cp)7C>p | 2440 | 2445.0 |
a The experimental values were obtained by MALDI-TOF MS from samples of the peaks obtained in experiments such as the one shown in Figure 2 ▶. See Materials and Methods for experimental details.
Analysis of the oligocytidylic acid cleavage pattern by RNase A and mutant enzymes
Figure 3 ▶ shows a typical chromatographic pattern for tetracytidylic acid digestion by native RNase A (Fig. 3A ▶) and the corresponding pattern obtained with the K7Q/R10Q-RNase A double mutant (Fig. 3B ▶) when ∼25% of the substrate had been consumed. The tetracytidylic acid is the smallest substrate that has phosphodiester bonds that can be submitted to either exo- or endonucleolytic activities (Table 2). From these chromatograms, it is readily apparent that the mutant acts on the substrate with a clearly different preference for the bond broken. Formation of trinucleotide with respect to dinucleotide predominates in the early stages of the action of the mutant, whereas the native enzyme produces all possible species more evenly. However, visual inspection of these chromatograms by itself can be deceptive as, for instance, two molecules of CpC>p are formed for each central bond broken instead of one, as is the case for (Cp)2C>p derived from the breaking of an external bond. On the other hand, the molar extinction coefficient of (Cp)2C>p is higher than that of CpC>p. These facts, together with the effect of a different diffusion according to the retention time, clearly influence the actual height and broadness of the peaks. Therefore, rigorous quantification is mandatory for meaningful results. The following results are from representative experiments; the reproducibility of the cleavage pattern is maintained and the variability between experiments is not >10%.
Fig. 3.
Separation by reversed-phase HPLC (Nova-Pak C18 column) of the products obtained from (Cp)3C>p digestion by RNase A (A) and K7Q/R10Q-RNase A (B). 100 μL of a (Cp)3C>p solution (A260 = 0.3) in 10 mM HEPES–KOH (pH 7.5) was digested with 10 μL of enzyme solution (0.1 nM in the case of RNase A and 1.17 μM in the case of the mutant) at 25°C for 15 min. 50 μL of the assay mixture was injected into the column. Elution was performed as described in Materials and Methods.
Table 2.
Possible distribution of products formed by the initial cleavage of oligocytidylic acidsa
| Substrate | Productsb | Type of cleavage |
| (Cp)3C>p | (Cp)2C>p + C>p | Exoc |
| 2 CpC>p | Endod | |
| (Cp)4C>p | (Cp)3C>p + C>p | Exo |
| (Cp)2C>p + CpC>p | Endo | |
| (Cp)5C>p | (Cp)4C>p + C>p | Exo |
| (Cp)3C>p + CpC>p | Endo | |
| 2 (Cp)2C>p | Endo |
a Characterization of the cleavage preference needs to be referred to the cleavage at zero time, as all products, except the mononucleotide, are also substrates for the enzyme.
b The products used to quantify the specificity of cleavage are indicated in bold.
c(Exo) Exonucleolytic cleavage pattern.
d (Endo) Endonucleolytic cleavage pattern.
Figure 4 ▶ shows the progress curves for the formation of di- and trinucleotide from the tetranucleotide by the native enzyme, the double mutant K7Q/R10Q-RNase A, and the single mutant K66Q-RNase A. The endonucleolytic or exonucleolytic cleavage preference was determined from the ratio of product formation at the beginning of the reaction (Table 2). For the tetranucleotide substrate, cleavage of the external bond adjacent to either the 5`- or 3`-terminal nucleotide (i.e., an exonucleolytic cleavage) gives rise to a trinucleotide plus a mononucleotide. Cleavage of the central bond (i.e., endonucleolytic cleavage) gives rise to only dinucleotides (two molecules per bond broken). For the pentanucleotide substrate, formation of tetranucleotide is the result of the exonucleolytic action, whereas formation of either tri- or dinucleotide represents endonucleolytic action. In the case of the hexanucleotide substrate, exonuclease action is represented by the formation of pentanucleotide, whereas tetra-, tri-, and dinucleotides derive from endonuclease action. Therefore, the ratio between these products will indicate the cleavage preference (see Discussion). However, this ratio varies with time as, with the exception of the mononucleotide, all the products formed are also substrates for the transphosphorylation reaction catalyzed by RNase A and the mutant enzymes. Thus, in the case of the tetranucleotide, formation of the dinucleotide at later stages of the reaction would not be the result of the endonucleolytic cleavage of the original tetranucleotide, but the result of exonucleolytic cleavage from the intermediate trinucleotide product. To know the real preference of the enzyme species on the intact substrate, an extrapolation to zero time of the ratios was performed graphically with the program GraFit v. 5 (Leatherbarrow 2001; Fig. 5 ▶). Figure 6 ▶ shows the results corresponding to all the substrates and enzyme species used with the value obtained by extrapolation to zero time.
Fig. 4.
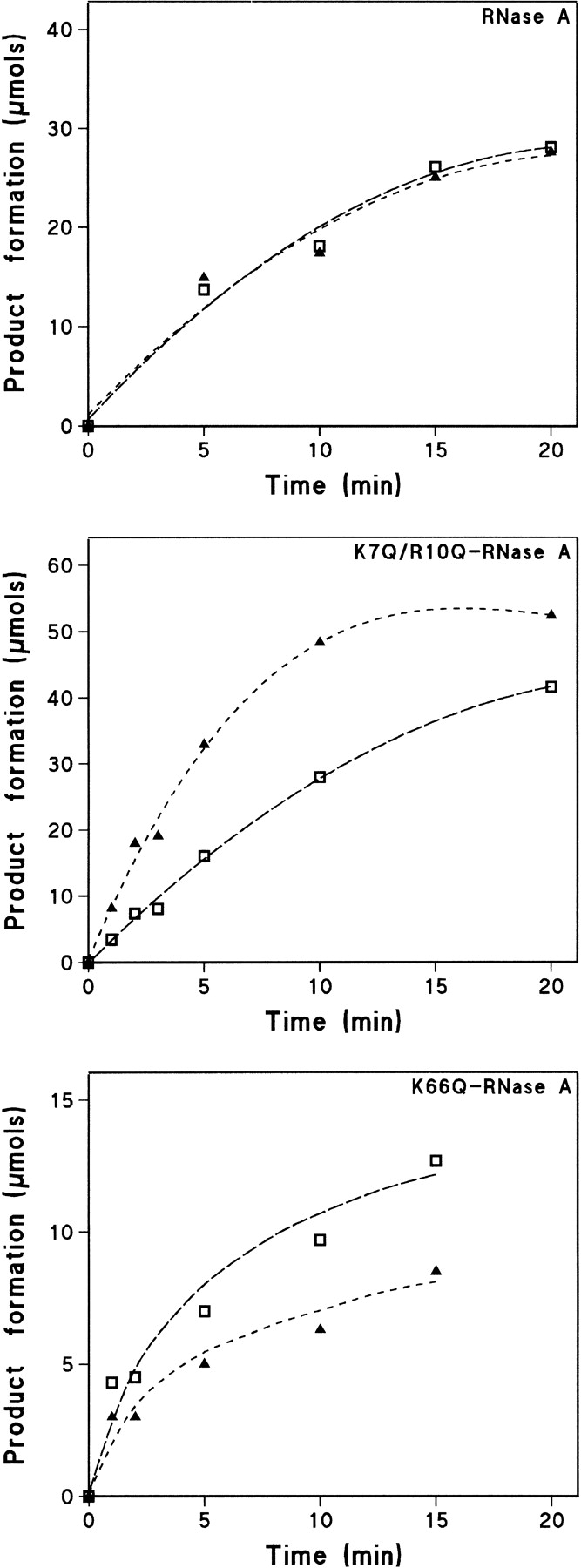
Progress curves of CpC>p (open squares) and (Cp)2C>p (filled triangles) produced from the cleavage of the oligocytidylic acid (Cp)3C>p substrate by RNase A, K7Q/R10Q-RNase A, and K66Q-RNas A. The experimental points were obtained by quantification, as described in Materials and Methods, of the peaks obtained in chromatograms of the type shown in Figure 3 ▶.
Fig. 5.
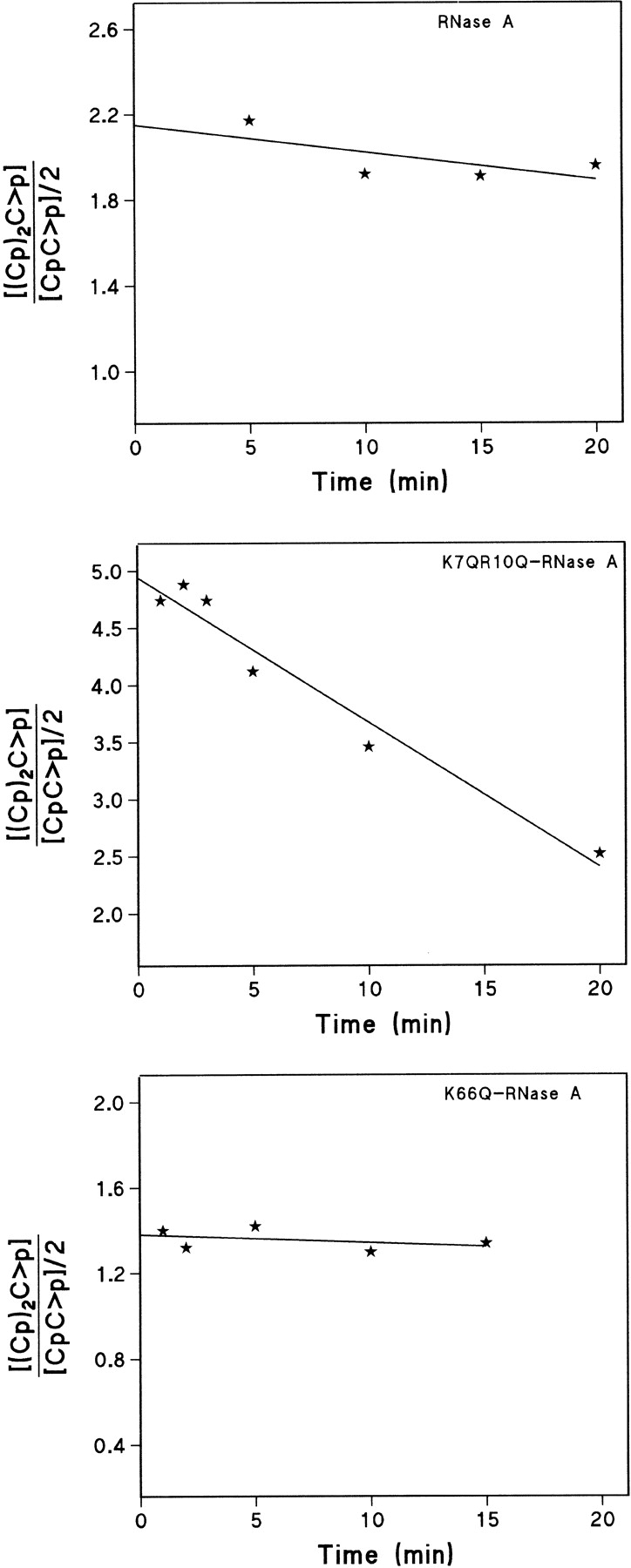
Trinucleotide/dinucleotide ratio for the cleavage of the oligocytidylic acid (Cp)3C>p substrate by RNase A, K7Q/R10Q-RNase A, and K66Q-RNase A. With this substrate, trinucleotide formation indicates exonucleolytic cleavage by the enzyme, whereas dinucleotide formation indicates endonucleolytic cleavage. The extrapolation of the ratios to zero time was determined with the program GraFit v. 5 (Leatherbarrow 2001) and indicates the preference of the enzymes on the intact substrate. The ratio decreases with time because the trinucleotide produced at the initial stages of the reaction is used later as a substrate by the enzyme producing more dinucleotide. The dinucleotide concentration has been divided by two because each endonucleolytically cleaved bond in the tetranucleotide yields two dinucleotide molecules, whereas each exonucleolytically broken bond only produces one trinucleotide molecule and one mononucleotide.
Fig. 6.
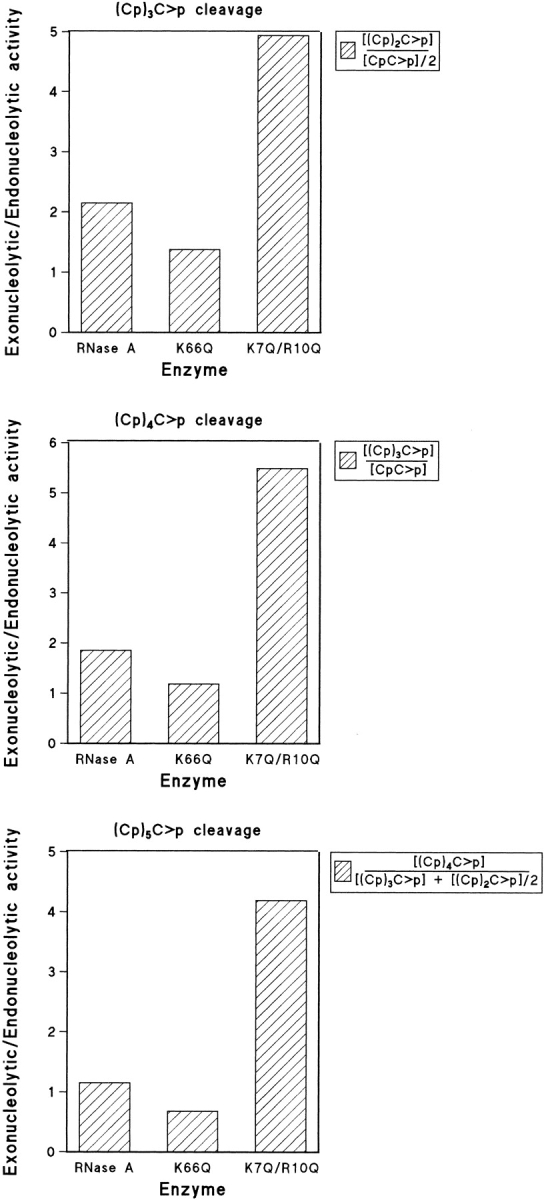
Exonucleolytic versus endonucleolytic activity of RNase A, K66Q-RNase A, and K7Q/R10Q-RNase A as determined from the ratios of the products at zero time. For the tetranucleotide substrate, the ratio of exonucleolytic to endonucleolytic cleavage was obtained as described in Figure 5 ▶. For the pentanucleotide substrate, the ratio was obtained by dividing the tetranucleotide formed by the dinucleotide formed, as each endonucleolytic cleavage gives rise to only one molecule of dinucleotide (the trinucleotide molecule could have been used as well). For the hexanucleotide substrate, endonucleolytic cleavage can result in either a tetranucleotide plus a dinucleotide or in two trinucleotides. Thus, the ratio was obtained by dividing the pentanucleotide formed (exonucleolytic cleavage) by the sum of tetranucleotide plus half of the trinucleotide formed.
Given that the mononucleotide elutes in a split peak (Figs. 2, 3 ▶ ▶) and the ambiguous identification by MALDI-TOF MS measurements, the quantification of the results was performed mainly with the oligonucleotide products. However, it was possible to check the accuracy of the results by adding up all the species, mononucleotide included (obtained in μM), according to the number of nucleotide residues using the formula
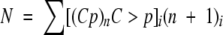 |
in which N is the total concentration of mononucleotide units (in μM) and i the different species (from 1 to n + 1). The same N value (±10%) was found at all digestion times with all the substrates used.
RNase A shows both endo- and exonucleolytic cleavage patterns (Fig. 6 ▶). Whether the exonucleolytic activity took place at the 5` end of the substrate or at the 3` end was determined by comparing the initial digestion patterns obtained with the oligonucleotides (Cp)4Cp and (Cp)4C>p (Table 3). The digestion of both substrates yielded an increase of the (Cp)3C>p product, no formation of (Cp)3Cp, and important differences in the profile of the 3`-CMP and C>p split peak. The formation of 3`-CMP and (Cp)3C>p from (Cp)4Cp is consistent with exonucleolytic cleavage by RNase A at the 3`-side of the substrate. The differences in the formation of (Cp)3C>p and C>p as it appears in Table 3 may be attributable to the fact that the (Cp)3C>p product is also an RNase A substrate contributing further to the formation of C>p. Preliminary studies of the cleavage of (Cp)4C>p by K7Q/R10Q-RNase A mutant indicate that the same cleavage pattern is maintained (data not shown).
Table 3.
Comparison of the terminal products obtained from (Cp)4Cp and (Cp)4C>p by RNase A at the initial steps of the reaction a
| Substrate | Relative product formationb | ||||
| (Cp)3C>p | (Cp)3Cp | 3`-CMP | C>p | 3`-CMP/C>p | |
| (Cp)4Cp | 0.45 | n.d.c | 0.41 | 0.08 | 5.12 |
| (Cp)4C>p | 0.5 | n.d. | 0.01 | 0.88 | 0.01 |
a Digestion products were determined when around 70% of the initial substrate remained undigested. Products were separated by reversed-phase HPLC. See Materials and Methods for digestion and separation conditions.
b Relative product formation was determined from the area percent of each chromatographic peak divided by the corresponding extinction coefficient at 260 nm (ɛ260): 7845 M−1cm−1 for C>p, 7145 M−1cm−1 for 3`-CMP and 24282 M−1cm−1 for (Cp)3C>p. Other products obtained from endonucleolytic activity are not indicated. The variability between experiments is not higher than 10%.
c (n.d.) Not detected.
Molecular modeling
Analysis of the molecular models obtained as described in Materials and Methods allowed us to measure the distance between the nearest oxygen on the phosphate group of the substrate and the tip of the amino acid in position 7. These calculations were performed for both the native enzyme and the K7Q-RNase A mutant (Fig. 7 ▶). The latter was chosen instead of the K7Q/R10Q-RNase A double mutant for simplicity of calculation. In addition, the tip of the residue in position 10 is too far from the phosphate and thus its influence on the results should be negligible. In both cases, the distance between the nearest oxygen of the 3`,5`-phosphodiester group and the residue at position 7 is longer than the corresponding value obtained for a terminal 2`,3`-cyclic phosphate (3.44 Å and 2.21 Å for the K7Q-RNase A mutant and 3.05 Å and 1.77 Å for native RNase A; Fig. 7 ▶). In the case of the p0 subsite, only the interaction between K66 or Q66 and the corresponding 3`,5`-phosphodiester group was analyzed because the location of the substrate's 2`,3`-cyclic phosphate group at this position results in nonproductive binding. As shown by X-ray crystallographic analysis, the p0 binding subsite is poorly defined because Lys 66 establishes a salt bridge with Glu 49 from a symmetry-related molecule. However, model building showed that by adopting a different conformation, Lys 66 could interact with the phosphate group adjacent, on the 5` side, to the active site, in which case the distance could be as short as 2.6 Å (Fontecilla-Camps et al. 1994). By molecular modeling, we have also estimated that, for the K66Q-RNase A mutant, the distance between the nearest oxygen of the 3`,5`-phosphodiester group and the residue at position 66 is 3.59 Å.
Fig. 7.
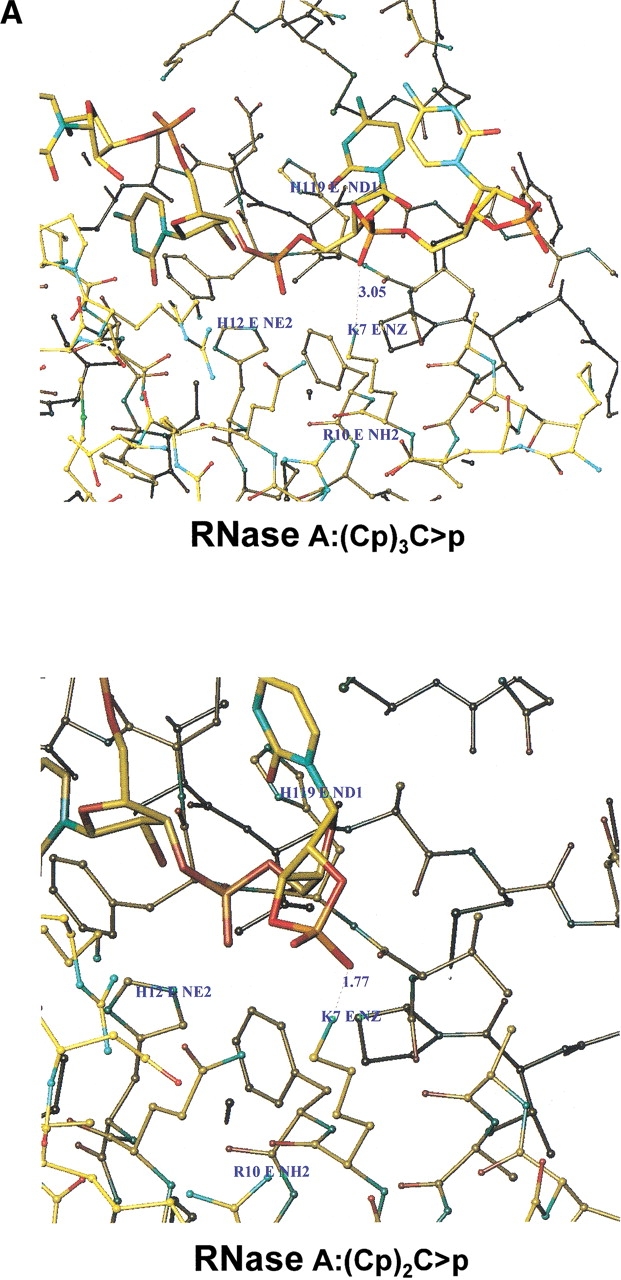
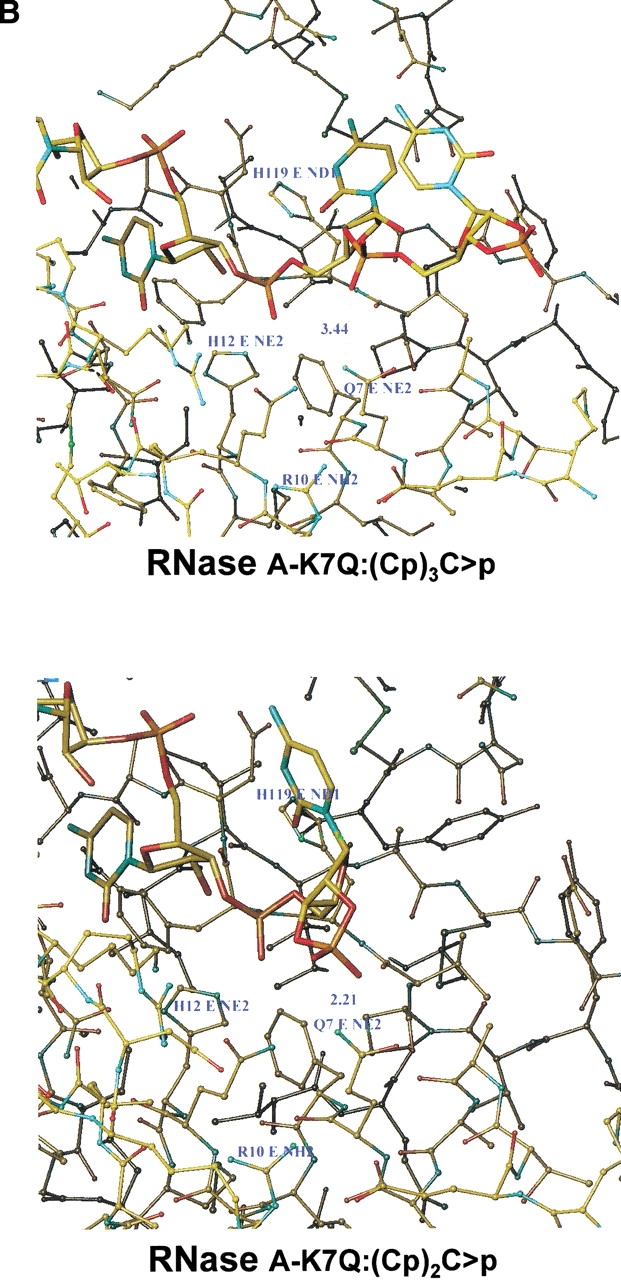
Molecular modeling of the interaction between oligocytidylic acids and native bovine pancreatic RNase A (A) or the K7Q-RNase A mutant (B). (Upper) The minimum distance between the tip of the amino acid in position 7 (Lys in RNase A, Gln in the mutant) and the nearest oxygen of the phosphate in the substrate involved in a 3`,5`-phosphodiester bond. (Lower) The corresponding distances between the tip of the amino acid in position 7 and the nearest oxygen of the terminal 2`,3`-cyclic phosphate of the substrate.
Discussion
Previous studies using oligocytidylic acids as substrates (Moussaoui et al. 1996, 1998) showed the important influence of substrate size on kinetic parameters. However, no attempt was made to determine the preferences for the bond cleaved. We thought that using these homo-oligonucleotides so that the only aspect that can influence the preference of the enzyme for a given bond is its distance to either end, the 5` end or the 3` end, and not the sequence could give some indication as to the role of nucleotide residues at both sides of the scissile bond. The same HPLC technique that had been used for the preparation of oligonucleotides can be applied successfully to the analysis of the products of the reaction (Fig. 2 ▶). However, the cyclizing nature of the enzyme action was the cause of an important ambiguity, namely the symmetrical nature of the products derived from both the 5` and the 3` end of the substrate (Fig. 8 ▶). Thus, for the oligocytidylic acids used as substrates in this study, the cleavage pattern for RNase for all possible bonds that can be broken is shown in Table 2. It is clear that, given the formation of a 2`,3`-cyclic phosphate on the 3` end of each product, as has been shown unambiguously by the MALDI-TOF MS data, there will be a symmetry such that the formation of a dinucleotide from the 5` end and a tetranucleotide from the 3` end (in the case of (Cp)5C>p) will be indistinguishable from the formation of a tetranucleotide from the 5` end plus a dinucleotide from the 3` end. This ambiguity, however, provides additional evidence for the cyclizing mechanism of action of RNase A. Based on studies with dinucleoside monophosphates, Cuchillo et al. (1993) and Raines and Thompson (1994) concluded that the so-called second, or hydrolysis, step in the reaction catalyzed by RNase A was actually an extreme case of the reverse of the transphosphorylation reaction in which the R-OH group (normally the 5` OH of a nucleoside or a polyribonucleotide molecule) was just H-OH. This was based on the fact that, when using dinucleoside monophosphates such as CpA or UpA, the formation of cytidine 3`-monophosphate or uridine 3`-monophosphate from the corresponding 2`,3`-cyclic monophosphates took place very slowly and only when practically all the original dinucleoside monophosphate substrate for the transphosphorylation step had been consumed. The MALDI-TOF MS analysis of the fractions obtained by RNase A digestion of poly(C) (Fig. 2 ▶; Table 1) demonstrated clearly that all the oligonucleotides formed had a molecular mass corresponding to the general formula (Cp)nC>p. No hydrolysis products with a non-cyclic 3`-terminal phosphate were found. The same result was seen with all products obtained from the oligonucleotide substrates. Thus, the claim that RNase A, and probably all other cyclizing RNases, should be reclassified as transferases instead of hydrolases gets substantial support (Cuchillo et al. 1993).
Fig. 8.

Pattern of products generated by the breaking of each of the phosphodiester bonds of (Cp)3C>p by RNase A. The exonucleolytic cleavage from either of the two outer bonds yields a mixture of C>p and (Cp)2C>p; thus, it is not possible to distinguish whether they arise from the breakage of one bond or the other. This ambiguity applies to all (Cp)nC>p used.
The results indicate that the native enzyme shows no clear preference for any bond, but the increase in the length of the substrate from the tetranucleotide to the hexanucleotide increases the preference for the endonucleolytic activity of RNase A, as seen in Figure 6 ▶ and Table 4. Theoretically speaking, as the length of the substrate increases, the number of possible endo products also increases in relation to the exo products. However, the results show that the relationship between both activities is not correlated directly with change in substrate size. This behavior is likely a consequence of a more satisfactory interaction owing to the binding to non-catalytic sites at either side of the scissile bond that gives rise to the formation of an optimized enzyme–substrate complex.
Table 4.
Effect of p0 (K66Q-RNase A mutant) and p2 (K7Q/R10Q-RNase A mutant) deletion on the exonucleolytic/endonucleolytic activity ratio of RNase
| Substrate | Enzyme | Exo/Endoa | Activity ratio respect to RNase Ab | Activity ratio (K7Q/R10Q)/K66Qc |
| (Cp)3C>p | RNase A | 2.15 ± 0.13 | — | — |
| K66Q-mutant | 1.38 ± 0.03 | 0.64 | — | |
| K7Q/R10Q-mutant | 4.94 ± 0.12 | 2.29 | 3.58 | |
| (Cp)4C>p | RNase A | 1.85 ± 0.11 | — | — |
| K66Q-mutant | 1.18 ± 0.05 | 0.64 | — | |
| K7Q/R10Q-mutant | 5.49 ± 0.4 | 2.96 | 4.64 | |
| (Cp)5C>p | RNase A | 1.15 ± 0.23 | — | — |
| K66Q-mutant | 0.68 ± 0.05 | 0.59 | — | |
| K7Q/R10Q-mutant | 4.19 ± 0.22 | 3.64 | 6.16 |
a Values taken from Figure 6 ▶.
b Activity ratio corresponds to the exonucleolytic/endonucleolytic activity values of the mutant related to those of the native enzyme.
c Activity ratio corresponds to the exonucleolytic/endonucleolytic activity values of the p2 mutant (K7Q/R10Q-RNase A) related to those of the p0 mutant (K66Q-RNase A).
These ratios change very markedly with the mutants, although the same pattern is observed for all three substrates. Mutant K66Q-RNase A shows a clear decrease in the ratio of external/internal bonds broken; this effect increases with the size of the substrate in parallel with the effect observed in the native enzyme (the substrate size does not modify the activity ratios of the K66Q-RNase A mutant with respect to the native enzyme; Table 4.). The double mutant K7Q/R10Q-RNase A shows a very clear increase in the ratio of external/internal bonds broken ratio, but no clear dependence on the substrate size is observed. The single mutant K7Q-RNase A (with a p2 site only partly deleted) gives intermediate values between those of the double mutant and the native enzyme (data not shown).
The effect of the deletion of specific phosphate-binding subsites on the distribution of the cleavage products is thus evident. The p0 binding subsite (absent in the K66Q-RNase A mutant) is important for exonucleolytic cleavage by RNase, as shown by the fact that its deletion favors the opposite effect; that is, it increases the endonucleolytic cleavage. On the contrary, the p2 binding subsite is important in favoring endonucleolytic cleavage; thus, its deletion (as in the double mutant K7Q/R10Q-RNase A) shifts the RNase action to a more exonucleolytic behavior. It has been shown that the p2 binding subsite is formed mainly by Lys 7, but in its absence Arg 10 can still contribute somehow to the binding of phosphate because of its positive charge (Boix et al. 1994; Fontecilla-Camps et al. 1994). Accordingly, the single mutant (K7Q-RNase A), in which p2 has been only partly deleted, shows intermediate behavior.
From the results shown in Figure 6 ▶ and Table 4, it is apparent that the cleavage pattern depends on the size of the substrate as the preference for exonucleolytic cleavage decreases with an increase in substrate size from the tetranucleotide to the hexanucleotide. On the other hand, the contribution of the two different subsites (p0 and p2) to the specific cleavage can be deduced from the ratios of exonucleolytic to endonucleolytic activities of K7Q/R10Q-RNase A and K66Q-RNase A mutants and comparison to those of the native enzyme. In all three substrates, the influence attributable to p2 in the strength of the interaction is higher than that of p0. This agrees with previous reports, in which the lack of p2 induces a more significant drop of activity than the lack of p0 (Nogués et al. 1998). Moreover, X-ray crystallographic studies show that whereas p2 is a very well-defined subsite with a clear interaction with the substrate, p0 is poorly defined (Fontecilla-Camps et al. 1994). No additional information was obtained from molecular modeling of this region.
There is, however, no obvious molecular explanation for this behavior. We thought that, at least in the case of p2, the ionic interaction between the negatively charged phosphate group of the substrate and the positive charge of Lys 7 would help to accommodate the substrate molecule. It is clear that the distance between the nearest oxygen of the phosphate, either in a 3`,5`-phosphodiester bond or in a 2`,3`-cyclic phosphate bond is close enough to Lys 7 for the ionic interaction to take place with approximate equal strength. Thus, for the tetranucleotide (Cp)3C>p substrate the phosphates could bind in i) p−1p0p1p2 giving rise to exonucleolytic cleavage, ii) p0p1p2p3 with endonucleolytic cleavage, or iii) p1p2p3p4 with exonucleolytic cleavage. As the main binding subsites are p1, p2 , and p0 (in order of strength of binding), whereas binding at p−1, p3, and p4 is rather weak, we hypothesize that the first two binding combinations are preferred. Nevertheless, the third combination would also be significant and, hence, RNase A shows an overall preference for exonucleolytic cleavage with the tetranucleotide substrate. In the case of larger oligonucleotides, the influence of weak binding subsites on both sides (upstream of p0 and downstream of p2) would tend to accommodate the substrate spanning the whole crevice (McPherson et al. 1986; De Llorens et al. 1989). Thus, the phosphodiester bond bound to p1 would be preferentially an inner bond. Therefore, predominantly endonucleolytic cleavage should be found.
To clarify the exonucleolytic cleavage position, we have analyzed the digestion pattern of the oligonucleotide (Cp)4Cp at the initial steps of the reaction (Table 3). We have selected this substrate because it does not contain an additional labeling group that might interfere with substrate binding and also can distinguish the 5`- and 3`-scission sides. The initial formation of C>p and (Cp)3Cp would indicate cleavage at the 5`-side, whereas the formation of 3`-CMP and (Cp)3C>p would be a consequence of cleavage at the 3`-side. From the results shown in Table 3, we conclude that the exonucleolytic scission takes place preferentially at the phosphodiester bond adjacent to the 3` side of the substrate and that the 3`-end phosphate group binds at the p2 subsite.
In the case of the p2 mutant, no ionic interaction is possible because the positive charges of both Lys 7 and Arg 10 have been removed. Instead, a hydrogen bond could be formed between the amide group of Gln 7 and an oxygen atom in the phosphate group of the substrate. Molecular modeling (Fig. 7 ▶) shows that formation of a hydrogen bond is possible only when there is a terminal (outer) 2`,3`-cyclic phosphate. In the case of a phosphate forming part of a 3`,5`-phosphodiester bond (inner bond), the allowed distance is too long for this hydrogen bond to be formed. This could explain the shift to a more exonucleolytic pattern when a functional p2 is absent. It should be noted, however, that in either case the binding to the mutant is much more inefficient than the binding to the native enzyme, as deduced from their respective kinetic parameters (Boix et al. 1994).
We can conclude that the exonucleolytic cleavage of the oligonucleotide substrates by RNase A takes place preferentially at the phosphodiester bond adjacent to the 3` side of the substrate and that deletion of either the p0 or the p2 binding sites drastically alters the distribution of the cleavage products. The p0 binding site contributes to the exonucleolytic preference of RNase A and the p2 binding subsite is involved directly in the endonucleolytic pattern shown by RNase A. This has been shown by the opposite effects on the product distribution pattern caused by deletion of either site.
Materials and methods
Bovine pancreatic ribonuclease A, poly(C), cytidine 2`,3`-cyclic phosphate, and HEPES were obtained from Sigma (St. Louis, MO). The oligonucleotide (Cp)4Cp was obtained from Genset Oligos (Paris, France). A reversed-phase Nova-Pack C18 column (4 μm, 3.9 × 150 mm; Waters, Milford, MA) was used for the HPLC separations. Acetonitrile (HPLC grade) was obtained from Carlo Erba (Milan, Italy). All other reagents were of analytical grade. Distilled water, treated with a MilliQ water purification system (Millipore Corp., Milford, MA), was used for the preparation of HPLC solutions. HPLC separations were performed with a Varian Star System with Star Chromatography Workstation v. 4.51 (Varian Associates Inc., Sunnyvale, CA).
Preparation of K7Q-, K7Q/R10Q- and K66Q-RNase A mutants
These mutants of RNase A were prepared according to the procedure of Boix et al. (1994).
Preparation of oligocytidylic acids ((Cp)nC>p) from poly(C) digestion
The oligocytidylic acids used as substrates were obtained by RNase A digestion of a poly(C) solution. In a typical experiment, 500 μL of a 10 mg/mL poly(C) solution in 10 mM HEPES–KOH (pH 7.5) was digested with 50 μL of 7μM RNase A at 25°C for 5 min. The reaction products were separated by reversed-phase HPLC (Moussaoui et al. 1996; Nogués and Cuchillo 2001) using a Nova Pak C18 column at a flow-rate of 1 mL/min. The pressure was maintained between 65 and140 atm. The column was washed for 20 min with MilliQ-water and equilibrated for 20 min with solvent A (10% (w/v) ammonium acetate and 1% (v/v) acetonitrile in water); 20 μL of the reaction mixture was injected into the column. The elution was performed with an initial 10 min wash and a 50 min linear gradient from 100% solvent A to 10% solvent A plus 90% solvent B (10% (w/v) ammonium acetate and 11% (v/v) acetonitrile in water). After each run, the system was washed for 5 min with water containing 1% acetonitrile, followed by a 10 min wash with 100% acetonitrile, and by a 5 min wash with water containing 1% acetonitrile. The column was then equilibrated with a 20 min wash with solvent A. Slight differences in the retention times of oligonucleotides can be produced depending on the equilibration time of the column. The intermediate washes with water are important to avoid contact between a highly concentrated saline solution with a concentrated organic solvent that can produce some precipitates that clog the column and increase the pressure of the system sharply. Products were monitored and quantified from the absorbance at 260 nm (Fig. 2 ▶). The elution positions of the oligonucleotides was deduced according to the method of McFarland and Borer (1979) and Moussaoui et al. (1996). Their identity was corroborated by MALDI-TOF MS analysis of the individual peaks. The fractions corresponding to the tetra-, penta-, and hexacytidylic acids from several chromatographic runs were pooled, freeze-dried, and kept at −20°C until use.
Analysis of products from the cleavage of oligocytidylic acids by RNase
Tetra-, penta-, and hexacytidylic acids were cleaved by either RNase A or specific mutants and the rate of formation of products at different digestion times was analyzed by means of reversed-phase HPLC. The general reaction conditions described for poly(C) digestion and separation of the reaction products were used. All assays were performed in 10 mM HEPES–KOH buffer (pH 7.5) at 25°C, the substrate solution used had an approximate absorbance (A260) of 0.3, and the enzyme concentration used depended both on the substrate and the enzyme species used. The enzyme concentration was selected in such a way that a progress curve covering the depletion of at least 25% of the initial substrate could be obtained. In general, the enzyme concentrations were extremely low (in the nanomolar range) and thus to avoid denaturation a more concentrated enzyme solution was prepared and the adequate dilution was made immediately before the assay. Given the time taken by each assay (∼2 h) and to avoid degradation owing either to contamination or to denaturing effects associated with continuous freezing and thawing, aliquots of the substrate stock solution were kept frozen in Eppendorf tubes and only thawed immediately before use.
The amount of each oligonucleotide product was calculated by first integrating the areas and dividing those values by the corresponding extinction coefficient at 260 nm (ɛ260): 7845 M−1cm−1 for C>p, 15175 M−1cm−1 for CpC>p, 20745 M−1cm−1 for (Cp)2C>p, 24282 M−1cm−1 for (Cp)3C>p, 28683 M−1cm−1 for (Cp)4C>p, 37711 M−1cm−1 for (Cp)5C>p, and 42428 M−1cm−1 for (Cp)6C>p. Previously, the number of integration counts per absorbance unit had been calculated with a standard nucleotide solution. The program GraFit v. 5 (Leatherbarrow 2001) was used to fit the plots.
To check the initial exonucleolytic cleavage position, either at the 3` or 5` end, the digestion patterns of the commercial oligonucleotide (Cp)4Cp by RNase A and by the K7Q/R10Q-RNase A mutant were analyzed. Minor contaminants of the commercial preparation were eliminated by reversed-phase HPLC according to the procedure used for the separation of oligocytidylic acids. General chromatographic conditions described for the separation of poly(C) and (Cp)nC>p digestion products were modified to avoid overlap between the substrate ((Cp)4Cp) and one of the products ((Cp)3C>p) and between 3`-CMP and C>p. The column was equilibrated for 20 min with solvent A' (10% (w/v) ammonium acetate and 0.3% (v/v) acetonitrile in water) and the elution was performed with an initial 10 min wash and a 20 min linear gradient from 100% solvent A` to 100% solvent B` (10% (w/v) ammonium acetate and 5% (v/v) acetonitrile in water).
Matrix assisted laser desorption ionization time of flying mass spectrometry (MALDI-TOF MS)
Mass determination by MALDI-TOF MS of oligocytidylic acids obtained from the HPLC separation of digestion products of poly(C) was performed with a Brucker Biflex mass spectrometer (Bremen, Germany) according to the protocol of Wang and Biemann (Wang and Biemannn 1994; Hahner et al. 1997). Oligonucleotides were freeze-dried and resuspended in deionized water. 1 μL of NH4+ cation exchange polymer beads (Dowex 50W-X8) was loaded to the target inert metal surface and excess solvent was removed. 1 μL of 0.3 M 3-hydroxypicolinic acid in 50% acetonitrile was added. Before solvent evaporation, 0.7 μL of 0.5–10 mM oligonucleotide was added. Ions were generated by irradiation with a 337 nm nitrogen laser with an acceleration ion voltage of 19 kV. All spectra were taken in the reflectron positive ion mode.
Molecular modeling
To clarify the effect of the p2 subsite on the cleavage pattern, we obtained a model of the enzyme–substrate interaction in this region. The model was obtained from the RNase A–d(ApTpApA) complex structure at 2.5 Å resolution (1RPG; Fontecilla-Camps et al. 1994). Purine and pyrimidine rings were removed and replaced by cytosine rings; one hydroxyl group was added to each deoxyribose at the 2` position. The rings of the bases were rotated to accommodate their position within the cavities of RNase A sites, avoiding van der Waals hindrances and yielding the optimal Coulombic energies. The molecular topology was defined for this system using the GROMOS 96 package and the mtb43b1 and ifp43b1 parameters (Scott et al. 1999). Bonds and angles were optimized using the SHAKE algorithm (Van Gunsteren and Berendsen 1977). The molecular topology of the previous system was modified manually to form the molecular topology of the cyclic nucleotide, the 2` hydroxyl group of the 3`-terminal cytidine was removed and the 2` methylene group was bonded to the closest oxygen of the 3`-phosphate group, thus cyclizing the last nucleotide. The phosphate group had to be rotated to approach the oxygen atom that had to bind the 2` carbon atom. Bonds and angles of the new tetranucleotide (Cp)3C>p were optimized by the SHAKE algorithm. The trinucleotide (Cp)2C>p was obtained by removing the 3`-terminal cytidine. The cyclic phosphate topology was also reproduced as for the previous tetranucleotide. In the case of the p0 binding site, only the interaction between Lys66 or Gln66 and the corresponding 3`,5`-phosphodiester group was analyzed.
Acknowledgments
We thank Dr. B. Oliva for his contribution on the molecular modeling of the interaction between oligocytidylic acids and ribonuclease A and Dr. F. Canals for his contribution to the MALDI-TOF MS analysis, both from the Servei de Proteòmica i de Bioinformàtica de l'Institut de Biotecnologia i Biomedicina, Vicent Villar Palasí, Universitat Autònoma de Barcelona. This work was supported by grants PB96-1172-C02-01 from the Dirección General de Enseñanza Superior of the Ministerio de Educación y Cultura, Spain; BMC2000-0138-C02-01 from the Dirección General de Investigación of the Ministerio de Ciencia y Tecnología, Spain; and SGR98-00065 from the Comissió Interdepartamental de Ciéncia i Tecnologia of the Generalitat de Catalunya. C.M.C. was a recipient of a Fellowship from the Ministerio de Educación y Cultura (Spain).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.13702.
References
- Boix, E., Nikolovski, Z., Moiseyev, G.P., Rosenberg, H.F., Cuchillo, C.M., and Nogués, M.V. 1999. Kinetic and product distribution analysis of human eosinophil cationic protein indicates a subsite arrangement that favors exonuclease-type activity. J. Biol. Chem. 274 15605–15614. [DOI] [PubMed] [Google Scholar]
- Boix, E., Nogués, M.V., Schein, C.H., Benner, S.A., and Cuchillo, C.M. 1994. Reverse transphosphorylation by ribonuclease A needs an intact p2-binding site. Point mutations at Lys-7 and Arg-10 alter the catalytic properties of the enzyme. J. Biol. Chem. 269 2529–2534. [PubMed] [Google Scholar]
- Cuchillo, C.M., Parés, X., Guasch, A., Barman, T., Travers, F., and Nogués M.V. 1993. The role of 2`,3`-cyclic phosphodiester in the bovine pancreatic ribonuclease A catalysed cleavage of RNA: Intermediates or products? FEBS Lett. 333 207–210. [DOI] [PubMed] [Google Scholar]
- D'Alessio, G. and Riordan, J.F. 1997. Ribonucleases. structures and functions. Academic Press, New York.
- De Llorens, R., Arús, C., Parés, X., and Cuchillo, C.M. 1989. Chemical and computer graphics studies on the topography of the ribonuclease A active site cleft. A model of enzyme-pentanucleotide substrate complex. Protein Eng. 2 417–429. [DOI] [PubMed] [Google Scholar]
- Fisher, B.M., Grilley, J.E., and Raines, R.T. 1998. A new remote subsite in ribonuclease A. J. Biol. Chem. 273 34134–34138. [DOI] [PubMed] [Google Scholar]
- Fisher, B.M., Ha, J-H., and Raines, R.T. 1998. Coulombic forces in protein–RNA interactions: Binding and cleavage by ribonuclease A and variants at Lys 7, Arg 10, and Lys 66. Biochemistry 37 12121–12132. [DOI] [PubMed] [Google Scholar]
- Fontecilla-Camps, J.C., de Llorens, R., le Du, M.H., and Cuchillo, C.M. 1994. Crystal structure of ribonuclease Ad(ApTpApApG) complex. Direct evidence for extended substrate recognition. J. Biol. Chem. 269 21526–21531. [DOI] [PubMed] [Google Scholar]
- Hahner, S., Lüdemann, H.C., Kirpekar, F., Nordhoff, E., Roepstorff, P., Galla, H.J., and Hillenkamp, F. 1997. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI) of endonuclease digests of RNA. Nucleic Acids Res. 25 1957–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie, M., Mikami, F., Monma, K., Ohgi, K., Watanabe, H., Yamaguchi, R., and Nagase, H. 1984. Kinetic studies on the cleavage of oligouridylic acids and poly(U) by bovine pancreatic ribonuclease A. J. Biochem. (Tokyo) 96 89–96. [DOI] [PubMed] [Google Scholar]
- Leatherbarrow R.J. 2001. GraFit Version 5. Erithacus Software Ltd., Hortley, U.K.
- McFarland, G.D. and Borer P. 1979. Separation of oligo-RNA by reverse-phase HPLC. Nucleic Acids Res. 7 1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson, A., Brayer, G.D., Cascio, D., and Williams, R. 1986. The mechanism of binding of a polynucleotide chain to pancreatic ribonuclease. Science 232 765–768. [DOI] [PubMed] [Google Scholar]
- Moussaoui, M., Guasch, A., Boix, E., Cuchillo, C.M., and Nogués, M.V. 1996. The role of non-catalytic binding subsites in the endonuclease activity of bovine pancreatic ribonuclease A. J. Biol. Chem. 271 4687–4692. [DOI] [PubMed] [Google Scholar]
- Moussaoui, M., Nogués, M.V., Guasch, A., Barman, T., Travers, T., and Cuchillo, C.M. 1998. The subsite structure of bovine pancreatic ribonuclease A accounts for the abnormal kinetic behavior with cytidine 2`,3`-cyclic phosphate. J. Biol. Chem. 273 25565–25572. [DOI] [PubMed] [Google Scholar]
- Nogués, M.V., Vilanova, M., and Cuchillo, C.M. 1995. Bovine pancreatic ribonuclease A as a model of an enzyme with multiple substrate binding sites. Biochim. Biophys. Acta 1253 16–24. [DOI] [PubMed] [Google Scholar]
- Nogués, M.V., Moussaoui, M., Boix, E., Vilanova, M., Ribó, M., and Cuchillo, C.M. 1998. The contribution of noncatalytic phosphate-binding subsites to the mechanism of bovine pancreatic ribonuclease A. Cell Mol. Life Sci. 54 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogués, M.V. and Cuchillo, C.M. 2001. Analysis by HPLC of distributive activities and the synthetic (back) reaction of pancreatic-type ribonucleases. In Methods in molecular biology: Nuclease methods and protocols (ed. C.H. Schein), Vol. 160, pp. 15–24. Humana Press Inc., Totowa, NJ. [DOI] [PubMed]
- Parés, X., Llorens, R., Arús, C., and Cuchillo, C.M. 1980. The reaction of bovine pancreatic ribonuclease A with 6-chloropurine riboside 5`-monophosphate. Evidence on the existence of a phosphate-binding sub-site. Eur. J. Biochem. 105 571–579. [DOI] [PubMed] [Google Scholar]
- Parés, X., Nogués, M.V., de Llorens, R., and Cuchillo, C.M. 1991. Structure and function of ribonuclease A binding subsites. In Essays in biochemistry (ed. K.F. Tipton), Vol. 26, pp. 89–103. Portland Press Ltd., London. [PubMed]
- Raines, R.T. 1998. Ribonuclease A. Chem. Rev. 98 1045–1065. [DOI] [PubMed] [Google Scholar]
- Scott, W.R.P., Hunenberguer, P.H., Tironi, I.G., Mark, A.E., Billeter, S.R., Fennen, J., Torda, A.E., Huber, T., Kruger, P., and van Gunsteren, W.F. (1999) The Gromos biomolecular simulation program package. J. Phys. Chem. 103 3596–3607. [Google Scholar]
- Thompson, J.E., Venegas, F.D., and Raines, R.T. 1994. Energetics of catalysis by ribonucleases: Fate of the 2`,3`-cyclic phosphodiester intermediate. Biochemistry 33 7408–7414. [DOI] [PubMed] [Google Scholar]
- Van Gunsteren, W.F. and Berendsen, H.J.C. 1977. Algorithms for macromolecular dynamics and constraint dynamics. Mol. Phys. 34 1311–1327. [Google Scholar]
- Wang, B.H. and Biemannn, K. 1994. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of chemically modified oligonucleotides. Anal. Chem. 66 1918–1924. [DOI] [PubMed] [Google Scholar]
- Witzel, H. and Barnard, E.A. 1962. Mechanism and binding sites in the ribonuclease reaction. II. Kinetic studies on the first step of the reaction. Biochem. Biophys. Res. Comm. 7 295–299. [DOI] [PubMed] [Google Scholar]