

Abstract
We examined the mechanisms by which two different types of photonic radiation, short wavelength UV (UV-C) and γ radiation, activate transcription factor NF-κB. Exposure of mammalian cells to either form of radiation resulted in induction with similar kinetics of NF-κB DNA binding activity, nuclear translocation of its p65(RelA) subunit, and degradation of the major NF-κB inhibitor IκBα. In both cases, induction of NF-κB activity was attenuated by proteasome inhibitors and a mutation in ubiquitin-activating enzyme, suggesting that both UV-C and γ radiation induce degradation of IκBs by means of the ubiquitin/proteasome pathway. However, although the induction of IκBα degradation by γ rays was dependent on its phosphorylation at Ser-32 and Ser-36, UV-C-induced IκBα degradation was not dependent on phosphorylation of these residues. Even the “super repressor” IκBα mutant, which contains alanines at positions 32 and 36, was still susceptible to UV-C-induced degradation. Correspondingly, we found that γ radiation led to activation of IKK, the protein kinase that phosphorylates IκBα at Ser-32 and Ser-36, whereas UV-C radiation did not. Furthermore, expression of a catalytically inactive IKKβ mutant prevented NF-κB activation by γ radiation, but not by UV-C. These results indicate that γ radiation and UV-C activate NF-κB through two distinct mechanisms.
Exposure of cells to different forms of radiation and other genotoxic stresses stimulates signaling pathways that activate transcription factors including AP-1, NF-κB, and p53 (1–4). These transcription factors elicit various biological responses through induction of target genes. For instance, p53 activation leads to induction of p21, an inhibitor of cyclin-dependent kinases, resulting in arrest at the G1 phase of the cell cycle (5–7). This cell cycle arrest is thought to provide affected cells with ample time to repair their damaged DNA before entering S phase (8). Although the role of AP-1 activation is somewhat contentious and needs to be investigated further, it appears that induction of c-Fos (9) and c-Jun (E. Shaulian and M.K., unpublished work) help cells exit the G1 checkpoint imposed by p53 and p21. Induction of NF-κB activity, on the other hand, appears to play an important antiapoptotic function (10–14).
The mechanism by which exposure to short wavelength UV radiation (UV-C and UV-B) results in activation of AP-1 has been investigated in detail. Exposure to UV-C, for instance, results in rapid c-fos and c-jun gene induction (15, 16) and phosphorylation of c-Jun at two N-terminal sites that potentiate its ability to activate transcription (17). These observations led to the identification of the c-Jun N-terminal kinases (JNKs), whose activity is rapidly stimulated by UV-C or UV-B exposure (18, 19). In addition to the JNKs, UV exposure also results in potent activation of the related p38 mitogen-activated protein kinases (MAPKs) and less efficient activation of the extracellular signal-regulated kinases (ERKs) (20–23). All of these protein kinases participate in c-jun (17, 18) and c-fos (20, 21, 23) induction through phosphorylation of distinct substrates (24).
JNK activation by UV does not require damage to nuclear DNA because it can be elicited in nucleus-free cytoplasts (25). Indeed, the earliest events elicited by UV exposure that can lead to MAPK activation include activation of the epidermal growth factor receptor and several other cell surface receptors, including interleukin 1 (IL-1) and tumor necrosis factor (TNF) receptors (26, 27). Two mechanisms were suggested to underlie UV-induced receptor activation, receptor clustering (27) and inhibition of receptor-inactivating phosphatases (22).
UV-C or UV-B also induce NF-κB activity (25, 28, 29). Like AP-1, induction of NF-κB does not require damage to nuclear DNA (25, 28). However, unlike AP-1, little is known regarding the mechanism by which UV exposure results in NF-κB activation. NF-κB is a dimeric transcription factor composed of members of the Rel family that is kept in the cytoplasm of nonstimulated cells through interaction with inhibitory proteins, the IκBs (30, 31). The IκBs retain NF-κB in the cytoplasm by masking the nuclear localization sequence embedded within the Rel homology domain (32). The most potent NF-κB activators are the proinflammatory cytokines IL-1 and TNF (33, 34), which cause rapid phosphorylation of IκBs at two sites within their N-terminal regulatory domain (35–38). This phosphorylation event, which in the case of IκBα occurs on Ser-32 and Ser-36, results in polyubiquitination of the IκBs and their degradation by the 26S proteasome (37, 39–43). This results in liberation of NF-κB, its nuclear translocation and activation of target genes (30, 31), which include those coding for inflammatory mediators and immunoregulatory molecules (33, 34). Recently, a protein kinase complex whose activity is stimulated by TNF or IL-1, which mediates IκB phosphorylation, was purified (44). Two of the subunits of this complex, named IKKα and IKKβ (IκB kinase α and β subunits), were molecularly cloned and found to contain an N-terminal protein kinase domain and a C-terminal regulatory domain with several protein interaction motifs (44–46). IKKα and IKKβ were also identified through a two hybrid screen as proteins that interact with the NF-κB-activating kinase NIK (47, 48). The IKK complex phosphorylates IκBα and IκBβ at each of the two N-terminal sites that trigger their ubiquitination and degradation. Expression of catalytically inactive IKKα or IKKβ prevents NF-κB activation (45–47).
In addition to short wavelength UV radiation, NF-κB activity is also induced by exposure to even shorter wavelength photons, γ rays or ionizing radiation (IR) (49, 50). We compared the mechanism by which UV-C and IR activate NF-κB. Although both radiations induce IκB degradation they operate through two distinct mechanisms. Whereas IR exposure results in IKK activation and IR-induced IκB degradation is phosphorylation dependent, exposure to UV-C does not result in IKK activation, and UV-induced IκB degradation occurs independently of its N-terminal serine phosphorylation.
MATERIALS AND METHODS
Reagents and Antibodies.
Proteasome inhibitors AcLLnL (N-Ac-Leu-Leu-norleucinal) (51) and clasto-lactacystin β-lactone (52, 53) were from Calbiochem, and Z-L3vs (Cbz-Leu-Leu-Leu-vinyl sulfone) (54) was kindly provided by Wei Jiang (The Salk Institute). They were dissolved in dimethyl sulfoxide. The peak emission wavelength of the UV-C lamp (American Ultraviolet Company, Lebanon, IN) used was 254 nm, and γ radiation was delivered by using a JL Shepherd Mark I Model 30 Irradiator with a 137Cs source. Anti-IκBα is a rabbit polyclonal antibody (41). Anti-IKKα is a monoclonal antibody raised against full-length IKKα (PharMingen). Anti-p65 (SC-372) was from Santa Cruz Biotechnology.
Cell Culture and Transfection.
HeLa and 293 cells were grown in DMEM with 10% fetal bovine serum (FBS) and transfected by using lipofectamine (GIBCO/BRL) as described (46). Pools of cells that stably express hemagglutinin (HA)-tagged IκBα or IκBα AA were obtained by selection in medium containing 1 mg/ml G418 for 2 weeks. The Chinese hamster cell line ts20 (55) was grown at 30°C in RPMI 1640 medium supplemented with 10% FBS.
Cell Extracts and Electrophoretic Mobility Shift Assay (EMSA).
Cell extracts were prepared as described (25, 56). Cells were harvested in hypotonic buffer and pelleted by centrifugation. The pellets were suspended in nuclear extract buffer. After 15 min on ice the suspensions were centrifuged and the supernatants were transferred to new tubes. For whole cell extracts, cells were lysed in cell lysis buffer containing proteinase and phosphatase inhibitors and after centrifugation the supernatants were used. EMSA was performed as described (25, 41, 56). Briefly, 5 μg of nuclear protein or 10 μg of whole cell extract was incubated with 2 μg of poly(dI-dC) and 5,000-10,000 cpm of labeled oligonucleotide probes (57). After 30 min at room temperature, the samples were analyzed on native 5% polyacrylamide gels.
Immunoprecipitation, Kinase Assay, Immunoblotting, and Immunofluorescence.
Cell lysates were incubated with anti-IKKα for 1 hr at 4°C, protein G-Sepharose beads were added, and the incubation continued for 1 hr. The beads were collected and washed and IKK activity was assayed at 30°C for 40 min in kinase buffer containing [γ-32P]ATP and 2 μg of glutathione S-transferase IκBα(1–54) as described (44). Immunoblotting was done as described (44, 46, 56). Indirect immunofluorescence was performed as described (46, 58).
RESULTS
UV and IR Induce NF-κB DNA Binding Activity and Nuclear Translocation.
To study NF-κB activation by UV-C and IR, we examined induction of NF-κB DNA binding activity by EMSA. Exposure of HeLa cells to UV-C resulted in dose-dependent induction of NF-κB binding activity that was maximal in cells receiving 20 J/m2 UV-C (Fig. 1A). Time course analysis revealed that NF-κB binding activity was detectable 1.5 hr after exposure to 40 J/m2 UV-C and increased until it reached a maximum 5–8 hr postirradiation. The UV-induced protein–DNA complex was competed by an excess of unlabeled κB oligonucleotide, but not by an unlabeled AP-1 oligonucleotide. Induction of NF-κB binding activity by UV-C was weaker and slower when compared with the response elicited by TNF (Fig. 1A). Exposure of HeLa cells to γ radiation also resulted in the induction of NF-κB binding activity (Fig. 1B). Maximal induction was produced by exposure to 20 Gy of IR and NF-κB binding activity was first detected 1 hr postradiation. Maximal induction was observed 2–3 hr after radiation exposure. The DNA–protein complex induced by UV-C and IR contained p65 (RelA) because it was “supershifted” by an anti-p65 antibody (data not shown). To confirm further that exposure to UV-C and IR activates NF-κB, we examined the nuclear translocation of p65 by indirect immunofluorescence. Similar to TNF treatment, exposure to both UV-C and IR enhanced nuclear staining of HeLa cells by anti-p65 (Fig. 1C). Although not visible in these photographs produced by an automatic exposure camera, the nuclear signals in cells exposed to UV-C or IR were weaker than those in cells treated with TNF, consistent with the relative capacities of these treatments to induce NF-κB DNA binding activity.
Figure 1.
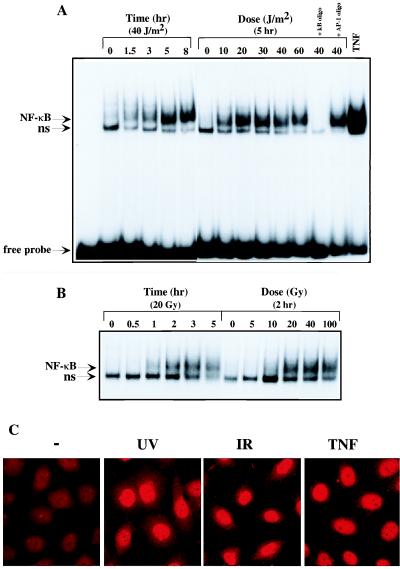
UV-C and IR activate NF-κB. Serum-starved HeLa cells were exposed to the indicated doses of UV-C (A) or IR (B). At the indicated times nuclear extracts were prepared and NF-κB DNA binding activity determined by EMSA. Specificity of binding was determined by competition with 100-fold excess unlabeled κB oligonucleotide or an AP-1 site oligonucleotide. As a positive control, EMSA was also performed with the nuclear extract of cells exposed to TNF (10 ng/ml) for 30 min. Migration positions of the NF-κB and nonspecific (ns) protein–DNA complexes and free probe are indicated. (C) UV-C and IR induce NF-κB nuclear translocation. HeLa cells grown on coverslips were untreated (−) or exposed to UV-C (40 J/m2), IR (20 Gy), or TNF (10 ng/ml) and fixed after 5, 3, or 0.5 hr, respectively. Cells were then incubated with anti-p65(RelA), stained with rhodamine-conjugated goat anti-rabbit IgG, and photographed through an epifluorescence-equipped microscope with a 63× oil objective and an automatic camera. Exposure times were considerably longer for the UV and IR irradiated samples to obtain exposure similar to the TNF treated samples.
The slow kinetics of the responses to UV-C and IR suggested that they may be mediated through induction of an intermediary protein. However, preincubation of HeLa cells with the protein synthesis inhibitor cycloheximide did not prevent or reduce the extent of NF-κB activation (data not shown). In addition, the induction of NF-κB by either UV-C or IR is unlikely to be mediated by secretion of TNF or activation of TNF receptor because it was not prevented by preincubation with neutralizing anti-TNF or anti-TNF receptor antibodies, which did inhibit the response to TNF (data not shown).
UV-C and IR Induce IκBα Degradation.
Activation of NF-κB by TNF or IL-1 is achieved through phosphorylation-dependent degradation of the IκBs, which results in the release and subsequent nuclear translocation of NF-κB (31). To understand how UV-C or IR activate NF-κB, we performed immunoblotting experiments with IκBα antibodies to examine whether these agents cause degradation of this most well characterized and abundant IκB protein. Although IκBα was almost completely degraded 20 min after TNF treatment, exposure to either UV-C or IR caused only partial and slow IκBα degradation (Fig. 2A). The kinetics of IκBα degradation correlated well with the kinetics of NF-κB activation by these stimuli. A retardation in the electrophoretic mobility of IκBα, which corresponds to its phosphorylation at Ser-32 and Ser-36, was easily observed in samples isolated from TNF-treated cells but not after exposure to UV-C or IR.
Figure 2.

UV-C and IR induce IκBα degradation. (A) HeLa cells were exposed to UV-C (40 J/m2), IR (20 Gy), or TNF (10 ng/ml) and whole cell extracts were prepared at the indicated times. Proteins (10 μg) were separated by electrophoresis on a 12.5% SDS/polyacrylamide gel and immunoblotted with anti-IκBα. Lane C, untreated cells. Positions of IκBα and phosphorylated IκBα (p-IκBα) are indicated. (B and C) Proteasome inhibitors block UV- and IR-induced NF-κB activation and IκBα degradation. HeLa cells were either untreated (control) or incubated with 100 μM AcLLnL, 50 μM Z-L3vs, or 10 μM clasto-lactacystin β-lactone (Lactacystin) for 1 hr before exposure to UV-C (40 J/m2), IR (20 Gy), or TNF (10 ng/ml). Cell extracts were prepared after 4 hr, 2 hr, and 15 min, respectively. NF-κB DNA binding activity was measured by EMSA (B) and IκBα degradation examined by immunoblotting with anti-IκBα (C). (D) Ubiquitination is required for NF-κB activation. ts20 cells were cultured at either the permissive (30°C) or restrictive (40°C) temperature for 6 hr to allow inactivation of the temperature-sensitive E1. Cells were then treated with TNF for 15 or 30 min or exposed to UV. Cell extracts were prepared and NF-κB (Upper) and NF-1 (Lower) DNA binding activities were measured by EMSA.
If the primary mechanism responsible for NF-κB activation depends on IκB degradation, inhibition of this process should block NF-κB activation. Indeed, induction of NF-κB binding activity was blocked when HeLa cells were treated with the proteasome inhibitors AcLLnL, Z-L3vs, and clasto-lactacystin β-lactone, which are known to inhibit NF-κB activation (41, 59, 60), before their exposure to UV-C, IR, or TNF (Fig. 2B). Immunoblotting analysis showed that these inhibitors blocked IκBα degradation induced by UV, IR, and TNF (Fig. 2C). Because the use of these inhibitors allows the accumulation of phosphorylated IκBα (37, 38), in their presence there was a large increase in the level of the slower migrating form of IκBα after TNF treatment, and that form was also detected after IR exposure (Fig. 2C). Nevertheless, UV-C irradiation did not cause a retardation of IκBα electrophoretic mobility even in the presence of proteasome inhibitors. If ubiquitination of IκBα is required for its degradation, inhibition of this modification should block NF-κB activation. To test this point, we examined NF-κB activation in the ts20 cell line, which has a temperature-sensitive ubiquitin-activating enzyme E1 (55). At the restrictive temperature, NF-κB activation induced by TNF or UV was abolished, whereas NF-1 DNA binding activity was not affected (Fig. 2D).
Ser-32 and Ser-36 Phosphorylation Is Required for IκBα Degradation Induced by IR but Not by UV-C.
Cytokine-inducible phosphorylation of IκBα at Ser-32 and Ser-36 is necessary for its degradation and subsequent NF-κB activation (35, 37, 38). To assess whether IκBα phosphorylation at these sites is also required for degradation induced by UV-C or IR, an IκBα mutant (IκBα AA) in which Ser-32 and Ser-36 were replaced by alanines was stably expressed as an HA-tagged protein in HeLa and 293 cells and its degradation was examined by immunoblotting. Similar experiments also were performed in cells that stably expressed HA-tagged wild-type IκBα. To visualize the HA-tagged proteins in HeLa cells the film was overexposed because their expression levels were lower than that of endogenous IκBα. The HA-tagged wild-type IκBα was degraded in response to UV-C, IR, or TNF with kinetics similar to those of endogenous IκBα (Fig. 3). Surprisingly, the IκBα AA mutant underwent degradation with the same kinetics as the wild-type protein after exposure to UV-C (Fig. 3A). However, both in HeLa and 293 cells (Fig. 3B and data not shown), this mutant was refractory to IR and TNF, which still induced the degradation of endogenous IκBα. Because 293 cells do not respond well to UV-C irradiation (unpublished results), we could not measure the effect of UV on IκBα degradation in these cells. These data strongly suggest that, although phosphorylation at Ser-32 and Ser-36 is required for IκBα degradation after exposure to IR or TNF, it is not a prerequisite for UV-C-induced degradation.
Figure 3.

Ser-32 and Ser-36 are required for IκBα degradation in response to IR, but not by UV-C. Pools of stably transfected HeLa cells expressing HA-tagged wild-type IκBα or the IκBα AA mutant were either untreated or exposed to UV-C (40 J/m2) (A), IR (20 Gy) (B), and TNF (10 ng/ml). At the indicated times, lysates were prepared, separated by SDS/PAGE, and immunoblotted with anti-IκBα. Lanes Un and C, untransfected and untreated transfected cells, respectively. The migration positions of endogenous IκBα and the transfected HA-IκBα are indicated. To visualize HA-tagged IκBα the film was overexposed.
IKK Is Activated in Response to IR but Not UV Irradiation.
Next we examined the effect of UV-C and IR on the activity of IKK, the TNF and IL-1 responsive protein kinase (44–48) that phosphorylates IκBα at Ser-32 and Ser-36. Proteins isolated at various intervals after exposure of HeLa cells to either UV-C or IR were immunoprecipitated with an IKKα antibody and then subjected to an immunecomplex kinase assay by using glutathione S-transferase–IκBα(1–54) as a substrate. Although this antibody is directed to IKKα, it precipitates the entire IKK complex (61). While TNF rapidly stimulated IKK activity by approximately 10-fold, IKK activity was slowly stimulated by exposure to IR, reaching a maximum of a 2.8-fold increase after 90 min (Fig. 4). Whereas the response to TNF is first detectable at 5 min (44, 46), the response to IR was first detected after 30–45 min. By contrast, exposure to UV-C had no effect on IKK activity (Fig. 4).
Figure 4.

IKK is activated by IR but not by UV-C irradiation. HeLa cells were either untreated (lane C) or exposed to UV-C (40 J/m2), IR (20 Gy), or TNF (10 ng/ml). At the indicated times after treatment (10 min for TNF), whole cell lysates were prepared. IKK activity was measured by the immunecomplex kinase assay (Upper). The amount of IKKα was determined by immunoblotting (Lower).
IKK Is Necessary for NF-κB Nuclear Translocation and IκBα Degradation Induced by IR but Not by UV-C.
The results described above suggest that IKK is required for NF-κB activation in response to IR but not UV-C. To confirm this point a catalytically inactive IKKβ mutant, HA-IKKβ (KA), in which the lysine residue that is crucial for ATP binding is replaced by an alanine, was employed. Expression of this mutant blocks NF-κB activation by TNF or IL-1 (46). HeLa cells grown on coverslips were transfected with an expression vector encoding HA-IKKβ (KA) and 36 hr later were exposed to UV-C, IR, or TNF. Cells were fixed and the subcellular distribution of p65 was examined by indirect immunofluorescence. Double staining with anti-HA and anti-p65 was used to identify transfected cells. Because staining with anti-HA produced a weak background nuclear fluorescence, only cells that exhibited strong cytoplasmic staining with anti-HA were considered as transfected. Expression of HA-IKKβ (KA) blocked the nuclear accumulation of p65 in cells exposed to IR or TNF but not in UV-C irradiated cells (Fig. 5A). Expression of wild-type HA-IKKβ did not prevent the nuclear entry of p65/RelA (46) (data not shown). HeLa cells were also transfected with HA-IκBα together with an empty vector or HA-IKKβ (KA) vector. After exposure to UV, IR, or TNF, cells were harvested and lysates analyzed by immunoblotting with anti-HA. Expression of HA-IKKβ (KA) blocked HA-IκBα degradation induced by IR and TNF but not by UV (Fig. 5B). Therefore, activation of NF-κB by IR depends on IKK activity, whereas the response to UV-C occurs through an IKK-independent mechanism.
Figure 5.
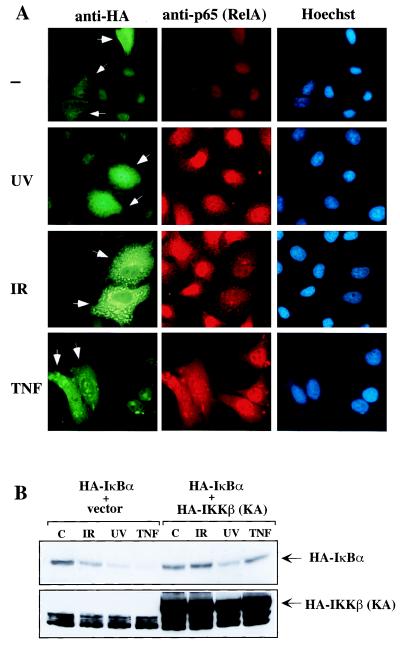
IKK is necessary for NF-κB nuclear translocation and IκBα degradation induced by IR but not by UV-C. (A) HeLa cells grown on coverslips were transfected with an expression vector encoding HA-IKKβ (KA). After 36 hr the cells were exposed to the indicated stimuli or left untreated (−) and fixed. Samples were then incubated with a mixture of anti-p65 (RelA) and anti-HA. After washing, samples were stained with rhodamine-conjugated goat anti-rabbit IgG, fluorescein isothiocyanate-conjugated goat anti-mouse IgG-IgM, and Hoechst dye and photographed through an epifluorescence-equipped microscope. Arrowheads indicate cells that express HA-IKKβ (KA). (B) HeLa cells were transfected with an expression vector encoding HA-IκBα together with an empty vector or with a vector encoding HA-IKKβ (KA). Twenty-four hours later, cells were left untreated (C) or treated with IR, UV, or TNF. After 2 hr, 4 hr, or 15 min, cells were harvested and lysates were analyzed by immunoblotting with anti-HA.
DISCUSSION
Because NF-κB activation can result in induction of antiapoptotic genes (4, 10, 12, 14, 62), it is an important stress response that can modulate the outcome of exposure to radiation and genotoxic drugs. We therefore compared the mechanisms through which exposure to UV-C and IR result in NF-κB activation. Our results indicate that these two forms of photonic radiation use different mechanisms to activate NF-κB. Although exposure to either UV-C or γ rays results in degradation of IκBα and nuclear entry of p65/RelA, the IR response depends on the phosphorylation of IκBα at Ser-32 and Ser-36, whereas the UV-C response does not. This conclusion rests on several pieces of evidence: (i) IR, but not UV-C, leads to IKK activation; (ii) substitution of the IKK phosphoacceptor sites of IκBα, Ser-32 and Ser-36, with nonphosphorylatable alanine residues abolishes IκBα degradation induced by IR but not by UV-C; (iii) a catalytically inactive IKKβ mutant, previously shown to inhibit NF-κB activation by TNF, blocks NF-κB activation by IR but not by UV-C.
Previous studies of the mammalian UV response were mostly focused on activation of AP-1 as an endpoint (17, 22, 26, 27). These studies indicated that a variety of MAPKs whose activities are stimulated by UV irradiation phosphorylate transcription factors, including c-Jun, ATF-2, Elk-1, and MEF-2C, that play an important role in the induction of c-jun and c-fos transcription (24). These MAPKs, which include JNK, p38, and ERK, are probably activated by signaling cascades that are usually triggered by occupancy of cell surface receptors (63, 64). It was also shown that UV irradiation activates the epidermal growth factor and TNF receptors (26, 27, 65). Thus, by mimicking the action of growth factors and cytokines short wavelength UV light can activate signaling pathways that stimulate the activities of MAPKs involved in induction of AP-1 activity. Two mechanisms were proposed to explain activation of cell surface receptors: (i) UV-induced receptor clustering (27) and (ii) inhibition of receptor-inactivating protein phosphatases (22). However, it was also proposed that certain MAPKs, such as the JNKs, are activated because UV-C radiation damages ribosomal RNA, which then triggers a yet-to-be identified signaling cascade (66). Regardless of the exact signaling pathway triggered by exposure to UV-C, it is somewhat surprising that the mechanism by which this form of radiation leads to NF-κB activation is so distinct from the one used by proinflammatory cytokines and other ligands that operate via cell surface receptors. Although UV-C and UV-B radiations induce association of TNF type I receptor (TNF-R1) with the signal transducer TRADD (27, 65), this signaling event is clearly insufficient for full activation of all the downstream responses usually elicited by TNF-R1 occupancy, which include JNK, p38, and NF-κB activation (11, 44).
The signaling event through which UV-C triggers IκBα degradation remains to be identified. Nevertheless, the use of proteasome inhibitors and the cell line that expressed a temperature-sensitive ubiquitin-activating enzyme (E1) suggest that UV-induced IκBα degradation still occurs via ubiquitin-mediated proteasomal degradation even though it is not dependent on phosphorylation of IκBα at Ser-32 and Ser-36. Similar observations were independently made by P. Herrlich and coworkers (personal communication). Neither group could detect any tyrosine phosphorylation of IκBα in response to UV-C (unpublished results). The phosphorylation of IκBα at Tyr-42 was detected upon reoxygenation of hypoxic cells, but instead of inducing IκBα degradation it causes its dissociation from NF-κB (67). Thus, UV-C-mediated NF-κB activation does not occur through the two previously described pathways, which depend on phosphorylation of IκBα at either serines or a tyrosine within its N-terminal regulatory domain.
Activation of NF-κB by IR, unlike the response to UV-C, does proceed via the classical activation pathway, which depends on phosphorylation of IκBα at Ser-32 and Ser-36. We find very good correlation between the kinetics and magnitude of IKK activation in γ irradiated cells and IκBα degradation, both of which parallel NF-κB activation. In addition to being dependent on phosphorylation of IκBα at Ser-32 and Ser-36, the activation of NF-κB by IR requires IKK activity and is prevented by a catalytically inactive IKKβ mutant. However, the signaling event initially triggered by IR leading to IKK activation remains to be identified. It is possible that IR may eventually activate cell surface receptors, which in this case is sufficient for IKK activation. Evidence for activation of signaling pathways normally initiated at cell surface receptors, such as the ERK MAPK cascade, by exposure to IR was presented (68). In addition, IR exposure results in activation of receptor-linked tyrosine kinases (69). The major chemical activity elicited by IR is generation of reactive oxidative intermediates (70). This activity accounts for induction of DNA damage (71). Interestingly, however, generation of reactive oxidative intermediates may not be involved in activation of NF-κB by IR, because this response is not modulated by drugs that alter the intracellular concentration of glutathione, the major cellular antioxidant (unpublished results).
The involvement of two different mechanisms in radiation-induced NF-κB activation has important implications to defense mechanisms that protect cells against various forms of radiation. For instance, inhibitors of IKK activity or expression of the “super-repressor” IκBα mutant (11, 62) should potentiate cell killing by IR because they prevent NF-κB activation and subsequent induction of an antiapoptotic response. These treatments, however, are unlikely to potentiate cell killing by UV-C, because they do not prevent activation of NF-κB by this type of photonic radiation.
Acknowledgments
We thank Joseph DiDonato, Mireille Delhase, David Rothwarf, and Ebi Zandi for helpful discussions and gifts of reagents, Joseph Aguilera in Dr. J. Ward’s lab for the use of the γ irradiator, Barbara Thompson for assistance with manuscript preparation, and Tony Hunter for the use of his microscope. N.L. was supported by a postdoctoral fellowship from the Cancer Research Institute. This work was supported by grants from the National Institutes of Health (ES04151 and CA76188) and the Department of Energy (DE-FG03–86ER60429).
ABBREVIATIONS
- JNKs
c-Jun N-terminal kinases
- MAPKs
mitogen-activated protein kinases
- TNF
tumor necrosis factor
- IL-1
interleukin 1
- IR
ionizing radiation
- HA
hemagglutinin
- EMSA
electrophoretic mobility shift assay
Note Added in Proof
While our paper was in press, the work from P. Herrlich’s group was published (72). They also demonstrated that the early phase of UV-induced NF-κB activation does not depend on phosphorylation of IκBα at Ser-32 and Ser-36.
References
- 1. Herrlich P, Ponta H, Rahmsdorf H J. Rev Physiol Biochem Pharmacol. 1992;119:187–223. doi: 10.1007/3540551921_7. [DOI] [PubMed] [Google Scholar]
- 2.Canman C E, Kastan M B. Nature (London) 1996;384:213–214. doi: 10.1038/384213a0. [DOI] [PubMed] [Google Scholar]
- 3.Ko L J, Prives C. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z-G, Baskaran R, Lea-Chou E T, Wood L D, Chen Y, Karin M, Wang J Y J. Nature (London) 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- 5.El-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 6.Levine A J. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 7.Deng C, Zhang P, Harper J W, Elledge S J, Leder P. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 8.Hartwell L H, Kastan M B. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber M, Bauman B, Cotten M, Angel P, Wagner E F. EMBO J. 1995;14:5338–5349. doi: 10.1002/j.1460-2075.1995.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beg A A, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z-G, Hu H, Goeddel D V, Karin M. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 12.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 13.Wang C-Y, Mayo M W, Baldwin A S., Jr Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 14.Wu M, Lee H, Bellas R E, Schauer S L, Arsura M, Katz D, FitzGerald M J, Rothstein T L, Sherr D H, Sonenshein G E. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 15.Buscher M, Rahmsdorf H J, Litfin M, Karin M, Herrlich P. Oncogene. 1988;3:301–311. [PubMed] [Google Scholar]
- 16.Devary Y, Gottlieb R A, Lau L, Karin M. Mol Cell Biol. 1991;11:2804–2811. doi: 10.1128/mcb.11.5.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devary Y, Gottlieb R, Smeal T, Karin M. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- 18.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 19.Dérijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 20.Cavigelli M, Dolfi F, Claret F X, Karin M. EMBO J. 1995;14:5957–5964. doi: 10.1002/j.1460-2075.1995.tb00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitmarsh A J, Shore P, Sharrocks A D, Davis R J. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 22.Knebel A, Rahmsdorf H J, Ullrich A, Herrlich P. EMBO J. 1996;15:5314–5325. [PMC free article] [PubMed] [Google Scholar]
- 23.Price M A, Cruzalegui F H, Treisman R. EMBO J. 1996;15:6552–6563. [PMC free article] [PubMed] [Google Scholar]
- 24.Karin M. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 25.Devary Y, Rosette C, DiDonato J A, Karin M. Science. 1993;261:1442–1445. doi: 10.1126/science.8367725. [DOI] [PubMed] [Google Scholar]
- 26.Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf H J. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 27.Rosette C, Karin M. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 28.Simon M M, Aragane Y, Schwarz A, Luger T A, Schwarz T. J Invest Dermatol. 1994;102:422–427. doi: 10.1111/1523-1747.ep12372194. [DOI] [PubMed] [Google Scholar]
- 29.Stein B, Rahmsdorf H J, Steffan A, Litfin M, Herrlich P. Mol Cell Biol. 1989;9:5169–5181. doi: 10.1128/mcb.9.11.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baeuerle P A, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 31.Verma I M, Stevenson J K, Schwarz E M, Van Antwerp D, Miyamoto S. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 32.Beg A A, Baldwin A S., Jr Genes Dev. 1993;7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 33.Barnes P J, Karin M. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 34.Siebenlist U, Franzoso G, Brown K. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 35.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485–1491. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 36.Brockman J A, Scherer D C, McKinsey T A, Hall S M, Qi X, Lee W Y, Ballard D W. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiDonato J A, Mercurio F, Rosette C, Wu-li J, Suyang H, Ghosh S, Karin M. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traenckner E B, Pahl H L, Henkel T, Schmidt K N, Wilk S, Baeuerle P A. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alkalay I, Yaron A, Hatzubai A, Jung S, Avraham A, Gerlitz O, Pashut-Lavon I, Ben-Neriah Y. Mol Cell Biol. 1995;15:1294–1301. doi: 10.1128/mcb.15.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Hagler J, Palombella V J, Melandri F, Scherer D, Ballard D, Maniatis T. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 41.DiDonato J A, Mercurio F, Karin M. Mol Cell Biol. 1995;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherer D C, Brockman J, Chen Z, Maniatis T, Ballard D. Proc Natl Acad Sci USA. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baldi L, Brown K, Franzoso G, Siebenlist U. J Biol Chem. 1996;271:376–379. doi: 10.1074/jbc.271.1.376. [DOI] [PubMed] [Google Scholar]
- 44.DiDonato J A, Hayakawa M, Rothwarf D M, Zandi E, Karin M. Nature (London) 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 45.Mercurio F, Zhu H, Murray B W, Shevchenko A, Bennett B L, Li J, Young D B, Barbosa M, Mann M, Manning A, Rao A. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 46.Zandi E, Rothwarf D, Delhase M, Hayakawa M, Karin M. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 47.Régnier C H, Yeong Song H, Gao X, Goeddel D V, Cao Z, Rothe M. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 48.Woronicz J D, Gao X, Cao Z, Rothe M, Goeddel D V. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 49.Singh S P, Lavin M F. Mol Cell Biol. 1990;10:5279–5285. doi: 10.1128/mcb.10.10.5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brach M A, Hass R, Sherman M L, Gunji H, Weichselbaum R, Kufe D. J Clin Invest. 1991;2:691–695. doi: 10.1172/JCI115354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rock K L, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg A L. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 52.Fenteany G, Standaert R F, Lane W S, Choi S, Corey E J, Schreiber S L. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 53.Dick L R, Cruikshank A A, Grenier L, Melandri F D, Nunes S L, Stein R L. J Biol Chem. 1996;271:7273–7276. doi: 10.1074/jbc.271.13.7273. [DOI] [PubMed] [Google Scholar]
- 54.Bogyo M, McMaster J S, Gaczynska M, Tortorella D, Goldberg A L, Ploegh H. Proc Natl Acad Sci USA. 1997;94:6629–6634. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kulka R G, Raboy B, Schuster R, Parag H A, Diamond G, Ciechanover A, Marcus M. J Biol Chem. 1988;263:15726–15731. [PubMed] [Google Scholar]
- 56.Li N, Schlessinger J, Margolis B. Oncogene. 1994;9:3457–3465. [PubMed] [Google Scholar]
- 57.Mercurio F, DiDonato J, Rosette C, Karin M. DNA Cell Biol. 1992;11:523–537. doi: 10.1089/dna.1992.11.523. [DOI] [PubMed] [Google Scholar]
- 58.Aronheim A, Engleberg D, Li N, Al-Alawi N, Schlessinger J, Karin M. Cell. 1994;78:949–961. doi: 10.1016/0092-8674(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 59.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;178:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 60.Delic J, Masdehors P, Omura S, Cosset J M, Dumont J, Binet J L, Magdelenat H. Br J Cancer. 1998;77:1103–1107. doi: 10.1038/bjc.1998.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothwarf, D. M., Zandi, E., Natoli, G. & Karin, M. (1998) Nature (London), in press. [DOI] [PubMed]
- 62.Wang X, Diener K, Jannuzzi D, Trollinger D, Tan T, Lichenstein H, Zukowski M, Yao Z. J Biol Chem. 1996;271:31607–31611. doi: 10.1074/jbc.271.49.31607. [DOI] [PubMed] [Google Scholar]
- 63.Cobb M, Goldsmith E J. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 64.Marshall C J. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 65.Tobin D, van Hogerlinden M, Toftgard R. Proc Natl Acad Sci USA. 1998;95:565–569. doi: 10.1073/pnas.95.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iordanov M S, Pribnow D, Magun J L, Dinh T H, Pearson J A, Chen S L, Magun B E. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imbert V, Rupec R A, Livolsi A, Pahl H L, Traenckner E B, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle P A, Peyron J F. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 68.Kasid U, Suy S, Dent P, Ray S, Whiteside T L, Sturgill T W. Nature (London) 1996;382:813–816. doi: 10.1038/382813a0. [DOI] [PubMed] [Google Scholar]
- 69.Uckun F M, Schieven G L, Tuel-Ahlgren L M, Dibirdik I, Myers D E, Ledbetter J A, Song C W. Proc Natl Acad Sci USA. 1993;90:252–256. doi: 10.1073/pnas.90.1.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rauth A M. In: Basic Science of Oncology. Tannock I F, Hill R P, editors. Elmsford, NY: Pergamon; 1987. pp. 106–124. [Google Scholar]
- 71.Friedberg E C. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 72.Bender K, Göttlicher M, Whiteside S, Rahmsdorf H J, Herrlich P. EMBO J. 1998;17:5170–5181. doi: 10.1093/emboj/17.17.5170. [DOI] [PMC free article] [PubMed] [Google Scholar]