

Abstract
Exposure to cyclopamine, a steroid alkaloid that blocks Sonic hedgehog (Shh) signaling, promotes pancreatic expansion in embryonic chicks. Heterotopic development of pancreatic endocrine and exocrine structures occurs in regions adjacent to the pancreas including stomach and duodenum, and insulin-producing islets in the pancreas are enlarged. The homeodomain transcription factor PDX1, required for pancreas development, is expressed broadly in the posterior foregut but pancreas development normally initiates only in a restricted region of PDX1-expressing posterior foregut where endodermal Shh expression is repressed. The results suggests that cyclopamine expands the endodermal region where Shh signaling does not occur, resulting in pancreatic differentiation in a larger region of PDX1-expressing foregut endoderm. Cyclopamine reveals the capacity of a broad region of the posterior embryonic foregut to form pancreatic cells and provides a means for expanding embryonic pancreas development.
Little is known about the mechanisms that pattern endoderm and mesoderm to form distinct organ boundaries along the gastrointestinal tract. Defects of patterning can manifest as organ agenesis and heterotopic organ formation (1). Epigenetic agents that promote these phenotypes have been critical for identifying the steps that control organ position, boundaries, and growth in development of other organ systems. For example, retinoic acid and its derivatives have been helpful in understanding steps regulating the segmentation of neuroectoderm (2) and axial patterning of the limb (3, 4). Characterization of molecules that modulate the genetic programs that establish gastrointestinal organ pattern may provide the basis for manipulating the neogenesis of vital cells like hepatocytes and pancreatic islet cells.
The pancreas, an organ with mixed exocrine and endocrine functions, arises from endodermal evaginations of duodenal endoderm in the posterior foregut, caudal to the stomach anlage. Human pancreatic hypoplasia and agenesis are rare (5), but pancreatic overgrowth, including annular pancreas, nesidioblastosis (islet cell hyperplasia), and heterotopic pancreas, is more common (6). Human pancreatic heterotopia can contain both mature endocrine and exocrine cells, and the majority (70–80%) occurs as asymptomatic overgrowths in organs that derive from endoderm adjacent to the pancreatic anlage, including the antrum of the stomach, duodenum, and small intestine (7, 8). The cause of these pancreatic malformations is presently unknown.
Endodermal expression patterns of Sonic hedgehog (SHH), a potent intercellular signaling protein, demarcate a molecular boundary between pancreas and adjacent stomach and duodenal anlagen (9–12). Shh is expressed at high levels in stomach and duodenal endoderm but repressed in pancreas endoderm. Misexpression of SHH in pancreas endoderm prevents normal pancreas morphogenesis and cell differentiation (9, 12). These observations show that maintaining the domain of SHH-nonexpressing endoderm in the pancreas anlage is required for normal development and suggests that expansion of this SHH-nonexpressing domain might allow expansion of the pancreas anlage.
Recently, a class of steroid alkaloid teratogens, including cyclopamine and jervine, from the plant Veratrum species (13), have been demonstrated to block SHH signaling effects in responding neural tissue (14). We report herein that cyclopamine addition in vitro and in vivo inhibits endodermal SHH signaling, and results in ectopic pancreas development in stomach and intestine. The results suggest that endodermal competence to form heterotopic pancreas correlates with embryonic endodermal expression of the homeodomain transcription factor PDX1 (10, 15–17). Factors like cyclopamine, which prevent Hedgehog signaling, present a way to expand pancreatic tissue.
MATERIALS AND METHODS
Stomach and Endoderm Growth and Cyclopamine Additions.
Chicken embryos were staged according to Hamburger and Hamilton (18). Stomachs were removed from Hamburger and Hamilton stage 15 chicken embryos and grown in Matrigel (Collaborative Research) as described (19). Separation of stomach from the pancreas anlage at this stage is demonstrated by absence of detectable glucagon or insulin by immunohistochemistry. In contrast, both these endocrine markers are detected when stomach with attached pancreas is grown in Matrigel for 60 h. Tomatidine (Sigma) and cyclopamine (a gift from W. Gaffield, U.S. Department of Agriculture, Albany, CA, and P. Beachy, Johns Hopkins University) dissolved in 95% ethanol were diluted in dimethyl sulfoxide and added in vitro at concentrations indicated in the results; appropriate additions of dimethyl sulfoxide were made in control samples. Hamburger and Hamilton stage 12 embryonic endoderm, which includes the stomach anlage, was isolated as described (11) and grown in Matrigel with 10 μM cyclopamine. Rudiments and endoderm were harvested and prepared for histology as described below. In vivo cyclopamine additions (0.125, 0.25, 0.5, 0.75, or 1.0 mg per embryo) to embryos at Hamburger and Hamilton stage 10, 11, 15, or 19 were made after windowing eggs as described (20). External phenotypes were more severe after cyclopamine exposure at earlier developmental stages. For example, exposure of five embryos to 1.0 mg at Hamburger and Hamilton stage 10 resulted in cyclopia in four embryos, in contrast to exposure at later stages that resulted in hypotelorism and microphthalmia but not cyclopia (n = 8). Severity of effects on foregut-derived organs after cyclopamine exposure was similarly reduced by adding cyclopamine at later stages. At higher doses, we observed 50% mortality rate by day 9, 80% by day 10, and 100% by day 11 of incubation with 0.5 mg of cyclopamine (n = 11); embryos exposed to 0.5–1.0 mg of cyclopamine were analyzed after 9 days.
Immunohistochemistry, in Situ Hybridization, and Proliferation Assays.
Immunohistochemistry on 7-μm sections was performed as described (21) except (i) exposure to primary antibodies was for 1 h, (ii) exposure to secondary antibodies was for 30 min, and (iii) appropriate secondary donkey antibodies (Jackson ImmunoResearch) conjugated to fluorescein isothiocyanate or Cy3 at a 1:200 dilution were used for fluorescence microscopy. Primary antibodies were guinea pig anti-insulin (Dako), rabbit anti-glucagon (Chemicon) and rabbit anti-carboxypeptidase A (Biogenesis, Bournemouth, U.K.). Methods for in situ hybridizations using chicken Pdx, Shh, and Patched (Ptc) probes have been described (11, 12).
Stomachs isolated at stage 15 and grown for 60 h in Matrigel were labeled for 3 h by using the cell proliferation labeling reagent (Amersham) diluted 1:100. Labeled explants were sectioned (7 μm), processed for detection of 5-bromodeoxyuridine (BrdUrd) as described by the manufacturer, counterstained in Gill’s hematoxylin (Polyscientific; Bayshore, NY) diluted 1:4 in water, and sealed with glass coverslips. Epithelial and mesenchymal cells in 10 random sections at ×40 magnification from cyclopamine-treated or control samples were counted and scored. The number of labeled epithelial or mesenchymal cells was divided by the total number of cells counted (6,856 control epithelium, 8,520 cyclopamine epithelium; 4,160 control mesenchyme, and 4,387 cyclopamine mesenchyme) to obtain the fraction of labeled cells. The results were found significant at P < 0.05 by Student’s t test.
To determine relative size of insulin-producing islets, pancreata from six cyclopamine-treated and five control embryos harvested after 9 days of incubation were completely sectioned at 7 μm and immunostained for the presence of insulin and glucagon. Measurements of the insulin-producing islets in every sixth section were obtained to avoid counting the same islet twice. Widths of 97 islets from cyclopamine-treated embryos and 72 islets from control embryos were averaged and presented as mean ± SEM. This method underestimates the relative change of islet size after cyclopamine exposure.
RESULTS
Insulin and Glucagon Expression in Stomach Endoderm After Cyclopamine Exposure.
An in vitro method for growing lung explants (19) was adapted to grow stomach explants in the presence of cyclopamine. Stomach endoderm and mesenchyme were dissected away from adjacent esophagus, liver, and dorsal pancreatic anlagen (Fig. 1A). The endoderm-derived epithelial layer in control stomach explants is a flattened monolayer of columnar cells with polarized nuclei and a homogenous adjacent mesenchyme. In contrast, stomach explants exposed for 60 h to 10 μM cyclopamine have an epithelium with multiple folds, containing several layers of nonpolarized cells adjacent to mesenchyme with numerous vascular lumen containing blood cells (Fig. 1B). It is not yet known whether vascularization promoted by cyclopamine is a direct or indirect effect on mesenchyme. The appearance of epithelium and mesenchyme of cyclopamine-treated stomach explants is similar to the nascent dorsal pancreas bud, described below.
Figure 1.
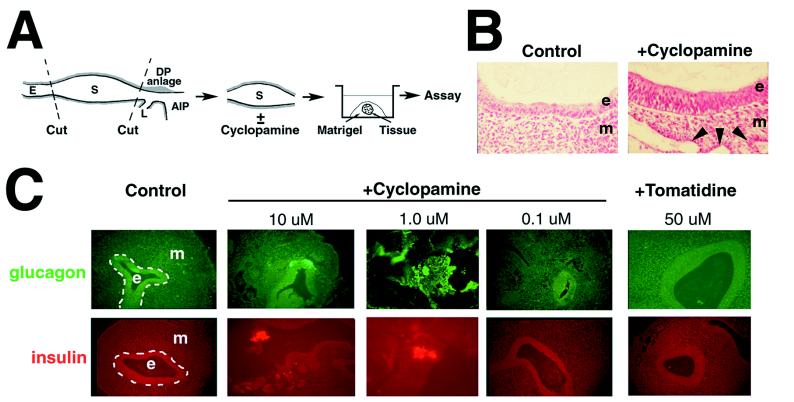
Cyclopamine-induced insulin and glucagon expression in isolated stomach explants grown in vitro. (A) Isolation and growth of stage 15 stomach. The stomach anlage is an easily identified bulge rostral to the anterior intestinal portal (AIP) and caudal to the esophagus (E) of stage 15 (23–27 somites) chicken embryos. The dorsal pancreatic (DP) anlage lies dorsal and caudal to the AIP and the ventral pancreatic anlage is within the liver (L) primordium, rostral and lateral to the AIP. Isolated stomachs were embedded in Matrigel and grown until assay by histological methods. (B) Cyclopamine-induced morphologic changes in stomach epithelium (e) and mesenchyme (m). Arrowheads mark the lumens of vascular structures that contained blood cells. Epithelial folds induced by cyclopamine are not evident in these panels. (C) Cyclopamine, at the indicated concentrations, induces insulin and glucagon in isolated stomach anlagen. All ectopic expression is detected in the epithelial cell layer. Tomatidine does not induce expression of insulin and glucagon.
Growth of control stomach explants in vitro for up to 60 h does not result in expression of detectable glucagon (n = 10) or insulin (n = 11) (Fig. 1C). Both markers are detected in the pancreas after growth of control explanted dorsal pancreas anlage in Matrigel (n = 6, data not shown). Both glucagon (8 of 11 samples) and insulin (10 of 11 samples) are induced in stomach endoderm by 10 μM cyclopamine. No glucagon or insulin are detected in the mesenchymal layer. Cyclopamine also induced endodermal expression of glucagon and insulin at 1.0 μM (n = 6) but not 0.1 μM (n = 6). Tomatidine, an alkaloid structurally similar to cyclopamine that is nonteratogenic and lacks inhibitory effects on SHH signaling (14), did not induce these markers even at 50 μM (n = 6) and did not alter epithelial or mesenchymal morphology compared with control explants. The concentration-dependent response of stomach explants between 0.1 and 10 μM is a range similar to that previously shown for the response of neuroectoderm to cyclopamine (14). These data indicate that cyclopamine exposure transforms stomach epithelium and mesenchyme toward a pancreatic phenotype and suggest that pancreatic heterotopia can form de novo, by ectopic differentiation. Similar results in explant culture were obtained with duodenal epithelium isolated from stage 18 embryos (data not shown).
Cyclopamine-Induced Ectopic Pancreas Development.
To test for later stage and in vivo effects of cyclopamine on stomach and possibly other organ anlagen, embryos were exposed to cyclopamine in ovo (n = 69). Exposure to cyclopamine resulted in visible external defects, including cyclopia, microphthalmia, proboscis formation, amelia, thoracic lordosis, and decreased body size (ref. 20 and unpublished results). Examination of gastrointestinal organs revealed severe deficits, including shortened length of the gut tube, and reduced mesenchymal cell numbers in foregut-derived organs (data not shown). In this context of generally reduced growth, however, we also found evidence of ectopic structures in duodenum, stomach, and dorsal pancreas as described below.
After 3 days of development in control embryos, a single dorsal pancreatic epithelial bud forms caudal to the stomach, surrounded by vascularizing mesenchyme that includes the splenic anlage, adjacent to the right omphalomesenteric vein (Fig. 2A). Thirteen percent of embryos exposed to cyclopamine (at stage 11) developed a second dorsal endodermal bud caudal to the normal bud (Fig. 2B), which expresses detectable insulin and glucagon (data not shown).
Figure 2.
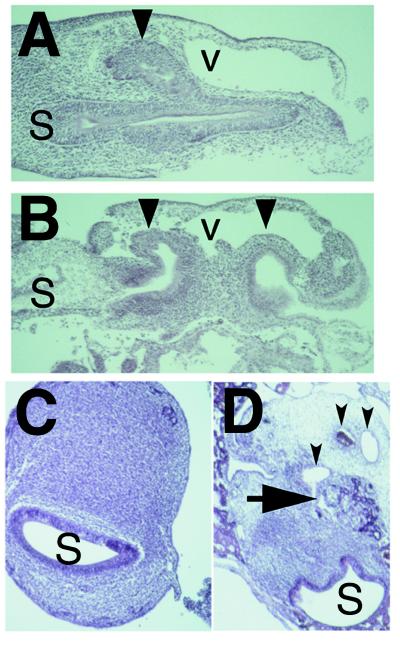
Cyclopamine induces heterotopic structures in duodenum and stomach. (A) Dorsal pancreatic bud (large arrowhead), stomach (S), and omphalomesenteric vein (v) from control embryo at 3 days. In addition to the vein, small vascular spaces containing blood cells in the dorsal mesenchyme surround the dorsal bud epithelium. (B) Two dorsal epithelial buds (large arrowheads) from a cyclopamine-exposed embryo grown for 3 days. The more posterior heterotopic dorsal bud bulges into the vascular space. (C) Transverse section through the antrum of the distal stomach (S) from a 9 day control embryo. (D) Transverse section through the corresponding antral portion of the stomach from an embryo treated with 1.0 mg of cyclopamine. Large arrow marks branched epithelial structures arising within the stomach mesenchyme. Three small arrowheads mark obvious vascular structures; the middle lumen is filled by red blood cells. As shown, cyclopamine exposure results in transformation of part, but not all, of the distal stomach toward a pancreatic phenotype. Dorsal is toward the top in A–D and anterior is toward the left in A and B.
Ectopic pancreatic development was also detected in 16% of cyclopamine-exposed embryos in the distal stomach (Fig. 2 C and D). The proximal stomach in chicken embryos normally develops into the proventricular, or glandular, stomach and the distal stomach develops into the muscular gizzard. By 9 days, the epithelium of the distal stomach is normally surrounded by a homogeneous thick mesodermal layer (Fig. 2C). Neither carboxypeptidase A nor insulin are normally expressed in the distal chicken stomach (Fig. 3 A and C and ref. 22). In contrast, the epithelium of the distal stomach in cyclopamine-treated embryos adjoins tissue with a characteristic pancreatic appearance, surrounded by vascular mesenchyme (Fig. 2D). Both carboxypeptidase A and insulin are expressed in these stomachs; in particular, insulin expression occurs in compact clusters of cells resembling islets of Langerhans (Fig. 3 B and D).
Figure 3.
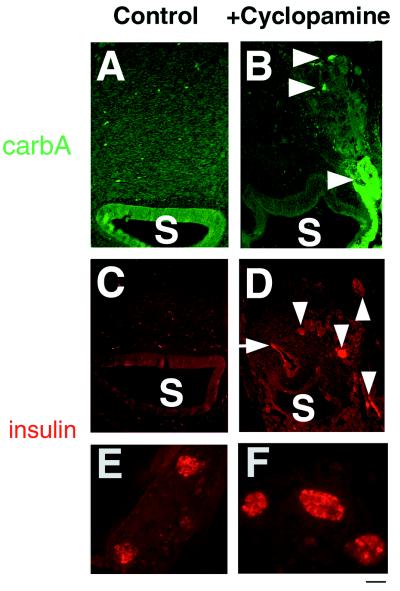
Carboxypeptidase A and insulin expression within heterotopic pancreas-like structures in stomach (S) after cyclopamine treatment. (A, C, and E) Tissues from 9-day control embryos. (B, D, and F) Tissues from 9-day cyclopamine-treated embryos. (A) No carboxypeptidase A staining in stomachs from control embryos. (B) Arrowheads mark epithelial cells expressing carboxypeptidase A. (C) No insulin staining in stomachs from control embryos. (C) Arrowheads mark cell clusters expressing insulin. Arrow marks the first portion of the duodenum that also expresses detectable insulin. (E) Insulin-expressing islets from the dorsal pancreas of 9-day control embryos with characteristic spacing and uniformity of size. (F) Insulin-expressing islets from the dorsal pancreas of cyclopamine treated embryos. Note smaller distance between islets and increased islet width, compared with E. [Bar = 50 μm (E and F).]
By 9 days of normal development, insulin-producing islets in dorsal pancreas have a uniform size, with an average width of 48.7 ± 6.3 μm (Fig. 3E); ventral insulin islets form later, by 13 days (23). In contrast, insulin-producing islets in the dorsal pancreas of cyclopamine-treated embryos have a wider range of sizes, appear more clustered, are larger, with an average width of 72.8 ± 12.2 μm, and stain more uniformly for insulin (Fig. 3F). Cyclopamine had no detectable effect on the number of dorsal (or ventral) pancreatic insulin-expressing islets, nor on the size, number, or distribution of dorsal and ventral pancreatic glucagon-expressing cells that form separate islets in the chicken (24).
The Competence to Form Heterotopic Pancreatic Tissue Corresponds to Embryonic Pdx Expression.
In embryos exposed to cyclopamine, organs where pancreas growth is promoted, including distal stomach, pancreas, and duodenum, have detectable endodermal Pdx expression (Fig. 4). Pdx specifies a homeodomain transcription factor required for pancreas development (15, 16). Other organs of cyclopamine-exposed embryos that did not express detectable insulin and/or carboxypeptidase A include the pharynx, esophagus, lungs, proximal stomach (proventriculus), and liver. Endodermal Pdx expression in the anlagen of these organs was not detected in cyclopamine-treated embryos. In one case, ectopic insulin and carboxypeptidase A in the jejunum were detected (Fig. 4).
Figure 4.
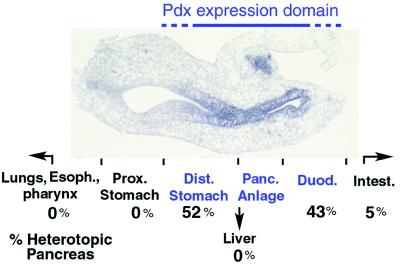
Summary of heterotopic pancreas structures and expression of immunodetectable insulin and carboxypeptidase A observed after cyclopamine exposure in vivo. Photomicrograph shows a sagittal view of the foregut region from a cyclopamine-exposed 3-day embryo after in situ hybridization with a probe that detects Pdx expression. Anterior is to the left, and dorsal is toward the top. Endodermal regions from foregut that express Pdx (“Pdx expression domain”) during the period of cyclopamine exposure are stained blue after in situ hybridization, are marked in blue type, and include the distal stomach, the pancreatic anlage, and the duodenum (see Fig. 2A for anatomic landmarks). Heterotopic pancreas cell types were observed in 21 of 69 treated embryos exposed to 0.5–1.0 mg of cyclopamine at stage 10–11; 52% (11 of 21) of these heterotopic structures were found in the distal stomach, and 43% (9 of 21) were in duodenum. In one embryo (1 of 21), insulin and carboxypeptidase A expression was detected in the both duodenum and the jejunal loop of the intestines (Intest.). Insulin, carboxypeptidase A, and pancreatic structures were not seen in lungs, pharynx, esophagus, proximal stomach, or liver after cyclopamine treatment.
Cyclopamine Effects on Epithelial–Mesenchymal Signaling.
Normal chicken stomach endoderm expresses high levels of Shh (10–12). Expression of Ptc, a candidate receptor for SHH, is induced by SHH signaling (25), and high levels of Ptc mRNA are detected in stomach and intestinal mesenchyme adjacent to Shh-expressing endoderm (Fig. 5A). Cyclopamine treatment in vivo reduces mesenchymal Ptc expression (Fig. 5B), consistent with the proposal that cyclopamine prevents normal SHH signaling (14). SHH expression and posttranslational processing in transfected cell lines are not affected by cyclopamine (14). However, we find that levels of stomach epithelial Shh mRNA are reduced after cyclopamine addition in vivo and in vitro. As shown in Fig. 5C, Shh expression is normally detected throughout the distal stomach epithelium but is repressed in focal patches in stomach rudiments grown in 1.0–10 μM cyclopamine (Fig. 5D). Mesenchymal removal and growth of isolated foregut epithelium in the presence of cyclopamine results in Shh expression throughout epithelium (n = 4; Fig. 5E). These data suggest that Shh repression in endoderm after cyclopamine exposure requires mesenchyme.
Figure 5.
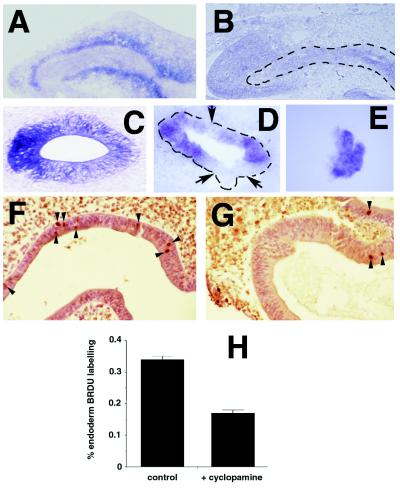
Changes in gene expression and cell proliferation in stomach endoderm and mesenchyme after cyclopamine treatment. (A) Ptc in situ hybridization of stomach in an embryonic day 3 chicken. (B) Ptc in situ hybridization of stomach of an embryonic day 3 chicken treated with cyclopamine. (C) Shh in situ hybridization of cultured stomach with no addition. (D) Shh in situ hybridization of cultured stomach treated with 10 μM cyclopamine. Note that portions of the stomach epithelium do not express Shh (arrows). (E) Shh in situ hybridization of cultured endoderm treated with 10 μM cyclopamine. (F) BrdUrd staining of control stomach endoderm; arrowheads mark nuclear staining. (G) BrdUrd staining of stomach endoderm treated with cyclopamine. (H) Bar graph showing percentage of labeled endoderm cells in controls or after cyclopamine addition.
Cyclopamine Effects on Epithelial Proliferation.
SHH has been shown to increase myoblast (26) and epidermal (27) proliferation, whereas PTC appears to function as a growth suppressor (25, 28, 29). To test the effect of cyclopamine on proliferation, we assayed incorporation of BrdUrd into DNA during in vitro growth of explanted stomachs (Fig. 5 F–H). There is a 2-fold reduction of BrdUrd incorporation by epithelial cells treated with cyclopamine, compared with control cultures. There is also a 3-fold reduction of BrdUrd incorporation by mesenchymal cells in cyclopamine-treated explants (8%) compared with controls (25%).
DISCUSSION
Heterotopic formation of pancreas cell types in stomach and intestinal anlagen after cyclopamine treatment reveals an unexpectedly wide area of gastrointestinal competence to differentiate as pancreas. Pancreatic heterotopia has been previously best described in humans, with highest frequencies in the stomach antrum, duodenum, and jejunum but also infrequently in ileum, liver, and biliary tree, and rarely in extraabdominal sites (for review, see ref. 6). PDX1, a homeodomain transcription factor required for pancreatic morphogenesis and cytodifferentiation (15, 16), is expressed at different times during embryogenesis in each of these extrapancreatic gastrointestinal sites with highest sustained expression in the antrum and duodenum (refs. 10, 15, and 16 and Fig. 4). Previous results showed that SHH is normally expressed throughout the gut, except in the pancreas (9, 10, 12, 17), and that SHH misexpression in pancreas prevents normal development (9, 12). Thus, these observations are consistent with a model that proposes that most pancreatic heterotopia occurs in regions where PDX1 expression and inactive SHH signaling coincide (Fig. 4).
Dorsal lobe islets containing insulin-producing β cells are enlarged after exposure to cyclopamine (Fig. 3F). Protein kinase A, a cAMP-stimulated kinase, and forskolin, an agent that increases intracellular cAMP levels, also negatively regulate SHH signaling (30, 31). Forskolin and other factors that increase intracellular cAMP including glucose, amino acids, prolactin, and growth hormone can promote β cell growth (32, 33). However, cyclopamine promotes both β cell development and pancreatic heterotopia.
Dominant and recessive mutations of Shh in humans, which can result (34, 35) in holoprosencephaly (HPE), have not yet been linked to an increased incidence of pancreatic heterotopia or nesidioblastosis. However, inherited defects of cholesterol synthesis, such as in Smith–Lemli–Opitz syndrome (SLOS), also result in HPE and are associated with nesidioblastosis and hypoglycemia (36, 37). To the extent that HPE implies perturbed embryonic Shh signaling in these SLOS patients (38), these observations indicate a possible role for Hedgehog proteins in human islet development and glucose regulation.
A recent study demonstrated that SHH signaling from notochord to neuroectoderm can be blocked by cyclopamine (14). Our results suggest that epithelial to mesenchymal signaling by SHH in the gut is also perturbed by cyclopamine. Previous work has shown that endodermal SHH regulates patterns of mesenchymal gene expression in pancreas (9, 12), and other organ anlage in foregut and hindgut (39, 40). Our data indicate that perturbation of SHH signaling from endoderm to mesenchyme results in a mesenchymal signal that represses endodermal Shh expression (Fig. 5 C–E). It is also possible that inhibition of SHH signaling can have direct effects on endoderm (12). Similarities in structure and posttranslational modification between SHH and other members of the Hedgehog family (38, 41, 42) also raise the possibility that cyclopamine’s effects on pancreas growth might result from signaling-blockade of additional Hedgehog proteins.
Cyclopamine blockade of Shh signaling in stomach promotes epithelial thickening and branching, and the mesenchymal formation of vascular structures and blood cells (Figs. 1B and 2D), which are characteristic of mesenchyme in the dorsal pancreatic/splenic anlage. Dorsal pancreas endoderm can direct hematopoiesis and vasculogenesis of heterologous mesenchyme (43), consistent with the location of the chicken splenic anlage in dorsal pancreatic mesenchyme. Cell proliferation studies (Fig. 5) indicate that the apparent increased thickness and folding of epithelium after cyclopamine exposure is not due to enhanced proliferation and instead may result from cyclopamine promotion of cell survival, movement, or transformation. These epithelial and mesenchymal phenotypes, resulting from blockade of Shh signaling, support previous models that proposed that spatially restricted Shh expression along the anterior–posterior axis generates distinct mesodermal boundaries for pancreas, spleen, and adjacent intestine (9, 12).
Cyclopamine effects described herein raise the possibility that other agents that block SHH activity, including cholesterol synthesis and sterol transport inhibitors (ref. 14 and references therein), can promote the growth of pancreatic cells. Class 2 sterol transport inhibitors include progesterone, an endogenous circulating hormone that can affect adult β cell growth (32). Understanding the mode of action of agents such as cyclopamine may aid the development of cell-replacement therapies for pancreatic diseases such as diabetes mellitus.
Acknowledgments
We are grateful to William Gaffield and Phil Beachy for cyclopamine samples and thank Brigid Hogan, Henk Roelink, Jeffery Porter, Benoit St.-Jacques, and members of the Melton laboratory for stimulating discussions. S.K.K. was supported by a physician-postdoctoral fellowship award from the Howard Hughes Medical Institute and D.A.M. is an investigator of the Howard Hughes Medical Institute.
References
- 1. Sadler T W. Langman’s Medical Embryology. 5th Ed. Baltimore: Williams & Wilkins; 1985. pp. 224–246. [Google Scholar]
- 2.Sasai Y, De Robertis E M. Dev Biol. 1997;182:5–20. doi: 10.1006/dbio.1996.8445. [DOI] [PubMed] [Google Scholar]
- 3.Helms J, Thaller C, Eichele G. Development (Cambridge, UK) 1994;120:3267–3274. doi: 10.1242/dev.120.11.3267. [DOI] [PubMed] [Google Scholar]
- 4.Johnson R L, Tabin C J. Cell. 1997;90:979–990. doi: 10.1016/s0092-8674(00)80364-5. [DOI] [PubMed] [Google Scholar]
- 5.Stoffers D A, Zinkin N T, Stanojevic V, Clarke W C, Habener J F. Nat Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 6.Hill I D, Lebenthal E. In: The Pancreas: Biology, Pathobiology, and Disease. Go V, Dimango E, Gardner J, Lebenthal E, Reber H, Scheele G, editors. New York: Raven; 1993. pp. 1029–1040. [Google Scholar]
- 7.Barbosa J J, Dockerty M B, Waugh J M. Surg Gynecol Obstet. 1946;82:527–542. [PubMed] [Google Scholar]
- 8.Dolan R V, ReMine W H, Dockerty M B. Arch Surg. 1974;109:762–765. doi: 10.1001/archsurg.1974.01360060032010. [DOI] [PubMed] [Google Scholar]
- 9.Apelqvist A, Ahlgren U, Edlund H. Curr Biol. 1997;7:801– 804. doi: 10.1016/s0960-9822(06)00340-x. [DOI] [PubMed] [Google Scholar]
- 10.Kim S K, Hebrok M, Melton D A. Cold Spring Harbor Symp Quant Biol. 1997;62:377–383. [PubMed] [Google Scholar]
- 11.Kim S K, Hebrok M, Melton D A. Development (Cambridge, UK) 1997;124:4243–4252. doi: 10.1242/dev.124.21.4243. [DOI] [PubMed] [Google Scholar]
- 12.Hebrok M, Kim S K, Melton D A. Genes Dev. 1998;12:1705–1713. doi: 10.1101/gad.12.11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaffield W. In: Studies in Natural Products Chemistry. Atta-ur-Ralman, editor. Vol. 21. Amsterdam: Elsevier; 1998. , in press. [Google Scholar]
- 14.Cooper M K, Porter J A, Young K E, Beachy P A. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 15.Jonsson J, Carlsson L, Edlund T, Edlund H. Nature (London) 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 16.Offield M F, Jetton T L, Labosky P A, Ray M, Stein R, Magnuson M A, Hogan B L M, Wright C V E. Development (Cambridge, UK) 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 17.Ahlgren U, Pfaff S L, Jessell T M, Edlund T, Edlund H. Nature (London) 1997;385:257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- 18.Hamburger V, Hamilton H L. J Exp Morph. 1951;88:49–92. [PubMed] [Google Scholar]
- 19.Bellusci S, Grindley J, Emoto H, Itoh N, Hogan B L M. Development (Cambridge, UK) 1997;124:4867–4878. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- 20.Bryden M M, Perry C, Keeler R F. Teratology. 1973;8:19–28. doi: 10.1002/tera.1420080104. [DOI] [PubMed] [Google Scholar]
- 21.Chiang M-K, Flanagan J G. Development (Cambridge, UK) 1996;122:2239–2250. doi: 10.1242/dev.122.7.2239. [DOI] [PubMed] [Google Scholar]
- 22.Kramer B, Andrew A, Rawdon B B, Becker P. Development (Cambridge, UK) 1987;100:661–671. doi: 10.1242/dev.100.4.661. [DOI] [PubMed] [Google Scholar]
- 23.Dieterlen-Lièvre F, Beaupain D. Gen Comp Endocrinol. 1974;22:62–69. doi: 10.1016/0016-6480(74)90088-4. [DOI] [PubMed] [Google Scholar]
- 24.Beaupain D, Dieterlen-Lièvre F. Gen Comp Endocrinol. 1974;23:421–431. doi: 10.1016/0016-6480(74)90040-9. [DOI] [PubMed] [Google Scholar]
- 25.Goodrich L V, Johnson R L, Milenkovic L, McMahon J A, Scott M P. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- 26.Duprez D, Fournier-Thibault C, Le Douarin N. Development (Cambridge, UK) 1998;125:495–505. doi: 10.1242/dev.125.3.495. [DOI] [PubMed] [Google Scholar]
- 27.Oro A E, Higgins K M, Hu Z, Bonifas J M, Epstein E H, Scott M P. Science. 1997;270:817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 28.Hahn H, Wicking C, Zaphiropoulous P G, Gailani M R, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden A B, Gillies S, et al. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 29.Johnson R L, Rothman A L, Xie J, Goodrich L V, Bare J W, Bonifas J M, Quinn A G, Myers R M, Cox D R, Epstein E H, Jr, et al. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 30.Hammerschmidt M, Bitgood M J, McMahon A P. Genes Dev. 1996;10:647–658. doi: 10.1101/gad.10.6.647. [DOI] [PubMed] [Google Scholar]
- 31.Fan C M, Porter J A, Chiang C, Chang D T, Beachy P A, Tessier-Lavigne M. Cell. 1995;81:457–465. doi: 10.1016/0092-8674(95)90398-4. [DOI] [PubMed] [Google Scholar]
- 32.Sorenson R L, Brelje T C. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 33.Schuppin G T, Pons S, Hugl S, Aiello L P, King G L, White M, Rhodes C J. Diabetes. 1998;47:1074–1085. doi: 10.2337/diabetes.47.7.1074. [DOI] [PubMed] [Google Scholar]
- 34.Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell H F, Donis-Keller H, Helms C, Hing A V, et al. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- 35.Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer S W, Tsui L C, Muenke M. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 36.McKeever P A, Young I D. J Med Genet. 1990;27:465– 466. doi: 10.1136/jmg.27.7.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lachman M F, Wright Y, Whiteman D A, Hesson V, Greenstein R M. Clin Genet. 1991;39:136–141. doi: 10.1111/j.1399-0004.1991.tb03000.x. [DOI] [PubMed] [Google Scholar]
- 38.Porter J A, Young K E, Beachy P A. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 39.Roberts D J, Johnson R L, Burke A C, Nelson C E, Morgan B A, Tabin C. Development (Cambridge, UK) 1995;121:3163–3174. doi: 10.1242/dev.121.10.3163. [DOI] [PubMed] [Google Scholar]
- 40.Roberts D J, Smith D M, Goff D J, Tabin C J. Development (Cambridge, UK) 1998;125:2791–2801. doi: 10.1242/dev.125.15.2791. [DOI] [PubMed] [Google Scholar]
- 41.Bitgood M J, Shen L, McMahon A P. Curr Biol. 1996;6:298–304. doi: 10.1016/s0960-9822(02)00480-3. [DOI] [PubMed] [Google Scholar]
- 42.Vortkamp A, Lee K, Lanske B, Segre G V, Kronenberg H M, Tabin C J. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 43.Dieterlen-Lièvre F. Dev Biol. 1970;22:138–156. doi: 10.1016/0012-1606(70)90011-4. [DOI] [PubMed] [Google Scholar]