

Abstract
The ability to use a vital cell marker to study mouse embryogenesis will open new avenues of experimental research. Recently, the use of transgenic mice, containing multiple copies of the jellyfish gene encoding the green fluorescent protein (GFP), has begun to realize this potential. Here, we show that the fluorescent signals produced by single-copy, targeted GFP in-frame fusions with two different murine Hox genes, Hoxa1 and Hoxc13, are readily detectable by using confocal microscopy. Since Hoxa1 is expressed early and Hoxc13 is expressed late in mouse embryogenesis, this study shows that single-copy GFP gene fusions can be used through most of mouse embryogenesis. Previously, targeted lacZ gene fusions have been very useful for analyzing mouse mutants. Use of GFP gene fusions extends the benefits of targeted lacZ gene fusions by providing the additional utility of a vital marker. Our analysis of the Hoxc13GFPneo embryos reveals GFP expression in each of the sites expected from analysis of Hoxc13lacZneo embryos. Similarly, Hoxa1GFPneo expression was detected in all of the sites predicted from RNA in situ analysis. GFP expression in the foregut pocket of Hoxa1GFPneo embryos suggests a role for Hoxa1 in foregut-mediated differentiation of the cardiogenic mesoderm.
Keywords: gene targeting/Hoxa1/Hoxc13
The jellyfish, Aequorea victoria, produces a protein, green fluorescent protein (GFP), which absorbs blue light and emits green light. In the years since the cDNA encoding this protein was cloned (1), it has become an important reporter gene in both prokaryotic and eukaryotic organisms. In contrast to β-galactosidase (β-gal) detection protocols, no exogenous substrates are required, only molecular oxygen and excitation light being needed to reveal the active chromophore (2). Therefore, GFP fusion genes can be detected in living organisms as they develop. In addition, GFP is small (26.9 kDa) and functions as a monomer, making it ideal for protein fusions (3), whereas the widely used reporter β-gal is much larger (135 kDa) and functions as a tetramer (4–5).
Use of GFP reporter genes in mammalian systems has been facilitated by the generation of derivatives optimized for mammalian codon usage. These alterations, which aid translational efficiency, may improve protein folding as well. In addition, several amino acid changes that shift the excitation spectrum toward red and increase fluorescent signal strength have been incorporated (6–7). To date, the use of optimized GFP variants in vertebrates have been limited to transgenic mice (8–10) and zebrafish (11–15) and to cell-labeling studies in Xenopus (16) and mouse (17) embryos. The mammalian studies have involved either injection of large numbers of plasmids encoding GFP or production of transgenic animals containing multiple-copy tandem arrays of the gene to obtain strong fluorescent signals. Notably, it has been possible to generate mice that express GFP in all cells (8). This strain can be used similarly to the lacZ-expressing Rosa26 mice in chimeric studies, with the additional advantage that individual chimeras can be examined over time (18–19). Importantly, ubiquitous expression of high levels of GFP appears to be nontoxic (8). In addition, it has been recently demonstrated that GFP can be used as a cell lineage marker in living mouse embryos (17). Although these GFP-expressing mice provide significant advances in the use of a vital marker for developmental studies, the aforementioned applications are subject to all the limitations inherent in transgenic methodology. For example, signal strength has varied widely among different transgenic lines (13); position effects may influence the ability to completely recapitulate all aspects of normal temporal and spatial regulation of gene expression, and transgenes are often subject to silencing.
Here, we demonstrate that GFP is detectable when a single-copy of the gene is targeted into two separate Hox genes. In addition, we show that single-copy GFP fusion gene expression can be examined both early in mammalian embryogenesis when the embryo is relatively clear and in organ preparations late in embryogenesis when the embryo is opaque. These targeted GFP-gene fusions provide the same benefits as targeted lacZ fusions (20) including retention of all cis-acting elements, lack of position effects, and labeling of the mutant cells in homozygotes but in addition, include the advantages of a vital marker. In particular, optical sectioning of GFP-expressing embryos by laser scanning confocal microscopy enables careful three-dimensional analysis of gene expression patterns without histological sectioning.
MATERIALS AND METHODS
Vector Construction.
The GFP fusion-gene cassette was constructed starting with the NcoI–AflII fragment containing the GFP-coding region from pEGFP (CLONTECH). This fragment was resected with S1 nuclease up to the first base of the second codon and cloned into a derivative of pSP72 (Promega). In addition, the NotI site between the GFP-coding region and Poly(A) addition signal was removed. Finally, an MC1neo cassette (21) flanked by loxP sites was cloned into the vector. The final vector allows the entire GFP loxP neo loxP cassette to be excised by using KpnI, SalI, or BamHI.
The Hoxa1 insertion vector was constructed from a 7.8-kb ClaI fragment containing the Hoxa1-coding region. The GFP loxP neo cassette was ligated into a unique AatII site present in exon 2. Verification of the correct reading frame across the Hoxa1/GFP junction was determined by manual dideoxy sequencing (United States Biochemical). Finally, the herpes simplex virus (HSV) thymidine kinase gene (TK1) (22) was inserted 5′ of the Hoxa1 sequences.
The Hoxc13 insertion vector was constructed by inserting the GFP fusion gene cassette into a previously described bacteriophage lambda clone (23), which carries an XhoI linker insertion at the BstEII site in the third helix of the homeodomain of Hoxc13, generating an in-frame fusion of the Hoxc13- and GFP-coding regions. The resulting fusion gene is directly analogous to the previously described Hoxc13lacZneo allele (23).
Generation of Targeted Cell Lines and Germline Transmitting Mice.
Targeting vectors were electroporated into R1 embryonic stem (ES) cells (24) and subjected to positive–negative selection as described (22). Targeted disruptions for each gene were detected by Southern transfer analyses. Targeted colonies were microinjected into C57BL/6J blastocysts to generate chimeric mice as previously described (23). Chimeric males were mated to C57BL/6J females, and resulting agouti progeny were tested for the presence of the mutation. Heterozygotes were mated to the Cre “deleter” mouse (25), and progeny were screened for removal of the neomycin gene by Cre-mediated site-specific recombination by using Southern transfer analyses as described in Figs. 1 and 2.
Figure 1.

Hoxa1GFPneo-targeting vector and Southern blot analysis. (A) Large solid boxes represent Hoxa1 exons, white box the GFP gene, and gray box the loxP flanked MC1neo cassette, AatII (A), ClaI (C), and PacI (P), and EcoRV (R). The position of the 3′-flanking probe used for Southern transfer analysis is indicated by the small solid box. The first line shows the wild-type genomic structure, the second line the targeting vector, and the third line the genomic structure resulting from homologous recombination. (B) Southern blot analysis of targeted cell line and chimera progeny. Shown are EcoRV digests of the parental R1 cell line, a targeted ES cell line, and offspring from a chimera. The wild-type allele is detected by a 19.6-kb restriction fragment, whereas the mutant allele is detected by a 13.2-kb restriction fragment. (C) Analysis of progeny of a Hoxa1GFPneo heterozygote crossed to a Cre deleter mouse. Shown are Southern transfer analyses of BamHI-digested DNA probed with an NcoI–AflII pEGFP fragment. A 2.2-kb fragment is detected in the Hoxa1GFPneo mice that shifts to 1.0 kb upon Cre-mediated removal of the neomycin resistance gene.
Figure 2.

Hoxc13GFPneo-targeting vector and Southern blot analysis. (A) Genomic structure and targeting vector. Symbols in A are as described in Fig. 1, except large solid boxes represent Hoxc13 exons, BamHI (B), BstEII (E), and HindIII (H). The position of the 5′-flanking probe used for Southern transfer analysis is indicated by the small solid box. (B) Southern blot analysis of targeted cell line and offspring from a chimera. Shown are BamHI digests of the parental R1 cell line, a targeted ES cell line, and an offspring from a chimera. (C) Analysis of progeny of a Hoxc13GFPneo heterozygote crossed to a Cre deleter mouse. Analysis is as described for Fig. 1C. (D) Genotypic analysis of a litter resulting from an intercross of Hoxc13GFPneo allele heterozygotes. Southern transfer analysis of BamHI-digested DNA was performed and shows two wild-type offspring carrying only the 6.5-kb fragment, three heterozygotes carrying both the 6.5- and 5.1-kb fragments, and three homozygous mutant offspring carrying only the 5.1-kb fragment.
Fluorescence Imaging.
Embryos derived from heterozygous intercrosses or crosses between heterozygous males and C57BL/6J females were harvested at gestational ages ranging from embryonic day (E) 7–9 for the Hoxa1GFPneo allele. Hoxc13GFPneo embryos were collected at E 11.5–17.5. Embryos were harvested, maintained, and imaged in Leibovitz’s L-15 media lacking phenol red (GIBCO/BRL).
Embryos or tissue samples were either pinned to Sylgard (Dow Corning) plates or placed in glass depression slides under coverslips before imaging. GFP fluorescence was recorded using a Bio-Rad MRC 1024 Laser Scanning Confocal Imaging System fitted to a Leitz Aristoplan microscope. Standard fluorescein isothiocyanate activation wavelengths were used for the excitation of the GFP fluorochrome with a digital Kalman averaging filter to reduce random noise.
RESULTS
Vector Construction and Generation of Mutant Mice.
We constructed a general cassette for use in generating GFP fusion genes by homologous recombination. The cassette contains an optimized GFP gene (6) that lacks the initiating methionine codon. Any one of three enzymes (see Materials and Methods) can be used to isolate the GFP gene, starting with the second codon, as well as the MC1neo gene (21) flanked by loxP sites. The initiation methionine codon of GFP was removed so that GFP expression requires fusion with the target gene. The GFP initiation methionine is not needed for GFP activity in fusion gene constructs (26–28). We used this cassette to generate targeting vectors for both Hoxa1 and Hoxc13. In brief, the Hoxa1 allele was designed to generate a protein fusion in the correct reading frame for all previously described Hoxa1 splice variants (refs. 29 and 30; see Fig. 1). The Hoxc13 allele generates a protein fusion at the same position that was used for the previously described lacZ fusion allele (ref. 23; see Fig. 2).
Targeting vectors were electroporated into R1 ES cells (24), which were subjected to positive–negative selection as described (22). Homologous recombination between the targeting vector and the Hoxa1 locus was assessed by Southern transfer analysis of EcoRV digested genomic DNA (Fig. 1). Ten of 143 independent cell lines demonstrated homologous recombination. Germline transmission was demonstrated in agouti offspring of chimeras by the same Southern transfer analysis (Fig. 1B). Finally, removal of the neomycin resistance gene by Cre-mediated site-specific recombination was confirmed by Southern transfer analysis (Fig. 1C).
Eleven of 144 independent cell lines surviving positive–negative selection (22) had undergone homologous recombination for the Hoxc13GFPneo allele as assayed by Southern transfer analysis (Fig. 2B). The removal of the neomycin gene by Cre-mediated site-specific recombination is shown in Fig. 2C. The same BamHI restriction fragments used to characterize the targeted ES cell line were diagnostic for genotyping progeny of heterozygous intercrosses (Fig. 2D).
Expression of the Hoxa1GFPneo Allele.
Fluorescence due to expression of the Hoxa1-GFP fusion protein was detectable as early as E 6.5 (data not shown). Some autofluorescence was visible in wild-type embryos in regions proximal to sites of Hoxa1GFPneo expression. However, while the extraembryonic endoderm autofluoresced at E 7.0, it became undetectable by E 8.25 (Fig. 3A–D). By E 8.25, the expression of the Hoxa1GFPneo fusion gene recapitulates the expression pattern of Hoxa1, labeling both the presumptive rhombomere 3/4 region of the developing hindbrain and the posterior regression of primitive streak into the caudal neural tube (Fig. 3C). Notably at E 8.75, GFP expression could no longer be detected in the hindbrain region of either heterozygous or homozygous embryos. This finding suggests that the Hoxa1GFPneo fusion gene is regulated similarly to the normal Hoxa1 gene, with no persistence of signal.
Figure 3.
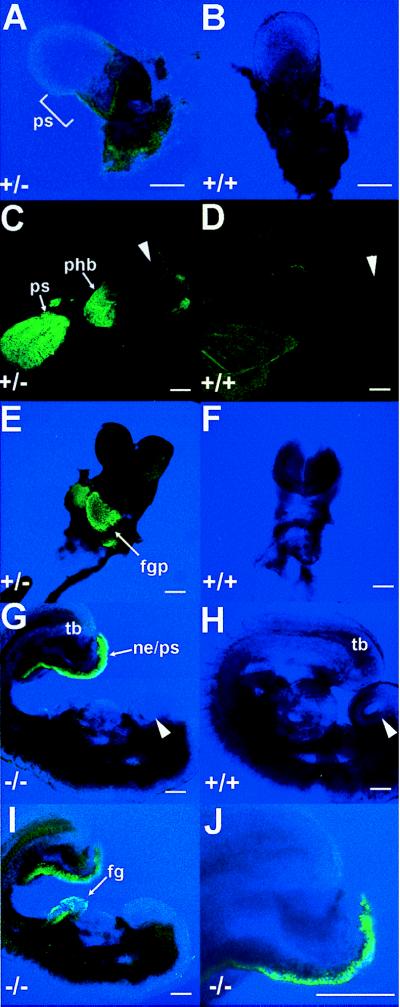
Embryonic expression of the Hoxa1GFPneo allele. (A) Expression in Hoxa1GFPneo heterozygote at E 7.0 in the primitive streak (ps). (B) Autofluorescence of extra-embryonic tissues in a wild-type embryo at E 7.0. Note same level of autofluorescence in a heterozygote (A). (C) Expression at E 8.25 in Hoxa1GFPneo heterozygote in rhombomere 3/4 of the presumptive hindbrain (phb), the regressing primitive streak (ps), and tail bud. Arrowhead indicates dorsal headfold region. (D) Lack of autofluorescence in a wild-type embryo at E 8.25 by using the same microscopic conditions as in C. Arrowhead indicates dorsal headfold region. (E) Expression in the developing foregut pocket (fgp) at E 8.75 in a Hoxa1GFPneo heterozygote. (F) Lack of fluorescence in a wild-type embryo at E 8.75 by using the same microscopic conditions as in E. (G) Expression in a Hoxa1GFPneo homozygote at E 9.0 in the caudal most region of the embryo in the nephrogenic duct (ne), tail bud (tb), and primitive streak (ps). Arrowhead indicates nasal placode. (H) Lack of fluorescence in wild-type embryo at E 9.0 by using the same microscopic conditions as in G and I. Arrowhead indicates nasal placode. (I) Expression in a Hoxa1GFPneo homozygote at E 9.0 in the foregut (fg) and tail bud (tb). (J) Higher magnification image of caudal expression showing single cell resolution. (For A–J, scale bar is 20 microns.)
Concomitant with reduction of the signal in the hindbrain at E 8.75, expression becomes detectable in the epithelium of the foregut pocket (Fig. 3E). In this region, differentiation of the cardiogenic mesoderm, gut-associated mesoderm, and foregut endoderm appear to be mediated by inductive processes of the foregut endoderm and/or visceral yolk sac endoderm (31–33). Hoxa1GFPneo expression also was detected in E 9.0 embryos in the most caudal regions of the neural tube and nephrogenic duct (Fig. 3G–J). GFP expression could not be detected in embryos older than E 9.5, confirming that the fusion gene again follows normal Hoxa1 expression and regulation in early embryos. A splice variant of the Hoxa1 gene expressed in adult intestine has been identified (34). Examination of adult intestines from wild-type and homozygous mutant mice revealed no GFP expression, although autofluorescence was seen in the intestinal vasculature in both wild-type and mutant samples. Initial experiments with Hoxa1GFPlox embryos (lacking the neomycin resistance gene) show the same level and pattern of expression as shown in Fig. 3C.
Expression of Hoxc13GFPneo and Hoxc13GFPlox Alleles.
Embryos were examined for expression in all spatial and temporal domains in which Hoxc13 expression was previously detected (23). At E 11.5 fluorescence in the tail was evident in the somites and the neural tube (Fig. 4A). By E 13.5, fluorescence in the tail was restricted to the outer layers of the tail and to a subregion of the somites (Fig. 4 B–D). The same laser intensity and detection conditions were used for the various genotypes and clearly show lack of significant background autofluorescence in the wild-type tail (Fig. 4B) and reduced fluorescence in the heterozygote (one Hoxc13GFPneo allele; Fig. 4C) as compared with the homozygote (two Hoxc13GFPneo alleles; Fig. 4D). Higher magnifications of E 13.5 tails showed expression in a sub-domain of the somites adjacent to the midline (Fig. 4E). In addition, the highest magnification of the tail (Fig. 4F) reveals that GFP expression can be resolved in single cells. The same pattern of expression in the tail was also detected using a Hoxc13GFPlox heterozygote (i.e., after removal of the neomycin gene; cf. Fig. 4 C and G). GFP expression also is seen in the nails and vibrissae at E 13.5 in a Hoxc13GFPlox heterozygote (Fig. 4H). At E 15.5 (Fig. 4 I–J), strong GFP expression fills much of the nail, although differences in signal width are already detectable depending on the focal plane of the laser. In addition, while 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-gal) staining of E 17.5, Hoxc13lacZneo embryos revealed an interesting talon-shaped expression pattern in the nails (23), optical sectioning of GFP embryos revealed that this area of Hoxc13 expression is restricted to a very thin cell layer. This cell layer is horizontal at the base of the nail and curves around the sides of the nail ventrally forming a curved planar sheet (data not shown). Furthermore, the restricted expression in the somites at E 13.5 (Fig. 4E) was masked by β-galactosidase (β-gal) staining in the outer layers of the tail (23). Thus, Hoxc13-GFP expression was detected in all domains previously described (23) including tail, nails, and vibrissae (see Fig. 4) as well as in the hair follicles and the filiform papillae of the tongue (data not shown). However, much higher resolution of the spatial aspects of Hoxc13 nail and tail expression was possible with the GFP allele.
Figure 4.

Expression of the Hoxc13GFPneo and Hoxc13GFPlox alleles. (A) Neural tube (nt) and somite (so) expression in the tail of a Hoxc13GFPneo heterozygote at E 11.5. (B) Tail expression in a wild-type embryo at E 13.5. The same microscopic conditions were used for B-D. (C) Tail expression in a Hoxc13GFPneo heterozygote at E 13.5. (D) Tail expression in a Hoxc13GFPneo homozygote at E 13.5. (E) Higher magnification of tail expression in Hoxc13GFPneo homozygote at E 13.5. (F) Higher magnification of tail expression in a Hoxc13GFPneo homozygote at E 13.5. (G) Tail expression in Hoxc13GFPlox heterozygote at E 13.5. (H) Vibrissae (arrowhead) and nail (arrow) expression in a Hoxc13GFPlox heterozygote at E 13.5. (I) Nail expression in a wild-type embryo at E 15.5. (J) Nail expression in a Hoxc13GFPneo heterozygote at E 15.5. The same microscopic conditions were used for I and J. Wild-type embryos showed no fluorescence under the same experimental conditions used for A, G, and H (data not shown). (Scale bar is 20 microns, except for H where it is 500 microns).
Hoxc13GFPlox homozygotes (i.e., from which the neomycin gene has been removed from both mutant alleles by CRE recombinase) are phenotypically indistinguishable from Hoxc13GFPneo homozygotes, as well as from the previously described Hoxc13neo and Hoxc13lacZneo homozygotes (Fig. 5A; ref. 23). This observation argues that all of the previously described mutant defects (23) in the formation of hair, nails, and filiform papillae are due to the absence of the Hoxc13 protein, rather than to interfering effects of the neomycin cassette on the transcription of neighboring genes. Interestingly, in contrast to Hoxc13neo or Hoxc13lacZneo heterozygotes (23), both Hoxc13GFPneo (Fig. 5C) and Hoxc13GFPlox (data not shown) heterozygotes have abnormal hair phenotypes. Notably, while the dorsal coat appears relatively normal, the ventral coat is sparse, especially in females. This suggests that some breakage of hair occurs in heterozygotes (Fig. 5C). In addition, at some stages of the hair cycle, lower portions of the face, especially below and behind the eyes, show patchy fur (Fig. 5D).
Figure 5.
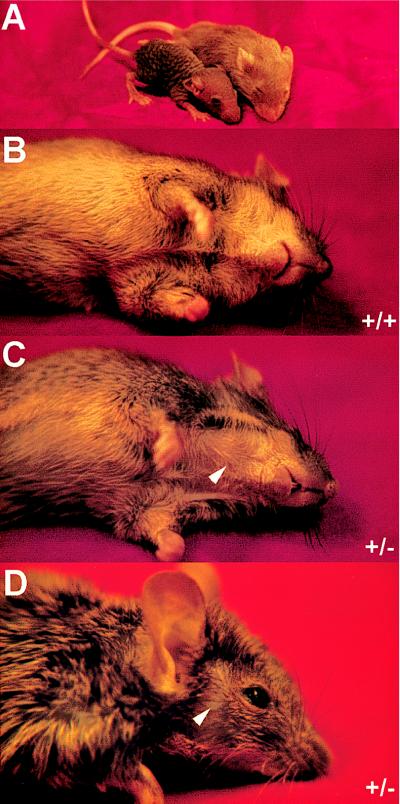
Hair phenotype of Hoxc13GFPneo and Hoxc13GFPlox mice. (A) Wild-type and Hoxc13GFPlox homozygous littermates at postnatal day 16. (B) Wild-type mouse ventral side. (C) Hoxc13GFPneo heterozygous littermate ventral side. Arrowhead indicates a region of fur that is patchy. (D) Hoxc13GFPneo heterozygous face. Arrowhead indicates a region of fur that is patchy.
It is possible that the phenotypic difference between the Hoxc13GFP and the Hoxc13lacZneo heterozygotes reflects the ability of the fusion proteins to interact with other proteins or the transcription complex. The large size of the β-gal fusion protein in either monomeric or tetrameric form may be sufficient to block a specific protein–protein interaction, while the smaller Hoxc13-GFP monomer can still interact leading to the observed hair phenotype in heterozygotes. An earlier study using a fibroblast growth factor receptor 1–β-gal fusion found transformation of cells due to receptor activation by β-gal tetramerization, while using fusion genes from which the tetramerization domain was removed did not transform cells (5). This example demonstrates that β-gal fusion protein tetramerization does have the potential to affect fusion protein function.
DISCUSSION
The use of targeted GFP fusion genes to track gene expression in living cells should enhance several avenues of research in murine development in general and Hox gene research in particular. For example, vital marking of cells containing targeted disruptions of specific genes will facilitate the analysis of cell autonomy of specific phenotypes, the analysis of cell migration, and examination of the contribution of cells expressing a given gene to the region affected by a mutation. In addition, since Leibovitz’s media appears to maintain embryo viability for at least 3 hr, analysis of gene expression in the same embryo at several time points may be feasible without special culture chambers. We have observed in early embryos (E 8–8.5) that sustained confocal laser imaging does exert some damage to embryonic tissues. Recently, we have found that combining digital imaging with the GFP 500/470 nm filter cube set available for the MZ12 dissecting microscope (Leica) or the SMZU stereo-microscope (Nikon) allows analysis of our single copy gene fusions without damage. Indeed, single copy Hoxc13GFPneo or Hoxc13GFPlox fusion gene expression is detectable in embryos even without the use of digital capture technology. However, the signal strength is greatly augmented with digital capture, e.g., using a 3CCD camera (Dage–MTI, Michigan City, IN). Finally, preliminary comparisons of GFPneo and GFPlox alleles for both Hoxa1 and Hoxc13 revealed that removal of the neomycin gene and its promoter/enhancer did not affect either the level or pattern of expression.
The Hoxa1 locus encodes two alternatively spliced mRNAs, which appear to be differentially expressed in the developing mouse embryo (34). From E 7–8.5, both transcripts are expressed equally within the neural tube in the presumptive rhombomere 3/4 region of the hindbrain. However, later in development (E 9.5) and continuing in the adult, only the non-homeodomain encoding mRNA is expressed within the developing foregut region and adult intestine. Aside from a uniform reduction in signal in heterozygotes, no temporal or spatial differences in Hoxa1GFPneo expression were detected between heterozygous and homozygous embryos. This finding suggests that the Hoxa1 gene product(s) are not required for maintenance of Hoxa1 expression.
The expression of the Hoxa1 fusion gene in the epithelium of the foregut pocket may provide a means to investigate inductive interactions between the endodermally derived gut epithelium and mesodermally derived cardiomyocytes. Of particular interest is the immediate proximity of the Hoxa1GFPneo-expressing cells and the cardiomyocytes within the foregut pocket. It is important to note that no cardiac malformations have been reported in mice with a targeted disruption of Hoxa1. However, given the role Hoxa1 has in patterning the hindbrain and lateral mesoderm, it is conceivable that this gene also might have either direct or indirect roles in patterning the foregut pocket and subsequent cardiac region. The fusion gene functions well as a vital marker early in hindbrain and foregut development. In particular, distinct tissue layers as well as individual cells expressing this vital marker are readily identifiable laying the foundation for future studies addressing cell autonomy within Hoxa1 mutants.
Because the Hoxc13lacZneo (23) and Hoxc13GFPneo fusion gene alleles disrupt the Hoxc13-coding region at exactly the same position, the two marker systems can be objectively compared. Both markers generate identical expression patterns that match RNA whole mount in situ analysis for Hoxc13 from E 9.5 to E 14.5 (23). Unexpectedly, the lacZ and GFP mutant alleles behave differently in heterozygous mice. This result illustrated that the marker gene itself can influence the behavior of the fusion gene allele by affecting parameters, such as accessibility to cellular components or mRNA and fusion protein stability. One major advantage of the GFP allele is the ability to reconstruct the detailed three-dimensional aspects of the expression patterns by using the optical sectioning capabilities of laser-based confocal systems, precluding the necessity to section or hemisect and clear β-gal-stained embryos. The Hoxc13GFPlox allele should prove useful not only for examining dynamic expression changes over time but also for isolating specific cell types expressing Hoxc13 for more detailed analysis.
In conclusion, use of targeted GFP fusion genes should add to the repertoire of techniques available for studies of gene expression and gene function. We have shown that expression from single-copy GFP-targeted alleles is detectable both early and late in embryogenesis. The ability to analyze the change in expression pattern over time in the same individual as well as the potential ability to use fluorescence-activated cell sorting to isolate pure populations of cells expressing a gene of interest fused to GFP should greatly aid understanding of Hox function.
Acknowledgments
We thank M. Allen, S. Barnett, J. Hayes, C. Lenz, G. Peterson, and M. Wagstaff for expert technical assistance, members of the Capecchi lab for critical reading of the manuscript and advice, and L. Oswald for help with preparation of the manuscript. A.R.G. and H.S.S. were associates of the Howard Hughes Medical Institute. A.R.G. also was supported, in part, by the G. Harold and Leila Y. Mathers Charitable Foundation.
ABBREVIATIONS
- GFP
green fluorescent protein
- E
embryonic day
- ES cell
embryonic stem cell
- Hox
homeobox containing
- HSV
herpes simplex virus
- β-gal
β-galactosidase
References
- 1. Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 2.Heim R, Prasher D C, Tsien R Y. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prasher D C. Trends Genet. 1995;11:320–323. doi: 10.1016/s0168-9525(00)89090-3. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson R H, Zhang X-J, DuBose R F, Matthews B W. Nature (London) 1994;369:761–766. doi: 10.1038/369761a0. [DOI] [PubMed] [Google Scholar]
- 5.Kouhara H, Kurebayashi S, Hashimoto K, Kasayama S, Koga M, Kishimoto T, Sato B. Oncogene. 1995;10:2315–2322. [PubMed] [Google Scholar]
- 6.Yang T-T, Cheng L, Kain S R. Nucleic Acids Res. 1996;24:4592–4593. doi: 10.1093/nar/24.22.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 8.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 9.Ikawa M, Kominami K, Yoshimura Y, Tanaka K, Nishimune Y, Okabe M. Dev Growth Differ. 1995;37:455–459. doi: 10.1046/j.1440-169X.1995.t01-2-00012.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhou L, Sun B, Zhang C-L, Fine A, Chiu S-Y, Messing A. Dev Biol. 1997;187:36–42. doi: 10.1006/dbio.1997.8601. [DOI] [PubMed] [Google Scholar]
- 11.Amsterdam A, Lin S, Moss L G, Hopkins N. Gene. 1996;173:99–103. doi: 10.1016/0378-1119(95)00719-9. [DOI] [PubMed] [Google Scholar]
- 12.Amesterdam A, Lin S, Hopkins N. Dev Biol. 1995;171:123–129. doi: 10.1006/dbio.1995.1265. [DOI] [PubMed] [Google Scholar]
- 13.Higashijima S-I, Okamoto H, Ueno N, Hotta Y, Eguchi G. Dev Biol. 1997;192:289–299. doi: 10.1006/dbio.1997.8779. [DOI] [PubMed] [Google Scholar]
- 14.Long Q, Meng A, Wang H, Jessen J R, Farrell M J, Lin S. Development (Cambridge, UK) 1997;124:4105–4111. doi: 10.1242/dev.124.20.4105. [DOI] [PubMed] [Google Scholar]
- 15.Meng A, Tang H, Ong B A, Farrell M J, Lin S. Proc Natl Acad Sci USA. 1997;94:6267–6272. doi: 10.1073/pnas.94.12.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zernicka-Goetz M, Pines J, Ryan K, Siemering K R, Haseloff J, Evans M J, Gurdon J B. Development (Cambridge, UK) 1996;122:3719–3724. doi: 10.1242/dev.122.12.3719. [DOI] [PubMed] [Google Scholar]
- 17.Zernicka-Goetz M, Pines J, McLean Hunter S, Dixon J P C, Siemering K R, Haseloff J, Evans M J. Development (Cambridge, UK) 1997;124:1133–1137. doi: 10.1242/dev.124.6.1133. [DOI] [PubMed] [Google Scholar]
- 18.Deng C, Bedford M, Li C, Xu X, Yang X, Dunmore J, Leder P. Dev Biol. 1997;185:42–54. doi: 10.1006/dbio.1997.8553. [DOI] [PubMed] [Google Scholar]
- 19.Friedrich G, Soriano P. Genes Dev. 1991;5:1213–1223. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 20.Mansour S L, Thomas K R, Deng C, Capecchi M R. Proc Natl Acad Sci USA. 1990;87:7688–7692. doi: 10.1073/pnas.87.19.7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas K R, Capecchi M R. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 22.Mansour S L, Thomas K R, Capecchi M R. Nature (London) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 23.Godwin A R, Capecchi M R. Genes Dev. 1998;12:11–20. doi: 10.1101/gad.12.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder J C. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwenk F, Baron U, Rajewsky K. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall J, Molloy R, Moss G W J, Howe J R, Hughes T E. Neuron. 1995;14:211–215. doi: 10.1016/0896-6273(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Zhang G, Ngo N, Zhao X, Kain S R, Huang C-C. J Biol Chem. 1997;272:28545–28549. doi: 10.1074/jbc.272.45.28545. [DOI] [PubMed] [Google Scholar]
- 28.Dopf J, Horiagon T M. Gene. 1996;173:39–44. doi: 10.1016/0378-1119(95)00692-3. [DOI] [PubMed] [Google Scholar]
- 29.Baron A, Featherstone M S, Hill R E, Hall A, Galliot B, Duboule D. EMBO J. 1987;6:2977–2986. doi: 10.1002/j.1460-2075.1987.tb02603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaRosa G J, Gudas L J. Mol Cell Biol. 1988;8:3906–3917. doi: 10.1128/mcb.8.9.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson A G, Sater A K. Development (Cambridge, UK) 1988;104:341–359. doi: 10.1242/dev.104.3.341. [DOI] [PubMed] [Google Scholar]
- 32.Stöhr P H. Wilhelm Roux’ Arch Entwicklungsmech Org. 1924;102:426–451. [Google Scholar]
- 33.Narita N, Bielinska M, Wilson D B. Dev Biol. 1997;189:270–274. doi: 10.1006/dbio.1997.8684. [DOI] [PubMed] [Google Scholar]
- 34.Murphy P, Hill R E. Development (Cambridge, UK) 1991;111:61–74. doi: 10.1242/dev.111.1.61. [DOI] [PubMed] [Google Scholar]