

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride ion channel, but its relationship to the primary clinical manifestation of CF, chronic Pseudomonas aeruginosa pulmonary infection, is unclear. We report that CFTR is a cellular receptor for binding, endocytosing, and clearing P. aeruginosa from the normal lung. Murine cells expressing recombinant human wild-type CFTR ingested 30–100 times as many P. aeruginosa as cells lacking CFTR or expressing mutant ΔF508 CFTR protein. Purified CFTR inhibited ingestion of P. aeruginosa by human airway epithelial cells. The first extracellular domain of CFTR specifically bound to P. aeruginosa and a synthetic peptide of this region inhibited P. aeruginosa internalization in vivo, leading to increased bacterial lung burdens. CFTR clears P. aeruginosa from the lung, indicating a direct connection between mutations in CFTR and the clinical consequences of CF.
Cystic fibrosis (CF) is caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR), a membrane protein involved in chloride ion secretion (1). Progressive loss of pulmonary function over many years caused by chronic bacterial infection with mucoid Pseudomonas aeruginosa is the hallmark of CF (2), yet the connection between lung infection and defects in chloride ion conductance has remained elusive. Most of the mutations in the gene encoding CFTR that lead to severe pulmonary disease result in a lack of CFTR protein in the membrane, although there are some rare exceptions (3). We have reported that CFTR modulated epithelial cell internalization of P. aeruginosa and not of other common bacterial respiratory pathogens (4), but these studies did not clearly identify the manner by which CFTR itself either interacted with P. aeruginosa or controlled cellular function to promote bacterial internalization. Internalization may promote bacterial clearance from the lung via cellular desquamation, but other responses subsequent to ingestion could also participate in the clearance of P. aeruginosa from the lung epithelium. Further understanding of the interaction of CFTR and this particular bacterial pathogen is clearly needed. Therefore, we extended our previous investigations into the role of CFTR in lung epithelial cell ingestion and clearance of P. aeruginosa to gain further insight into mechanisms of host defense against infection.
MATERIALS AND METHODS
Epithelial Cell Ingestion Assays and Bacterial Strains.
P. aeruginosa ingestion by parental and transfected murine C127 epithelial cells (5), transformed human CFT1-LCFSN epithelial cells (6, 7), WI-38 human embryonic lung cells, A549 human lung carcinoma cells, and suspensions of mouse lung cells was accomplished by using gentamicin-exclusion assays as described previously (4, 8). In these assays, addition of gentamicin to cell cultures 3 hr after infection with P. aeruginosa kills extracellular but not intracellular bacteria, which can be quantified by viability assays after washing away the antibiotic and lysing the cells with 0.05% Triton X-100 to release the intracellular bacteria. P. aeruginosa strains were PAO1, a laboratory strain, and 149 and 324, clinical isolates from infected CF patients obtained early in the course of infection [i.e., lipopolysaccharide (LPS) smooth, nonmuocid strains (9)].
Reagents Used in Inhibition of Ingestion Assays.
Membranes from transfected C127 cells and wild-type CFTR from transfected C127 cells were prepared and purified as described (10). Membranes and CFTR were suspended in the previously described solubilization buffer (10). mAbs (all IgM) used as inhibitors of ingestion were kindly provided by George Banting (University of Bristol, Bristol, U.K.) (11). mAb CF2 was raised to peptide 1467–1480 at the carboxy-terminal end of CFTR; mAb CF3 was raised to peptide 103–117 in the first predicted extracellular domain of CFTR; and mAb CF4 was raised to peptide 881–896 in the fourth predicted extracellular domain of CFTR. Percentage inhibition was determined in comparison with wells lacking any mAb. mAbs were tested for specificity of binding to CFTR by immunoblot using both cognate and noncognate peptides as inhibitors. Synthetic peptides were obtained commercially. Peptide GRIIASYDPDNKEER (15 aa) represents amino acids 103–117 of mature CFTR and the first predicted extracellular domain; peptide GKDPNYRDEAIRSIE is a scrambled version of peptide 103–117; peptide LWLLGNTPLQDKGNSTHSRNNSYAVIITST (30 aa) represents amino acids 881–910 of mature CFTR and the fourth predicted extracellular domain. A carboxy-terminal biotinylated version of peptide 103–117 was synthesized with an extra lysine (E) residue at the carboxy-terminus to which the biotin group was coupled via the free amino group of the additional lysine residue (biotin labeling the amino terminus abrogated biologic activity of this peptide in internalization–inhibition assays). Peptides were made up as stock solutions containing 1–10 nM peptide per μl of RPMI 1640 medium and diluted in medium before use in assays. All reagents were tested for toxicity on epithelial cells and on P. aeruginosa, and no evidence of toxicity was found.
Peptide Binding to P. aeruginosa LPS and Cells.
To assess the binding of CFTR-related peptides to the O-side-chain deficient, core-oligosaccharide-portion of the P. aeruginosa LPS, previously identified as the bacterial ligand for epithelial cell ingestion (4), LPS with a complete outer-core oligosaccharide (containing 9 monosaccharides) but no O side chains was isolated from P. aeruginosa strain PAC557. As a control, LPS lacking the outer portion of the core oligosaccharide, designated incomplete outer-core oligosaccharide (containing 4 monosaccharides), was isolated from strain PAC1RalgC::tet, which is isogenic to strain PAC557 (4). The structure of these oligosaccharides is shown in figure 2 of ref. 12. LPS (1 μg) and suspensions of P. aeruginosa or Escherichia coli HB101 cells (≈109 cfu) were immobilized on nylon membranes in 0.04 M phosphate buffer, pH 7.4. Membranes were blocked with 1% BSA and then probed with the biotinylated CFTR peptide 103–117. Avidin-alkaline phosphatase was used as a secondary reagent followed by the enzyme–substrate indicator reagent.
Mouse Infection Model.
Groups of 6–7 mice were infected with P. aeruginosa strain PAO1 as described (4, 13). Ten nanometers of CFTR peptide was added to the bacterial inoculum before its application to the nares of the mice. Determination of the total and internalized cfu per lung was as described (4).
Electron Microscopy.
Three hours after P. aeruginosa was added to epithelial cell cultures, cells were released from monolayers by trypsin treatment and collected by centrifugation, and a portion was resuspended in methanol at −20°C for 5 min for permeabilization and fixation. An additional portion of cells was placed directly in 2% glutaraldehyde fixative and then processed for electron microscopy. Cells for antibody treatment were washed three times with PBS containing 1% BSA; a 1:10 dilution of mAb CF8 (11) raised to peptide 1035–1050 of CFTR was then added to cells. Controls had no added antibody. After 1 hr at 37°C, cells were washed and goat antibodies to mouse IgM labeled with 30-nm gold particles were added, and this preparation was incubated for another hour. One aliquot of cells was treated with gold-labeled secondary antibody only. After three washes, cells were suspended in 2% glutaraldehyde and processed for electron microscopic observation.
Fluorescent and Light Microscopy.
Lungs from mice were placed in 2% glutaraldehyde immediately after removal from the chest cavity, embedded in paraffin, and sectioned for hematoxylin and eosin staining. Comparable sections were also used for fluorescence microscopy after sectioning and removal of the paraffin embedding. Twenty microliters of mAb CF3 (6 μg/ml in PBS) was placed on each section and incubated in a humidified chamber for 90 min. Some sections received mAb CF3 incubated for 30 min with 1 nM of its cognate peptide (amino acids 103–117 of CFTR) before placing the mAb on sections of lungs from infected mice for 90 min. After three washings with PBS, rabbit-anti mouse IgM conjugated to fluorescein isothiocyanate was added and incubated for 90 min in the humidified chamber. After three additional PBS washes the slides were covered with 0.1M N-propyl gallate to reduce photobleaching (14), sealed and examined at ×100 magnification by using a Nikon inverted microscope. Film was exposed using manual exposure settings to allow a sufficient amount of the background autofluorescence to reveal lung tissue structure and form a visible photograph.
RESULTS
CFTR Is an Epithelial Cell Receptor for Ingestion of P. aeruginosa.
Our previous studies showed that maximal ingestion of P. aeruginosa occurred in epithelial cells expressing functional membrane CFTR but did not specifically determine the mechanism underlying the enhanced cellular uptake. One possibility was that CFTR itself was the epithelial cell ligand for P. aeruginosa. To evaluate this hypothesis, we exposed murine C127 cells transfected with cDNA for wild-type human CFTR to three strains of P. aeruginosa and measured bacterial ingestion using gentamicin-exclusion assays wherein the antibiotic kills extracellular bacteria but allows intracellular bacteria to survive (4). Murine cells expressing wild-type human CFTR had a markedly enhanced ability to ingest P. aeruginosa compared with control cells lacking any CFTR or transfected with cDNA for mutant ΔF508 CFTR (Fig. 1A). When membranes from these cells were probed in an ELISA with mAb CF3, which binds to both human and mouse CFTR (see results in Figs. 2, human CFTR, and 6A, mouse CFTR), only membranes from cells expressing wild-type human CFTR bound the antibodies, confirming expression of only this human CFTR by the appropriately transfected C127 cell line (data not shown). Both cell membranes containing CFTR and highly purified (≈85%) recombinant CFTR inhibited uptake of P. aeruginosa PAO1 by the CFT1-LCFSN cell line derived from a CF patient homozygous for the ΔF508 mutation and subsequently transfected with wild-type CFTR cDNA (6, 7) (Fig. 1B). No effect on epithelial cell internalization of P. aeruginosa was observed when either insect cell membranes containing the CFTR-related protein, P glycoprotein from Leishmania, protein solubilization buffer, or doses of membranes with CFTR lower than those shown in Fig. 1B were added to assays. These results implicated CFTR as the epithelial cell ligand for endocytosis of P. aeruginosa.
Figure 1.

Determination that CFTR is the likely epithelial cell receptor for internalizing P. aeruginosa. (A) Enhanced bacterial uptake by murine epithelioid cells transfected with cDNA for wild-type human CFTR compared with uptake by cells expressing either no CFTR (parental) or ΔF508 human CFTR, as indicated in the legend. (B) Inhibition of uptake of P. aeruginosa ingestion by human CFT1-LCFSN-transformed airway epithelial cells in the presence of membranes from the C127 cells or purified wild-type human CFTR, as indicated in the legends. PgP indicates insect cell membranes expressing P-glycoprotein from Leishmania, and control represents insect cell membranes not expressing foreign proteins. Bars represent the means of 6–9 replicates, and error bars represent SD. Asterisks (∗) indicate points significantly different from others in the same groups by ANOVA (P < .001; P < .01 for all pairwise comparisons by Fisher’s probable least-squares difference).
Figure 2.

Effect of mAbs to CFTR on inhibition of internalization of three strains of P. aeruginosa. Legend indicates specificity of the mAbs. (A) P. aeruginosa strain 324. (B) P. aeruginosa strain PAO1. (C) P. aeruginosa strain 149. Bars indicate the means of 6–9 replicate determinations, and error bars represent SD. Asterisks (∗) indicate the inhibitors shown by multiple comparisons to significantly decrease internalization (P ≤ 0.001, ANOVA; P ≤ 0.01 Fisher’s probable least-squares difference for pairwise comparisons).
Identification of the First Extracellular Domain of CFTR as the Ligand for P. aeruginosa.
To confirm that CFTR was the cellular ligand for P. aeruginosa ingestion and to identify the extracellular domain of CFTR that interacts with P. aeruginosa, we performed inhibition-of-ingestion assays by using mAbs raised to synthetic peptides corresponding to the first (mAb CF3) and fourth (mAb CF4) predicted extracellular domains of CFTR as well as a control mAb specific to a peptide representing the intracellular carboxy-terminal 14 aa of mature CFTR (mAb CF2) (11). These mAbs were added in various concentrations to cultures of CFT1-LCFSN cells before addition of the P. aeruginosa organisms (4). A concentration-dependent inhibition of P. aeruginosa internalization was observed only with mAb CF3 (Fig. 2), indicating that the first predicted extracellular domain of CFTR, comprising amino acids 103–117, is a binding site on CFTR for P. aeruginosa.
To verify this interaction, synthetic peptides corresponding to the first (amino acids 103–117) and fourth (amino acids 881–910) extracellular domains were used in internalization–inhibition assays, as was a scrambled version of the first domain peptide. Only the peptide containing the correct sequence of the first predicted extracellular domain of CFTR inhibited epithelial cell internalization of P. aeruginosa (Fig. 3A–C). When we used both the mAbs and the same peptides in studies with two additional cell lines homozygous for wild-type CFTR (WI-38 diploid human embryonic lung cells and A549 human lung carcinoma cells), we found that ingestion of P. aeruginosa could be inhibited by the same reagents as those that inhibited uptake by CFT1-LCFSN cells (data not shown).
Figure 3.

Effect of synthetic peptides of CFTR on inhibition of internalization of three strains of P. aeruginosa. Legend indicates peptide used as inhibitor. (A) P. aeruginosa strain 324. (B) P. aeruginosa strain PAO1. (C) P. aeruginosa strain 149. Points indicate the means of 6–9 replicate determinations, and error bars represent SD. Asterisks (∗) indicate the inhibitors shown by multiple comparisons to significantly decrease internalization (P ≤ 0.001; ANOVA, P ≤ 0.01 Fisher’s probable least-squares difference for pairwise comparisons). (D) Binding of carboxy-terminal biotinylated peptide comprising amino acids 103–117 of CFTR to purified complete or incomplete-core LPS from P. aeruginosa, P. aeruginosa cells, or control E. coli cells.
Binding of CFTR Peptide to P. aeruginosa LPS and Cells.
When immuno-dot blots were used to visualize direct binding of biotinylated CFTR peptide 103–117 to P. aeruginosa cells and the bacterial ligand for epithelial cells, complete-core LPS (4), we observed binding to these entities but not to incomplete-core LPS, which does not inhibit internalization of P. aeruginosa by epithelial cells, or to control E. coli cells (Fig. 3D). Binding of the biotinylated peptide was completely inhibited by unlabelled peptide 103–117 but not by the scrambled 15-aa peptide (data not shown). This confirmed the ligand–receptor nature of the interaction of P. aeruginosa complete-core LPS and CFTR residues 103–117.
Blocking of CFTR Binding to P. aeruginosa in Vivo Decreases Lung Clearance.
Tang et al. (13) showed that placing P. aeruginosa onto the nares of neonatal BALB/C mice results in lung infection along with extensive epithelial-cell uptake of bacteria. Thus, this model would be useful to determine whether binding and endocytosis of P. aeruginosa by the first predicted extracellular domain of CFTR is important in resistance to lung infection and would not likely be confounded by ingestion of P. aeruginosa by phagocytic cells, because this results in rapid bacterial killing (15, 16). Because the first extracellular domain of CFTR differs between humans and mice at two positions (D112E and V115E, human to mouse), we first confirmed that the human peptide inhibited P. aeruginosa ingestion by mouse lung cells, by using single-cell suspensions derived from whole mouse lungs (4). Although we could not verify which cell type ingested P. aeruginosa, including the first-extracellular-domain peptide of human CFTR in the bacterial inoculum reduced mouse lung cell uptake of P. aeruginosa by >95% (from ≈105 cfu of P. aeruginosa ingested per 106 mouse lung cells to <5 × 103 per 106 cells). None of the other control peptides inhibited mouse cell uptake of P. aeruginosa. Thus, it is likely that intact mouse lung cells efficiently ingest P. aeruginosa using murine CFTR in a manner analogous to cells expressing human CFTR.
To determine whether CFTR-mediated ingestion of P. aeruginosa in vivo affected bacterial burdens in the lung, we nasally inoculated 7-day-old BALB/C mice with 108 cfu of P. aeruginosa strain PAO1 mixed with either nothing, with 10 nM of the peptide corresponding to the first predicted extracellular domain of human CFTR, with 10 nM of the scrambled version of the first domain peptide, or with 10 nM of the fourth predicted extracellular domain of human CFTR. Twenty-four hours after infection, mice inoculated with bacteria plus the first-extracellular-domain peptide had virtually no internalized P. aeruginosa, whereas mice receiving the bacteria along with either the fourth extracellular domain peptide, no peptide, or the scrambled version of the first-domain peptide had a median of >105 cfu of P. aeruginosa internalized per gram of lung tissue (Fig. 4A). As a consequence of this inhibition of internalization, mice infected with bacteria and the first extracellular domain peptide had ≥3.3 × 106 more cfu of P. aeruginosa per gram of lung tissue (Fig. 4B) than the mice infected with bacteria plus either no peptide or the control peptides. Smaller but still significant differences in total bacterial counts per gram of lung tissue were found 14 hr and 48 hr after infection (data not shown). Thus, inhibition of P. aeruginosa internalization by blocking the bacterial interaction with murine CFTR in the lung led to increased bacterial counts in this tissue.
Figure 4.

Effect of addition of synthetic peptides to the bacterial inoculum on P. aeruginosa infection in neonatal mice. (A) Amount of P. aeruginosa internalized by lung cells 24 hr after infection. (B) Total amount of P. aeruginosa found in lungs 24 hr after infection. Box plots indicate (from bottom to top) the 10th, 25th, 50th (median), 75th, and 90th percentiles. Circles above or below the 90th or 10th percentile indicate individual points outside this range. There were 12–14 total lung samples used in each group. For both groups of comparisons (A and B), the overall differences were significant at P < 0.001 using the Kruskal–Wallis nonparametric ANOVA test, and the difference between the group receiving the first-domain peptide and the other three groups was significant at P < .001 using the Dunn procedure for pairwise comparisons.
Microscopy Reveals CFTR Accumulating in Cell Membranes and Binding to P. aeruginosa.
Because expression of CFTR in uninfected lung epithelium appears to be low, we used electron microscopy to investigate how CFTR could function in binding and internalizing P. aeruginosa. Previous results (4) showed that the initial binding of P. aeruginosa to lung cells is not inhibited by the free bacterial ligand (LPS) that inhibits epithelial cell ingestion of P. aeruginosa, indicating that the initial anchoring of bacteria to cell membranes does not involve LPS binding to CFTR. Therefore, CFTR must accumulate in the cell membrane subsequent to the initial bacterial attachment to a level sufficient to bind and internalize P. aeruginosa. To substantiate this idea we permeabilized CFT1-LCFSN cells ingesting P. aeruginosa with methanol then added mAb CF8 followed by anti-mouse IgM labeled with 30-nm gold particles to visualize the interaction of bacteria and CFTR. Cells ingesting P. aeruginosa showed heavy accumulation of the gold label in clusters at a single contact point on the outer surface of the bacteria (Figs. 5A–B). Epithelial cells not in close contact with P. aeruginosa had much less CFTR visualized in their membranes as well as gold label binding to immunoreactive material in cytoplasmic vesicles (Fig. 5C). Controls lacking primary antibodies were devoid of gold label (Fig. 5D). Because methanol permeabilization disrupts the fine internal structure of the cells, we used standard electron microscopic techniques to visualize the vesicles containing P. aeruginosa within CFT1-LCFSN cells. Consistent with the observations shown in Fig. 5 A–B that CFTR accumulates on a small portion of the P. aeruginosa cell surface, bacteria in cytoplasmic enclosures had only a portion of the surface in contact with the vacuolar membrane (Figs. 5 E–G). The parental CFT1 cells, homozygous for the ΔF508 CFTR gene, poorly ingest P. aeruginosa, and we saw no bacterial interaction with these cells by electron microscopy (Fig. 5H).
Figure 5.

Electron microscopic visualization of CFTR interacting with P. aeruginosa. (A and B) P. aeruginosa cells are seen with accumulations of CFTR at a single point on the bacterial surface (arrow). (Bar = 0.1 μM; gold particles are 30 nm.) (C) Epithelial cells not ingesting P. aeruginosa showed primarily intracytoplasmic CFTR (open arrows) and membrane CFTR usually only bound to one or two gold particles (closed arrows) and, on a rare occasion, small aggregations of gold particles (closed arrowhead). (Bar = 0.5 μM; gold particles are 30 nm.) (D) Control where the primary antibody to CFTR was omitted showing epithelial cell with internalized P. aeruginosa bacteria (indicated by “b” on figure). (Bar = 0.1 μM.) (E–G) Epithelial cells not treated with methanol (used to permeabilize the cells in A–D for intracytoplasmic antibody reactions) showed P. aeruginosa in intracytoplasmic enclosures with only a portion of the bacterial cell surface attached to the vesicle membrane (arrow), similar to the accumulation of CFTR on the bacterial surface (A and B). (Bar = 0.1 μM in E; 0.5 μM in F and G.) (H) Epithelial cells homozygous for the ΔF508 CFTR mutation could not be seen ingesting P. aeruginosa. (Bar = 1.0 μM.)
Further documentation that CFTR accumulates in airway epithelial cells as part of the host response to P. aeruginosa infection was obtained by fluorescent microscopic analysis of fixed sections of lungs removed from infected neonatal mice. The sections were first reacted with mAb CF3, which binds to the first extracellular domain of CFTR, then with fluorescein-conjugated anti-mouse IgM. Mice infected with P. aeruginosa had brightly stained areas of fluorescence in bronchial epithelial cells (Fig. 6A), and the tissues surrounding the airways were highly congested because of the accumulation of host inflammatory cells that accompanies acute P. aeruginosa lung infection (Fig. 6B). As a specificity control, we incubated mAb CF3 with the first extracellular domain peptide of CFTR before staining lung sections, and this resulted in no visible fluorescence in airway epithelial cells (Fig. 6C). In addition, mAb CF3 showed virtually no staining of bronchial epithelial cells in sections of lungs from mice infected 24 hr previously with P. aeruginosa along with the first extracellular domain peptide that inhibited CFTR-mediated cellular internalization of the bacteria in vivo (Fig. 6D). Sections of lung from uninfected neonatal mice showed little to no CFTR present in the airway epithelium and a healthy-appearing histologic profile (Figs. 6 E and F).
Figure 6.
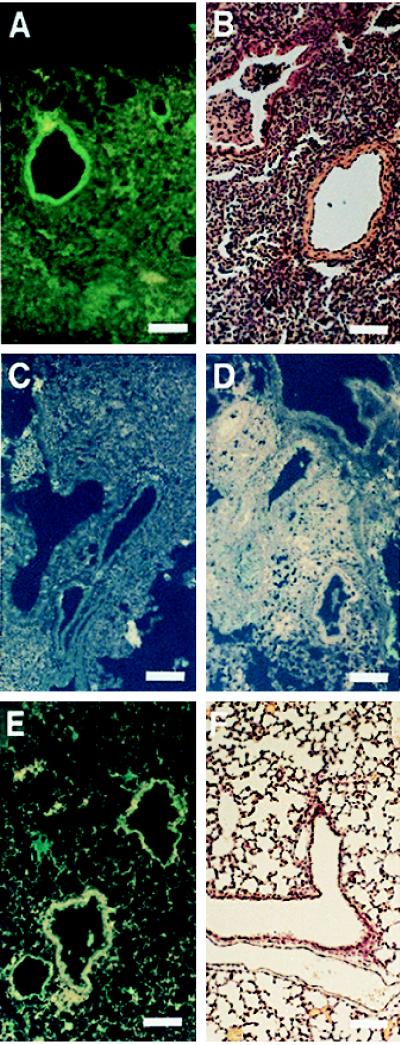
Immunofluorescence of CFTR in lung sections of mice infected with P. aeruginosa. (Bars = 100 μM.) (A) CFTR visualized by mAb CF3 and fluoresceinated secondary antibody in the airway epithelium of mice infected with P. aeruginosa 24 hr previously. (B) Hematoxylin and eosin stain of a lung section from a neonatal mouse infected with P. aeruginosa. (C) Visualization of CFTR by mAb CF3 and fluoresceinated secondary antibody in the airway epithelium of mice infected with P. aeruginosa 24 hr previously was inhibited if the mAb was first incubated with the first extracellular domain synthetic peptide (amino acids 103–117 of CFTR). (D) CFTR could not be visualized by mAb CF3 and fluoresceinated secondary antibody in the airway epithelium of mice infected 24 hr previously with P. aeruginosa along with a synthetic peptide corresponding to the first predicted extracellular domain of CFTR. This synthetic peptide inhibited CFTR-mediated epithelial cell internalization of P. aeruginosa and the subsequent clearance of the bacterium from the lung (see Fig. 4). (E) Staining of uninfected mouse lung section with mAb CF3 and fluoresceinated secondary antibody. Little to no CFTR is seen in the epithelium. (F) Hematoxylin and eosin stain of a lung section from an uninfected neonatal mouse.
DISCUSSION
The identification of CFTR as a cellular receptor for internalization of P. aeruginosa indicates an additional function for this molecule, which to date has been characterized primarily as an ion channel. We are unaware of any other reports indicating that an ion channel can also serve as a receptor for bacterial binding and endocytosis. Interestingly, the amino terminus of CFTR up to amino acid 150, including the first predicted extracellular domain, can be deleted from the molecule without markedly affecting its ability to function as a chloride ion channel (17). Thus, CFTR-mediated cellular internalization of P. aeruginosa is unrelated to the ion-channel properties of this molecule. CFTR is not likely to be the initial cellular receptor for P. aeruginosa binding; the normal level of membrane expression of CFTR appears to be too low for effective initial binding of bacteria, and factors that inhibit or modulate P. aeruginosa internalization by CFTR have little effect on bacterial adherence to epithelial cells (4, 12). P. aeruginosa probably binds initially to cells using multiple bacterial ligands, including pili, flagella, LPS, and a nonpilus adhesin (18–20), and likely a variety of cellular receptors such as asialoganglioside 1 (21, 22). Once P. aeruginosa is bound to epithelial cells, CFTR accumulates in the cell membrane at a specific point of contact with the bacterial surface.
At present we do not know the exact mechanism whereby CFTR-mediated cellular interactions with P. aeruginosa leads to bacterial clearance; we have postulated that cellular uptake followed by shedding could clear the organisms from the airways (4), but we have no specific data that indicate to what degree this particular aspect of the binding and ingestion process promotes P. aeruginosa clearance from lungs. CFTR-mediated ingestion of P. aeruginosa may evoke additional cellular and tissue inflammatory responses that participate in bacterial removal from the lung by other mechanisms. For example, the activity of the human Kv1.5 potassium channel, like CFTR, is regulated by phosphorylation, and it has recently been shown that the potassium channel associates with Src tyrosine kinase via SH3 domains, indicating that an ion channel can form a signaling complex with protein kinases (23). If a signaling complex were formed by CFTR and other cellular regulatory factors when P. aeruginosa was present in the airway, this could be a critical part of the respiratory epithelium’s response to this pathogen that results in its elimination from the lung.
The binding to P. aeruginosa and accumulation of additional CFTR in bronchial epithelial cells could also be part of the mechanism recently proposed by Smith et al. (24) for eliminating bacterial pathogens inhaled onto the airway epithelium. These workers showed that in normal airway fluid there is a putative antimicrobial factor that kills a variety of bacterial pathogens and this killing activity is inhibited in the extracellular airway fluid of CF patients due to increased concentrations of NaCl. To achieve maximal bactericidal activity even on normal airway surfaces, it may be necessary to increase the amount of CFTR in the apical membrane of the epithelial cells to keep the NaCl concentration sufficiently low for the antimicrobial factor to be effective. This requirement would be consistent with the increased levels of CFTR observed in response to P. aeruginosa infection. Obviously, mutations in CFTR that limit or prevent the appearance of functional protein in the cell membrane (e.g., the common ΔF508 mutation) could result in defective clearance of P. aeruginosa, as could mutations in CFTR genes that result in the expression of CFTR protein in the membrane that could bind P. aeruginosa (i.e., G551D), but either fail to accumulate in response to P. aeruginosa infection or, because they are defective in ion-channel activity, cannot maintain the airway epithelium in a state that resists bacterial infection. Such a scenario would require that CFTR be both intact and have functional ion-channel activity for removal of P. aeruginosa from the airways.
Further evaluation of these concepts in transgenic mice either lacking murine CFTR (25) or bearing a CFTR gene with the ΔF508 mutation (26) would provide additional confirmation of the findings reported here. We have been breeding ΔF508 CFTR mice initially produced by Colledge et al. (26) and have performed some preliminary experiments in these animals. Neonatal mice carrying either one or two copies of the ΔF508 mutant CFTR from these breedings are not susceptible to P. aeruginosa lung infection following nasal application, as opposed to BALB/C mice. We also have observed that other inbred strains of mice such as C57BL/6 mice are not susceptible to infection by this route (unpublished observation). It is of interest that the MHC haplotype of the resistant and transgenic ΔF508 CFTR mice (H-2b) is different from that of the susceptible BALB/C mice (H-2d), suggesting a possible immunologic basis for these results. Further evaluations in older ΔF508 CFTR mice infected by intratracheal instillation of P. aeruginosa are in progress, but we have already noted a high level of variability in bacterial levels in the lungs of individual animals from different litters, which would represent mice with different genetic compositions. This finding is consistent with the observations that genetic factors in addition to CFTR control the host response to P. aeruginosa infection. Indeed, differences in overall survival between ΔF508 CFTR mice bred in different backgrounds have been reported (26, 27), and the wide variation in pulmonary phenotype of humans homozygous for this CFTR mutation is well documented (28).
Overall, it appears that a specific interaction between P. aeruginosa and the first extracellular domain of CFTR triggers CFTR-mediated resistance to infection in individuals who have wild-type CFTR. Lack of this interaction and lack of a functional CFTR protein in most CF patients could contribute significantly to the respiratory manifestations of CF. Documenting that CFTR is a receptor that specifically clears P. aeruginosa but not other common respiratory pathogens from the lung (4) provides a clear connection between the most common mutations in CFTR leading to a lack of functional membrane protein and the pulmonary infection typical of severe CF. We expect further details of the mechanisms of CFTR-mediated clearance of P. aeruginosa to come from studies of cell lines and animals with mutations such as G551D in the CFTR gene (29). This mutation results in membrane protein that is defective in chloride ion conductance and, in humans often, but not always, is associated with severe pulmonary disease (30). Determining that CFTR has an important function in addition to that of a chloride ion channel extends the basic knowledge about this protein and also indicates that ion channels may be capable of having multiple, seemingly unrelated activities that are critical for the overall normal physiologic function of an organism.
Acknowledgments
We thank Alan Smith and colleagues at Genzyme (Cambridge, MA) for the C127 cell lines and purified recombinant CFTR; James Croop of the Dana–Farber Cancer Hospital (Boston, MA) for membranes containing P glycoprotein; George Banting of the University of Bristol (Bristol, U.K.) for mAbs CF2, CF3, CF4, and CF8; Gloria Meluleni of the Channing Laboratory and Bonnie Meek of the Harvard School of Public Health (Boston, MA) for electron microscopic services; and Ervin Meluleni of the Harvard School of Public Health for processing samples for light and fluorescence microscopy. This work was supported by the National Institute of Allergy and Infectious Diseases (AI 22806) and by the Cystic Fibrosis Foundation.
ABBREVIATIONS
- CF
cystic fibrosis
- CFTR
CF transmembrane conductance regulator
- LPS
lipopolysaccharide
Footnotes
To whom reprint requests should be addressed at: Channing Laboratory, 181 Longwood Avenue, Boston, MA 02115-5899. e-mail: gpier@channing.harvard.edu.
References
- 1.Welsh M J, Anderson M P, Rich D P, Berger H A, Sheppard D N. In: The CFTR Chloride Channel. Guggino W B, editor. San Diego: Academic; 1994. pp. 153–171. [Google Scholar]
- 2.Cystic Fibrosis Foundation. Patient Registry 1995 Annual Data Report. MD: Bethesda; 1996. [Google Scholar]
- 3.Kerem E, Reisman J, Corey M, Canny G J, Levison H. N Engl J Med. 1992;326:1187–1191. doi: 10.1056/NEJM199204303261804. [DOI] [PubMed] [Google Scholar]
- 4.Pier G B, Grout M, Zaidi T S, Olsen J C, Johnson L G, Yankaskas J R, Goldberg J B. Science. 1996;271:64–67. doi: 10.1126/science.271.5245.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng S H, Fang S L, Zabner J, Marshall J, Piraino S, Schiavi S C, Jefferson D M, Welsh M J, Smith A E. Am J Phys Lung Cell Mol Physiol. 1995;12:L615–L624. doi: 10.1152/ajplung.1995.268.4.L615. [DOI] [PubMed] [Google Scholar]
- 6.Sarkadi B, Bauzon D, Huckle W R, Earp H S, Berry A, Suchindran H, Price E M, Olsen J C, Boucher R C, Scarborough G A. J Biol Chem. 1992;267:2087–2095. [PubMed] [Google Scholar]
- 7.Olsen J C, Johnson L G, Stutts M J, Sarkadi B, Yankaskas J R, Swanstrom R, Boucher R C. Hum Gene Ther. 1992;3:253–266. doi: 10.1089/hum.1992.3.3-253. [DOI] [PubMed] [Google Scholar]
- 8.Fleiszig S M J, Zaidi T S, Pier G B. Infect Immun. 1995;63:4072–4077. doi: 10.1128/iai.63.10.4072-4077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pier G B, DesJardins D, Aguilar T, Barnard M, Speert D P. J Clin Microbiol. 1986;24:189–196. doi: 10.1128/jcm.24.2.189-196.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Riordan C R, Erickson A, Bear C, Li C H, Manavalan P, Wang K X, Marshall J, Scheule R K, Mcpherson J M, Cheng S H, Smith A E. J Biol Chem. 1995;270:17033–17043. doi: 10.1074/jbc.270.28.17033. [DOI] [PubMed] [Google Scholar]
- 11.Walker J, Watson J, Holmes C, Edelman A, Banting G. J Cell Sci. 1995;108:2433–2444. doi: 10.1242/jcs.108.6.2433. [DOI] [PubMed] [Google Scholar]
- 12.Zaidi T S, Fleiszig S M J, Preston M J, Goldberg J B, Pier G B. Invest Ophthalmol Visual Sci. 1996;37:976–986. [PubMed] [Google Scholar]
- 13.Tang H, Kays M, Prince A. Infect Immun. 1995;63:1278–1285. doi: 10.1128/iai.63.4.1278-1285.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giloh H, Sedat J W. Science. 1982;217:1252–1255. doi: 10.1126/science.7112126. [DOI] [PubMed] [Google Scholar]
- 15.Eshima I, Mathes S J, Paty P. Plast Reconstr Surg. 1990;86:541–547. doi: 10.1097/00006534-199009000-00026. [DOI] [PubMed] [Google Scholar]
- 16.Pierangeli S S, Sonnenfeld G. Clin Exp Immunol. 1993;93:165–171. doi: 10.1111/j.1365-2249.1993.tb07960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carroll T P, Morales M M, Fulmer S B, Allen S S, Flotte T R, Cutting G R, Guggino W B. J Biol Chem. 1995;270:11941–11946. doi: 10.1074/jbc.270.20.11941. [DOI] [PubMed] [Google Scholar]
- 18.Saiman L, Prince A. J Clin Invest. 1993;92:1875–1880. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson D A, Ramphal R, Lory S. Infect Immun. 1992;60:3771–3779. doi: 10.1128/iai.60.9.3771-3779.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simpson D A, Ramphal R, Lory S. Infect Immun. 1995;63:2950–2957. doi: 10.1128/iai.63.8.2950-2957.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krivan H C, Roberts D D, Ginsburg V. Proc Natl Acad Sci USA. 1988;85:6157–6161. doi: 10.1073/pnas.85.16.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imundo L, Barasch J, Prince A, Al-Awqati Q. Proc Natl Acad Sci USA. 1995;92:3019–3023. doi: 10.1073/pnas.92.7.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes T C, Fadool D A, Ren R, Levitan I B. Science. 1996;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 24.Smith J J, Travis S M, Greenberg E P, Welsh M J. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 25.Snouwaert J N, Brigman K K, Latour A M, Malouf N N, Boucher R C, Smithies O, Koller B H. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 26.Colledge W H, Abella B S, Southern K W, Ratcliff R, Jiang C W, Cheng S H, Macvinish L J, Anderson J R, Cuthbert A W, Evans M J. Nat Genet. 1995;10:445–452. doi: 10.1038/ng0895-445. [DOI] [PubMed] [Google Scholar]
- 27.Van Doorninck J H, French P J, Verbeek E, Peters R H P C, Morreau H, Bijman J, Scholte B J. EMBO J. 1995;14:4403–4411. doi: 10.1002/j.1460-2075.1995.tb00119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerem E, Corey M, Kerem B S, Rommens J, Markiewicz D, Levison H, Tsui L C, Durie P. N Engl J Med. 1990;323:1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 29.Delaney S J, Alton E W F W, Smith S N, Lunn D P, Farley R, Lovelock P K, Thomson S A, Hume D A, Lamb D, Porteous D J, Dorin J R, Wainwright B J. EMBO J. 1996;15:955–963. [PMC free article] [PubMed] [Google Scholar]
- 30.Parad R B. J Med Genet. 1996;33:711–713. doi: 10.1136/jmg.33.8.711. [DOI] [PMC free article] [PubMed] [Google Scholar]