

Abstract
The pufferfish Fugu rubripes has a genome ≈7.5 times smaller than that of mammals but with a similar number of genes. Although conserved synteny has been demonstrated between pufferfish and mammals across some regions of the genome, there is some controversy as to what extent Fugu will be a useful model for the human genome, e.g., [Gilley, J., Armes, N. & Fried, M. (1997) Nature (London) 385, 305–306]. We report extensive conservation of synteny between a 1.5-Mb region of human chromosome 11 and <100 kb of the Fugu genome in three overlapping cosmids. Our findings support the idea that the majority of DNA in the region of human chromosome 11p13 is intergenic. Comparative analysis of three unrelated genes with quite different roles, WT1, RCN1, and PAX6, has revealed differences in their structural evolution. Whereas the human WT1 gene can generate 16 protein isoforms via a combination of alternative splicing, RNA editing, and alternative start site usage, our data predict that Fugu WT1 is capable of generating only two isoforms. This raises the question of the extent to which the evolution of WT1 isoforms is related to the evolution of the mammalian genitourinary system. In addition, this region of the Fugu genome shows a much greater overall compaction than usual but with significant noncoding homology observed at the PAX6 locus, implying that comparative genomics has identified regulatory elements associated with this gene.
The pufferfish Fugu rubripes has a compact genome of 400 Mb, some 7.5 times smaller than that of mammals but with a similar number of genes. This compaction is caused by intron sizes being smaller as well as much reduced intergenic distances, i.e., distances between transcription units. For this reason, Fugu has been proposed as a model organism to study genes both at the level of the individual gene and in a genomic context (1, 2). The Fugu genome exhibits homology with other species across critical regulatory segments of some genes (3), and it has been established that synteny is conserved across a number of small regions between pufferfish and mammals. To date, linkage of two or three genes has been demonstrated for certain synteny groups such as FOS/S31iii125/s20i15 (4), PDGFRβ/CSF1R (5), MAP-2/MYL-1/CPSIII (6), and TSC2/PKD1/SSTR5 (7) although these examples consist of a limited number of genes covering relatively small distances in terms of human DNA. Additionally, these studies did not provide complete Fugu DNA sequence. It is becoming apparent that not all regions of the human genome are suitable for modeling in Fugu; for example, even the tightest mammalian gene cluster, that of the surfeit genes, does not exhibit complete conservation of synteny between mammals and Fugu (8). With accelerating progress in the Human Genome Project, the biological importance of long range genomic organization is now beginning to emerge, and it is important that conservation of synteny, genomic organization, and noncoding sequence homology be identified across vertebrate species to explore genomic function.
We chose to investigate possible conservation of synteny between Fugu and the 11p13 region of the human genome for several reasons: (i) The human region contains two well studied disease genes, PAX6 and WT1, each of which is essential for normal mammalian development. Their heterozygous deletion gives rise to WAGR syndrome - Wilms’ tumor, Aniridia, Genitourinary abnormalities, and mental Retardation (9). PAX6 and WT1 are conserved through evolution, can generate several different protein isoforms, and have complex regulatory systems, which orchestrate tissue-specific and temporally regulated expression patterns. Certain forms of aniridia arise as a result of a translocation outside the PAX6 gene presumably through a “position effect” (10) and it is thought that both WT1 and PAX6 use long-range genetic regulatory elements. Therefore, a study of this region should lead to a greater understanding of the structure and regulation of these complex, developmentally important human disease genes; (ii) There are several well characterized mouse mutants in this region, both at specific gene loci (11, 12) and deletions up to several megabases (13, 14), which will facilitate in vivo functional analyses by gene replacement/complementation studies in the mouse; (iii) There is extensive coverage of this region in both human and mouse with YAC/PAC contigs (15) although the associated gene maps are not complete. This will allow the degree of synteny to be accurately assessed while allowing the assembly of better gene maps across this region; (iv) Other human disease-associated loci, e.g., those associated with loss of heterozygosity in breast and bladder cancers (16), mapping to this region may be defined through homology studies. In addition, the cause(s) of mental retardation associated with WAGR deletions has yet to be determined. (v) Sequencing of 225 kb of the human PAX6 region has been completed [http://www.sanger.ac.uk/cgi-bin/hummap?map = Chr_11 misc] and that of the WT1 region is underway, allowing direct “base-by-base” comparisons to be made.
The PAX6 and WT1 genes are separated by approximately 750 kb in the human genome. Recently, a human homolog of reticulocalbin (RCN1) was identified at a CpG island site between WT1 and PAX6 (14), thereby providing another reference point with which to compare Fugu. If this region shows synteny conservation and the expected level of compaction for the Fugu genome, a search for new genes and for long distance regulatory elements within 11p13 could be undertaken by using comparative genomics in addition to allowing detailed studies of the structure and regulation of individual genes.
METHODS
The Tetraodon fluviatilis probe was generated by degenerate PCR of 50 ng of genomic DNA, by using the following oligonucleotide primer pair (5′-3′): exon 8 (sense) CAG TGT GA(C/T) TT(C/T) A(A/C)(A/G) GAC TG and exon 10 (antisense) GT(T/C) TCT (C/T)TG (G/A)TG CAT (G/C)TT CTG.
The reaction profile of the PCR consisted of an initial 5-min denaturation at 94°C, followed by 30 cycles of 10 s at 94°C, 45 s at 50°C, 2 min at 72°C with a final extension step of 10 min at 72°C.
PCR products of ≈900 bp were subcloned and sequenced to verify clones corresponding to WT1. A gridded cosmid library of Fugu rubripes genomic DNA [Greg Elgar, Human Genome Mapping Project Resource Centre (HGMP-RC)] was probed with the pufferfish (Tetraodon fluviatilis) WT1 genomic fragment (exon 8-exon 10) at high stringency.
Sequence analysis of cosmid 151-J19: Cosmid DNA was sonicated and ligated into EcoRV digested pBluescript. Recombinants were PCR amplified with limiting dilutions of dNTPs and primers. PCR products were sequenced by using dye terminator chemistry on an Applied Biosystems 377 sequencer. Gaps were filled in by using walking primers. All coding sequence has been sequenced in both directions, and all regions have been covered by at least two independent clones. Assembly was performed by using seqman (DNAStar). Sequence analysis was performed by using clustal w (17) alignments, and dotter for Fugu:human genomic DNA comparisons (18).
PCR sequencing was used to verify the presence of amino acids KPS in multiple cosmid clones and independent samples of Fugu rubripes genomic DNA. The probes used for this were: sense oligo (5′-3′) CTG TGA AAC CTC CAC CAC and antisense oligo (5′-3′) GAG TGA TTT CAG CTC CAG.
Reaction conditions were 5-min denaturation at 94°C, followed by 30 cycles of 5 s at 94°C, 30 s at 58°C, 30 s at 72°C, and a final 5-min elongation step at 72°C. The resulting 240-bp PCR products were directly sequenced by using the Sequenase kit as directed by the manufacturer.
RESULTS AND DISCUSSION
To provide an entry point for analysis of the Fugu homolog of the WAGR region, we screened the HGMP Fugu cosmid library (Greg Elgar, HGMP-RC) by using a PCR-generated genomic Tetraodon WT1 probe. One cosmid, 151J19, was selected for further analysis because it contained the largest Fugu insert. Sequence scanning demonstrated, in addition to WT1, the presence of Fugu homologs of both RCN1 and PAX6 on this cosmid. As the gene content of this Fugu cosmid corresponded well to the WAGR region, we decided to sequence 151J19 in its entirety to allow comparison with human and other species data for the region. Cosmid 151J19 contains 45,565 bp of Fugu DNA with the 5′ end of the WT1 exon 1 at base 191 and of PAX6 exon 12 at base 44,736. As in the human genome, the Fugu RCN1 gene is transcribed in the opposite direction to its neighbors, demonstrating conservation of both gene order and orientation between Fugu and mammals. Screening of the HGMP Fugu cosmid library with probes isolated from 151J19 resulted in the isolation of overlapping cosmids at each end (Fig. 1). Analysis of the PAX6 overlapping cosmid has located part of the Fugu homolog of a putative gene (expressed sequence tag contig) telomeric to PAX6. The WT1-overlapping cosmid has revealed homology with a human CpG island expressed sequence tag (19) mapping ≈500 kb centromeric to WT1. The size reduction of this region in Fugu compared with man is 15-fold, with Fugu sequences in three contiguous cosmids covering ≈1.5 Mb of the human genome. More importantly, the relative positions of five genes has been conserved between Fugu and man thereby fixing these loci and enabling the intervening sequences to be directly compared as they become available. These data predict that RCN1 may be the only gene between PAX6 and WT1 in the human genome, leaving >600 kb of DNA between transcription units. Until the complete sequence of human 11p13 becomes available, the presence of other genes between WT1 and PAX6 remains a possibility. To date, however, despite intense study (15, 19–21), no other genes have been found in this region in man. It is interesting that the Fugu homolog of this human region, which exhibits a low density of CpG islands, shows greater than average size reduction, consistent with Fugu counterparts of gene-poor regions exhibiting the greatest compaction (2). The complete sequencing of this Fugu region will facilitate studies probing the function of noncoding DNA in gene sparse regions of the human genome. Given that known gene order and orientation across 1.5 Mb has been conserved, this example provides the most compelling evidence to demonstrate the validity of the Fugu genome project away from the context of gene families.
Figure 1.
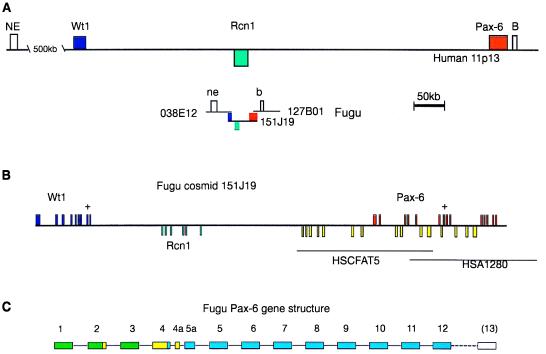
(A) Scale representation of the human chromosome 11p13 region encompassing the WT1, RCN1, and PAX6 genes compared with the equivalent genomic region in Fugu cosmids. Colored boxes indicate the positions of the three genes relative to each other in the two genomes. Genes in the reverse orientation are marked below the sequence line. Fugu cosmid 151J19 has been completely sequenced (see text), and the approximate positions of two overlapping Fugu cosmids are indicated. The two overlapping cosmids have been sequence scanned. Cosmid 038E12 contains a sequence (ne) homologous to a NotI end clone (NE, GenBank accession no. HSMFRAI) lying 500 kb from the WT1 gene in man. There is an expressed sequence tag cluster (B) 6 kb 3′ of the human PAX6 gene, which has a Fugu homolog (b) found on cosmid 127B01. (B) Detailed diagram of Fugu cosmid 151J19. Fugu genomic DNA (45,565 kb) is represented. WT1 and PAX6 are transcribed in the forward direction whereas RCN1 is transcribed from the reverse strand. The complete coding sequence of the WT1 gene is represented by the nine blue exons, RCN1 by six green exons, and PAX6 by the first 12 exons in red. The last exon of PAX6 (exon13) is not present on this cosmid clone. A “+” on top of an exon indicates that this exon is alternatively spliced in man. Human genomic sequence is available across the human PAX6 gene, and this is represented by HSCFAT5 and HSA1280. Additional regions of DNA similarity between human and Fugu is indicated by yellow boxes. (C) Exon structure of the Fugu PAX6 gene. The human PAX6-coding region (represented in blue) starts toward the end of exon 4, with exons 1–3 being entirely noncoding. Fish (including the blind cavefish and zebrafish) have additional coding sequences located in exon 2, exon 4, and exon 4a (yellow), which combine with the blue exons to form a series of additional isoforms. Note that the beginning of the Fugu-coding sequence results from the splicing together of exons 2 and 4, skipping exon 3.
The Fugu WT1, RCN1 and PAX6 genes show very high similarity to their well-characterized human orthologs. The intron/exon organization of both the WT1 and PAX6 genes is conserved and that of the reticulocalbin gene has been predicted (Fig. 2) to be the same as in the mouse. The Fugu PAX6 gene also contains a homolog of the alternatively spliced human exon 5a (Fig. 2), although this exon in Fugu consists of 12 amino acids instead of the 14 amino acids found in other vertebrates. Comparisons with PAX6 cDNA sequences from zebrafish (22) and the blind cavefish Astyanax (23) suggest that the fish proteins can initiate upstream of the mammalian ATG site to generate an N-terminal extension, which would involve skipping the Fugu sequence homologous to the noncoding mammalian exon 3 (Fig. 1C).
Figure 2.

(A) Clustal alignment of WT1 genes from human, mouse, Xenopus, and Fugu. Exons are numbered and their boundaries marked by a vertical line in all three alignments. Both Xenopus and Fugu genes lack the polyproline tract in the middle of exon 1 and the whole of the alternatively spliced exon V. The alternative splice donor sites at the end of exon IX are marked by open circles on the boundary markers. (B) Clustal alignment of mouse, human, and Fugu RCN1 genes. Intron/exon boundaries are marked for Fugu as these are not published for the mouse or human genes. It is likely that the mammalian genes have a very similar or identical genomic organization. (C) Clustal alignment of PAX6 genes from Fugu, blind cavefish, zebrafish, Xenopus, mouse, and human. In the three fish species shown, there are additional coding sequences at the 5′ end of the gene giving rise to a variety of transcripts. The most 5′ transcripts in fish are formed by splicing together exons II and IV, skipping exon III. There is also an additional exon found in cavefish and (by homology) Fugu that has not been found in higher vertebrates and does not appear to be present in human genomic sequence for this region (HSCFAT5 and HSA1280). Exon 13 has not yet been sequenced in Fugu.
The human WT1 gene has the potential to generate 16 different protein isoforms by a combination of alternative splicing, RNA editing, and alternative translational start site usage (24). We predict that the Fugu WT1 gene can generate only two different isoforms by the introduction of three amino acids between the two most C terminal zinc fingers (Fig. 2). The retention of this alternative splice implies that WT1 could play a dual role in both transcription and RNA processing in nonmammalian, as well as mammalian, vertebrates (25). In Fugu, this amino acid triplet consists of KPS (verified in several independent cosmid clones and samples of Fugu rubripes genomic DNA, not shown) in contrast to all other known WT1 homologs where it is KTS. The change from threonine to proline converts a noncharged polar amino acid to a noncharged nonpolar amino acid. Given the general level of amino acid conservation of the zinc finger region of Fugu WT1 and the conservation of the alternative splice donor and acceptor sequences, it would appear that the ±3 amino acid alternative splice is functional in Fugu and that this function is not strictly reliant on KTS. The role of the ±KPS alternative splicing could be to generate protein isoforms with different target-binding abilities by altering the spacing of zinc fingers 3 and 4. Fugu WT1 lacks the 17 amino acid exon 5, consistent with this feature being mammalian-specific (26). Mammalian WT1 translation can initiate at a CTG codon, adding 68 amino acids to the N terminus of the protein (27). The sequence upstream of the ATG codon in Fugu shows no evidence of an alternative start site resembling that found in mammals, suggesting this may represent another mammalian specific isoform. The final element creating the diversity of human WT1 proteins is RNA editing (28), which results in an amino acid change at position 281 from leucine to proline. Intriguingly, the Fugu genome encodes a proline at position 281 (Fig. 2). The cloning of the Fugu WT1 gene opens up the possibility of investigating the evolution of genitourinary development at a functional level in terms of both evolving protein structure and the regulation of gene expression, i.e., to what extent the evolution of WT1 protein isoforms and genetic regulatory elements have contributed to the evolution of the mammalian genitourinary system.
To test the usefulness of Fugu for identifying conserved regulatory elements, we performed dot plot analyses between Fugu PAX6 and the corresponding human genomic sequence (Fig. 3). Several noncoding, potential regulatory, sequences were identified, which are represented in homologous genomic positions along the genome in Fugu and in man. The first two clusters of these regions are >5 kb 5′ of the noncoding exon 1 of human PAX6. The third, most highly conserved, region is the proposed retinal specific enhancer (29) in intron 4, while a fourth conserved noncoding cluster lies in intron 7 (Fig. 3), the largest intron in both Fugu and man. Human intron 7 has the features of a CpG island, suggesting the possible presence of another gene; however, none of the sequences conserved between Fugu and man, nor any neighboring tracts, reveal ORF. The lack of “wobble” at what might be the third position of the triplet codon also suggests a regulatory function for these sequences. We are considering the possibility that these sequences may constitute an alternative start site, which would give rise to a PAX6 isoform lacking a paired domain. A similar paired domain lacking PAX6 product has been found at the genetic locus mab-18 (male abnormal) in C. elegans (30) as an alternative form to the full length protein observed in vab-3 chemotactic mutants (31). N-terminally truncated forms of PAX6, lacking the paired domain, also have been reported in quail (32). By comparing genomic data from species as divergent as Fugu and man in this way, it has been possible to identify a number of conserved regions, with characteristics of regulatory elements, which may now be studied further to assess their role in gene expression and function.
Figure 3.

(A) Dot plot alignment of cosmid 151J19 (x axis) with human genomic sequence HSA1280 (y axis) from the human PAX6 region. The program dotter was used to align the sequences with a window size of 21. Regions of similarity are identified as a concentration of dots (identical bases) generating a line. Exonic regions of similarity are marked by numbers. Three other regions of high DNA similarity also are marked (i–iii). (B) The weakest of these is magnified by using the dotter program, and a nucleotide alignment across this region is represented in C, which illustrates the lack of third base “wobble” associated with these noncoding segments of homology between Fugu and man.
In conclusion, we have demonstrated conserved synteny between Fugu and 1.5 Mb of human chromosome 11p13—a region where the identified genes are not members of gene families and yet their order and orientation is not scrambled compared with Fugu. There are important implications of such a finding for the comparative analysis of a large genomic region, given that the full human chromosome 11p13 sequence of ≈15 Mb may be available soon and excellent biological resources exist for functional studies in several animal species as well as in cell culture. Despite near complete coverage of this region with human YAC/PAC contigs, the identification of genes remains difficult, although there is some evidence (such as relatively low CpG island density) that there may be few genes in this region. This study suggests that it will now be possible to use comparative genomics between Fugu and man to assist the designation of new genes and the identification of regulatory elements and, more fundamentally, increase our understanding of the nature of large segments of intergenic DNA in the human genome.
Acknowledgments
Thanks to Linda McCarthy and Alan Schafer for Tetraodon DNA. We are grateful to the team at the Sanger Centre for the PAX6 contig sequencing. C.M. is a Medical Research Council Research Fellow. D.J.K. is supported by European Union Grant BMH4-CT96-1428. V.v.H. and N.H. were International Research Scholars of Howard Hughes Medical Institute.
ABBREVIATIONS
- WAGR syndrome
Wilms’ tumor, Aniridia, Genitourinary abnormalities and mental Retardation
- HGMP-RC
Human Genome Mapping Project Resource Centre
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AL021531 for the sequence of cosmid 151-J19).
References
- 1. Brenner S, Elgar G, Sandford R, Macrae A, Venkatesh B, Aparicio S. Nature (London) 1993;366:265–268. doi: 10.1038/366265a0. [DOI] [PubMed] [Google Scholar]
- 2.Elgar G, Sandford R, Aparicio S, Macrae A, Venkatesh B, Brenner S. Trends Genet. 1996;12:145–150. doi: 10.1016/0168-9525(96)10018-4. [DOI] [PubMed] [Google Scholar]
- 3.Aparicio S, Morrison A, Gould A, Gilthorpe J, Chaudhuri C, Rigby P, Krumlauf R, Brenner S. Proc Natl Acad Sci USA. 1995;92:1684–1688. doi: 10.1073/pnas.92.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trower M K, Orton S M, Purvis I J, Sanseau P, Riley J, Christodoulou C, Burt D, See C G, Elgar G, Sherrington R, et al. Proc Natl Acad Sci USA. 1996;93:1366–1369. doi: 10.1073/pnas.93.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.How G-F, Venkatesh B, Brenner S. Genome Res. 1996;6:1185–1191. doi: 10.1101/gr.6.12.1185. [DOI] [PubMed] [Google Scholar]
- 6.Schofield P, Elgar G, Greystrong J, Lye G, Deadman R, Mickelm G, King A, Brenner S, Vaudin M. Genomics. 1997;45:158–167. doi: 10.1006/geno.1997.4913. [DOI] [PubMed] [Google Scholar]
- 7.Sandford R, Sgotto B, Burn T, Brenner S. Genomics. 1996;38:84–86. doi: 10.1006/geno.1996.0596. [DOI] [PubMed] [Google Scholar]
- 8.Gilley J, Armes N, Fried M. Nature (London) 1997;385:305–306. doi: 10.1038/385305a0. [DOI] [PubMed] [Google Scholar]
- 9.Francke U, Holmes L B, Atkins L, Riccardi V M. Cytogenet Cell Genet. 1979;24:185–192. doi: 10.1159/000131375. [DOI] [PubMed] [Google Scholar]
- 10.Fantes J, Redeker B, Breen M, Boyle S, Brown J, Fletcher J, Jones S, Bickmore W, Fukushima Y, Mannens M, et al. Hum Mol Genet. 1995;4:415–422. doi: 10.1093/hmg/4.3.415. [DOI] [PubMed] [Google Scholar]
- 11.Hill R E, Favor J, Hogan B L, Ton C C, Saunders G F, Hanson I M, Prosser J, Jordan T, Hastie N D, Van Heyningen V. Nature (London) 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- 12.Kreidberg J A, Sariola H, Loring J M, Maeda M, Pelletier J, Housman D, Jaenisch R. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 13.Glaser T, Lane J, Housman D. Science. 1990;250:823–827. doi: 10.1126/science.2173141. [DOI] [PubMed] [Google Scholar]
- 14.Kent J, Lee M, Schedl A, Boyle S, Fantes J, Powell M, Rushmere N, Abbott C, Van Heyningen V, Bickmore W A. Genomics. 1997;42:260–267. doi: 10.1006/geno.1997.4706. [DOI] [PubMed] [Google Scholar]
- 15.Fantes J A, Oghene K, Boyle S, Danes S, Fletcher J M, Bruford E, Williamson K, Seawright A, Schedl A, Hanson I M, et al. Genomics. 1995;25:447–461. doi: 10.1016/0888-7543(95)80045-n. [DOI] [PubMed] [Google Scholar]
- 16.Huff V, Saunders G F. Biochim Biophys Acta. 1993;1155:295–306. doi: 10.1016/0304-419x(93)90011-z. [DOI] [PubMed] [Google Scholar]
- 17.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonnhammer E L L, Durbin R. Gene-combis. 1995;167:1–18. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- 19.Thate C, Pongratz J, Konig A, Klamt B, Tsaoussidou S, Higgins M, Shows T, Jones C, Gessler M. Mamm Genome. 1995;6:421–425. doi: 10.1007/BF00355644. [DOI] [PubMed] [Google Scholar]
- 20.Rose E A, Glaser T, Jones C, Smith C L, Lewis W H, Call K M, Minden M, Champagne E, Bonetta L, Yeger H, Housman D E. Cell. 1990;60:495–508. doi: 10.1016/0092-8674(90)90600-j. [DOI] [PubMed] [Google Scholar]
- 21.Ton C C T, Hirvonen H, Miwa H, Weil M M, Monaghan P, Jordan T, van Heyningen V, Hastie N D, Miejers-Heijboer H, Drechsler M, et al. Cell. 1991;67:1059–1074. doi: 10.1016/0092-8674(91)90284-6. [DOI] [PubMed] [Google Scholar]
- 22.Puschel A W, Gruss P, Westerfield M. Development (Cambridge) 1992;114:643–651. doi: 10.1242/dev.114.3.643. [DOI] [PubMed] [Google Scholar]
- 23.Behrens M, Langecker T G, Wilkens H, Schmale H. Mol Biol Evol. 1997;14:299–308. doi: 10.1093/oxfordjournals.molbev.a025765. [DOI] [PubMed] [Google Scholar]
- 24.Hastie N D. Annu Rev Genet. 1994;28:523–558. doi: 10.1146/annurev.ge.28.120194.002515. [DOI] [PubMed] [Google Scholar]
- 25.Larsson S H, Charlieu J P, Miyagawa K, Engelkamp D, Rassoulzadegan M, Ross A, Cuzin F, Van Heyningen V, Hastie N D. Cell. 1995;81:391–401. doi: 10.1016/0092-8674(95)90392-5. [DOI] [PubMed] [Google Scholar]
- 26.Kent J, Coriat A-M, Sharpe P T, Hastie N D, Van Heyningen V. Oncogene. 1995;11:1781–1792. [PubMed] [Google Scholar]
- 27.Bruening W, Pelletier J. J Biol Chem. 1996;271:8646–8654. doi: 10.1074/jbc.271.15.8646. [DOI] [PubMed] [Google Scholar]
- 28.Sharma P M, Bowman M, Madden S L, Rauscher F J, Sukumar S. Genes Dev. 1994;8:720–731. doi: 10.1101/gad.8.6.720. [DOI] [PubMed] [Google Scholar]
- 29.Plaza S, Dozier C, Langlois M C, Saule S. Mol Cell Biol. 1995;15:892–903. doi: 10.1128/mcb.15.2.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Emmons S. Nature (London) 1995;377:55–59. doi: 10.1038/377055a0. [DOI] [PubMed] [Google Scholar]
- 31.Chisholm A D, Horvitz R. Nature (London) 1995;377:52–55. doi: 10.1038/377052a0. [DOI] [PubMed] [Google Scholar]
- 32.Carriere C, Plaza S, Martin P, Quatannens B, Bailly M, Stehelin D, Saule S. Mol Cell Biol. 1993;13:7257–7266. doi: 10.1128/mcb.13.12.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]