

Abstract
Numerous inherited human genetic disorders are caused by defects in pre-mRNA splicing. Two recent studies have added a new twist to the link between genetic variation and pre-mRNA splicing by identifying SNPs that correlate with heritable changes in alternative splicing but do not cause disease. This suggests that allele-specific alternative splicing is a mechanism that accounts for individual variation in the human population.
Genomic variation
It is inescapable that we are all different from one another. Whether it is in a laboratory or sitting at home with your family, when you look around, you notice that everyone (with the exception of identical twins) is different. However, the mechanisms by which the unique sequence of our individual diploid genomes manifest themselves as phenotypic variation are largely unknown. Two recent papers by Hull et al. [1] and Kwan et al., [2] have identified single nucleotide polymorphisms (SNPs) that are common in the human population that correlate with inheritable differences in the splicing patterns between individuals.
Although the main impetus for the human genome project was to identify genes involved in human diseases, the genome sequence has provided significant insight into much more [3,4]. For instance, from an anthropologic sense, comparison of the human genome to the genomes of other primates (including Neanderthals), vertebrates and other forms of life has already changed our understanding of human evolution [5,6]. Additionally, functional studies of the human genome through the ENCODE project have radically altered our definition of a gene and changed our view of gene regulation [7]. However, one of the most interesting aspects of the human genome project has been the exploration of the extent of genetic variation within the human population.
Splicing and human disease
It is well established that several human diseases are caused by mutations that disrupt the normal pattern of splicing [8]. In fact, up to 50% of the mutations that cause human disease alter the efficiency and pattern of splicing. Well-known human diseases that are associated with splicing defects include cystic fibrosis and cancer. The mutations in cases such as these typically occur within the sequences that direct the splicing reaction themselves (Box 1). The most basic of these sequence elements are located within the intron and include the 5′ splice site, the branchpoint, the polypyrimidine tract and the 3′ splice site, which are all recognized in a sequence-specific manner by components of the spliceosome. Although these sequence elements span at least 20 nucleotides, only the GT and AG dinucleotides at the 5′ and 3′ ends of the intron, respectively, are essentially invariant. As a result, mutations in these four nucleotides have the most profound impact on splicing and typically result in skipping of the exon linked to the mutation. However, a variety of diseases such as spinal muscular atrophy and multiple sclerosis are caused by mutations in exonic splicing enhancers (ESEs) or silencers (ESSs) that modulate the efficiency of exon inclusion.
Box 1.
Sequences and proteins involved in pre-mRNA splicing. Pre-mRNAs contain a variety of cis-acting sequences required for splicing (Figure I). The intron upstream of an exon contains a branchpoint sequence, polypyrimidine tract (Py tract) and an AG dinucleotide at the 3′ splice site, whereas the downstream intron contains a conserved sequence at the 5′ splice site. The branchpoint sequence is recognized by U2 small nuclear ribonuclear protein particle (snRNP), the Py tract and AG dinucleotide are recognized by the 65-and 35-kDa subunits of U2AF, and the 5′ splice site is recognized by U1 snRNP. Several types of auxiliary sequences modulate the efficiency of splicing. These included exonic splicing enhancers (ESEs) and silencers (ESSs) and intronic splicing enhancers (ISEs) and silencers (ISSs). ESEs are typically recognized by members of the SR protein family, which increase the frequency of exon inclusion, whereas ESSs interact with hnRNP proteins that usually repress splicing. ISEs and ISSs interact with a variety of splicing regulatory proteins that act in a positive or negative manner, respectively.
More than just diseases
Although these examples serve to demonstrate that genetic mutations can result in human disease by disrupting splicing, they have much broader implications. Most common SNPs are not thought to cause human disease. However, it is possible that they are not aphenotypic. Perhaps these SNPs are at the root of individual variation in the human population. If this is true, however, it is unclear how these SNPs manifest themselves phenotypically.
The recent studies by Hull et al. [1] and Kwan et al. [2] have explored this area to search for variations in alternative splicing within the human population. The two groups have taken very different approaches, but both have made use of the HapMap Project data (Box 2) to identify common SNPs that correlate with the observed splicing changes.
Box 2.
The HapMap project. In 2002, the International HapMap Project began with the goal of cataloging common genetic variants that occur in the human genome and the distribution of these variants among populations throughout the world. To do this, 1 million single nucleotide polymorphisms (SNPs) that were evenly distributed throughout the human genome were genotyped for 269 samples from four populations [10]. These samples included 90 individuals from Ibadan, Nigeria, 90 individuals from Utah, USA, 45 individuals from Bejing, China, and 44 individuals from Tokyo, Japan. An important outcome of this project was the identification of haplotype blocks – regions of the genome in which closely located SNPs are co-inherited. This allows for the patterns of inheritance for large regions of the genome to be traced by analyzing only a subset of the known SNPs in each haplotype block. The information derived from the HapMap project, along with the use of genome-wide genotyping assays, has dramatically increased the pace at which researchers can identify genes associated with inherited human disorders such as myocardial infarction [11], diabetes [12], and asthma [13]. Once these disease genes are identified, it is important to understand the mechanisms by which the causative mutations give rise to the disease state.
First, Hull et al. [1] took a directed approach by logically selecting exons to study that are likely to be differentially spliced in the human population. They reasoned that, for any exon that had this property, the mRNA isoforms containing and lacking this exon should both be commonly observed. They identified 250 exons that satisfied their criteria and verified that, for 70 of these exons, both isoforms were indeed expressed in lymphoblastoid cell lines (LCLs) generated from the CEPH HapMap project. A comparison of the splicing pattern and genotypes of 22 LCLs revealed that six of these exons were spliced in an allele-specific manner.
By contrast, Kwan et al. [2] took a more global approach. They analyzed splicing in two LCLs from the CEPH HapMap project on microarrays containing 1.4 million probe sets to query 1 million known and/or predicted exons. They identified nine exons that are differentially spliced between the two LCLs and subsequently showed that three exons within the genes encoding 2’, 5′-oligoadenylate synthetase (OAS1), calplastinin (CAST), and cartilage-associated protein (CRTAP) are spliced in an allele-specific manner.
Although some of the SNPs linked to these splicing changes are located far from the affected exons identified in these two studies, others are located in close proximity or even within the alternatively spliced exon, providing an opportunity to predict the mechanisms by which the sequence differences could impact the splicing pattern. Kwan et al. [2] found an SNP located at the 5′ splice site of the affected exon in CAST, suggesting that this SNP most likely impacts the efficiency of U1 small nuclear ribonuclear protein particle (snRNP) binding. Four of the genes identified by Hull et al. [1] contained SNPs within the affected exon. In SH3YL1 and RBM23, these SNPs are located just downstream of the 3′ splice site, suggesting that they modulate the efficiency of 3′ splice site recognition. However, for ZDHHC6 and CASP3, the SNPs are located in the middle of the exon and most likely create or destroy either an ESE or an ESS. Finally, the SNPs within CD46 and IFI16 are located within the flanking introns. The CD46 SNP is located just downstream of the 5′ splice site and most likely impacts the efficiency of 5′ splice site recognition. Interestingly, in IFI16, the SNP is located 1300 nucleotides (nt) upstream of the 3′ splice site, making it hard to predict how this SNP might impact splicing. Nonetheless, these examples illustrate that identifying SNPs that modulate splicing can provide clues into the mechanisms by which they exert their effect.
The fact that these two studies identified only nine exons that are differentially spliced in an allele-specific manner that correlates with SNPs that are common in the human population suggests that allele-specific splicing is uncommon in the human population. However, it is more likely that the number of exons identified from these studies is a vast underestimate of the true extent of this phenomenon. First, Hull et al. [1] selected only 250 exons from the human genome to study. Second, although Kwan et al. [2] used exon arrays, which should provide a genome-wide view of this phenomenon, the authors proceeded to experimentally validate only a small subset (20 of ~1000 candidate events). Furthermore, the authors only examined RNA isolated from two different cell lines on these arrays, and therefore, many of the common human haplotypes were not examined. Second, and perhaps most importantly, although these arrays can identify alternative splicing changes, there are several issues that complicate interpretations of the results. First, the algorithms used to deconvolute the microarray results require one to assume the splicing patterns for each gene, which if wrong, can lead one astray. Second, these arrays have difficulty in identifying cases where the splicing changes are subtle, even though they might be significant, both statistically and functionally. Third, the arrays can be ‘noisy’ or have a high degree of false positives and false negatives – the study by Kwan et al. [2] had a 55% false-discovery rate. Several of these issues could be improved on in the future by the use of similar arrays that are supplemented with splice junction probes or with even newer technologies. Although these are things that need to be kept in mind, this approach did allow the authors to identify bona fide examples of allele-specific splicing events.
Looking forward
At the onset of the HapMap project, it was thought that any two copies of the human genome would be 99.9% identical – there would be one difference every 1000 nucleotides. Recently the first diploid human genome sequence has been published – that of Craig Venter [9]. One of the most significant aspects of Dr. Venter’s genome sequence is that the extent of genetic variation between humans is significantly greater than previously thought—99.5% rather than 99.9% identity. What about alternative splicing? How much variation in alternative splicing truly exists within the human population, and how much of this plays a role in our phenotypic diversity? In the near future, it will be possible to sequence the diploid genome of a single individual and to sequence their transcriptome at sufficient depth to identify allele-specific differences in transcription and alternative splicing. These types of studies will facilitate a true exploration of the variability of alternative splicing between individuals and might open up the field of haplo-spliceo-transcriptomics.
Figure I.
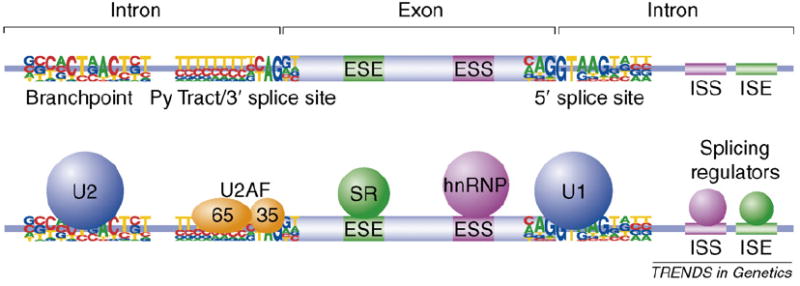
The cis- and trans-acting players of pre-mRNA splicing. The sequences required for splice site recognition and splicing regulation are shown on top, and the proteins that recognize these sequences are shown on the bottom.
Acknowledgments
The author thanks Michael Duff and David Kulp for discussions and comments on the manuscript. Work on alternative splicing in my laboratory is funded by grants from the NIH (GM067842, GM062516, and HG004271) and the Connecticut State Stem Cell Research Fund.
Glossary
- Exonic Splicing Enhancer (ESE)
A sequence element located in an exon that is recognized by a protein that enhances the efficiency of exon inclusion. ESEs are typically recognized by members of the SR protein family
- Exonic Splicing Silencer (ESS)
Exonic sequences that are typically recognized by members of the hnRNP family that decrease the efficiency of exon inclusion
- Haplotype
A set of single nucleotide polymorphisms on a single chromatid that is statistically associated
- HapMap
The map or catalog of the common genetic variants that occur in the human genome (www.hapmap.org)
- Intronic Splicing Enhancer (ISE)
Intronic sequences that are recognized by proteins that enhance the splicing of adjacent exons
- Intronic Splicing Silencer (ISS)
Intronic sequences that are recognized by proteins that repress the splicing of adjacent exons
- Pre-mRNA
A primary transcript that is a precursor to an mRNA
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author’s institution, sharing with colleagues and providing to institution administration. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Hull J, et al. Identification of common genetic variation that modulates alternative splicing. PLoS Genet. 2007;3:e99. doi: 10.1371/journal.pgen.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwan T, et al. Heritability of alternative splicing in the human genome. Genome Res. 2007;17:1210–1218. doi: 10.1101/gr.6281007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 5.Green RE, et al. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- 6.Noonan JP, et al. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006;314:1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper TA, Wang GS. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 9.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helgadottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 12.Hakonarson H, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 13.Moffatt MF, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]