

Abstract
Ethanol-induced excitation of ventral tegmental area dopamine (DA VTA) neurons is thought to be critical for the reinforcing effects of ethanol. Although ligand-gated ion channels are known to be the targets of ethanol, ethanol modulation of voltage-dependent ion channels of central neurons has not been well studied. We have demonstrated that ethanol excites DA VTA neurons by the reduction of sustained K+ currents and recently reported that M-current (IM) regulates action potential generation through fast and slow afterhyperpolarization phases. In the present study we thus examined whether ethanol inhibition of IM contributes to the excitation of DA VTA neurons using nystatin-perforated patch current- and voltage-clamp recordings. Ethanol (20–120 mM) reduced IM in a concentration-dependent manner and increased the spontaneous firing frequency of DA VTA neurons. Ethanol-induced increase in spontaneous firing frequency correlated positively with ethanol inhibition of IM with a slope value of 1.3. Specific IM inhibition by XE991 (0.3–10 μM) increased spontaneous firing frequency which correlated positively with IM inhibition with a slope value of 0.5. In the presence of 10 μM XE991, a concentration that produced maximal inhibition of IM, ethanol still increased the spontaneous firing frequency of DA VTA neurons in a concentration-dependent manner. Thus we conclude that, although ethanol causes inhibition of IM and this results in some increase in the firing frequency of DA VTA neurons, another effect of ethanol is primarily responsible for the ethanol-induced increase in firing rate in these neurons.
INTRODUCTION
The ventral tegmental area dopamine (DA VTA) neurons are cells of origin of the mesolimbic dopamine pathway and provide their projections to the nucleus accumbens (NAcb) (Oades and Halliday 1987). Earlier behavioral and pharmacological studies showed that the mesolimbic dopamine pathway is critical for the reinforcing effects of ethanol (Appel et al. 2004; DiChiara and Imperato 1988; Weiss et al. 1993). Pharmacological study with dopamine agonists and antagonists also supports the involvement of dopamine in ethanol reinforcement (Samson et al. 1990). It was previously reported that rats self-administer ethanol directly into the VTA (Rodd-Henricks et al. 2000) and ethanol excites dopamine cells in the VTA (Gessa et al. 1985). Thus ethanol-induced excitation of DA VTA neurons would increase dopamine release in the NAcb and mediate the reinforcing effects of ethanol.
There are many studies on the ethanol modulation of ligand-gated ion channels in central neurons. In hippocampal neurons, N-methyl-d-aspartic acid (NMDA)-receptor–mediated current is inhibited by ethanol (Hendricson et al. 2004; Lovinger et al. 1989; Peoples and Weight 1995). γ-Aminobutyric acid type A (GABAA)-receptor–mediated response is potentiated by ethanol in hippocampal neurons (Aguayo and Pancetti 1994; Proctor et al. 1992). Glycine-receptor–mediated response is potentiated by ethanol in midbrain dopamine neurons (Ye et al. 2001) and hypoglossal motoneurons (Eggers and Berger 2004). Adenosine (P2X) receptor–mediated current is inhibited by ethanol in hippocampal neurons (Li et al. 2000). Although ethanol modulates ligand-gated ion channels and affects the excitability of central neurons, the effect of ethanol on modulation of voltage-dependent ion channels in the CNS has not been well studied. DA VTA neurons have intrinsic pacemaker activity with the average firing frequency of 0.5–5 Hz and several voltage-dependent ionic currents contribute to the excitability of these neurons (Appel et al. 2003, 2006; Brodie and Appel 1998; Brodie et al. 1990, 1999; Koyama and Appel 2006a,b; Koyama et al. 2005; Mueller and Brodie 1989; Neuhoff et al. 2002; Okamoto et al. 2006). Therefore the effects of ethanol on voltage-dependent ion channels and the subsequent change in the excitability of these neurons are important cellular mechanisms involved in the reinforcing effects of ethanol.
We previously reported that ethanol increases the spontaneous firing frequency of acutely dissociated DA VTA neurons, in which the influence of synaptic activity on the excitability of these neurons was eliminated (Brodie et al. 1999). Our current-clamp studies suggest that ethanol excites DA VTA neurons by reducing a K+ current, which contributes to the action potential (AP) afterhyperpolarization (AHP) (Appel et al. 2003; Brodie and Appel 1998). In addition, our voltage-clamp studies showed that ethanol reduces the sustained K+ currents of DA VTA neurons (Appel et al. 2003; Brodie et al. 2000). Thus it is likely that ethanol excites DA VTA neurons directly by the reduction of K+-channel conductance.
M-current (IM) is a sustained K+ current and activated at the subthreshold range of membrane potential, regulating AP generation (Aiken et al. 1995; Brown and Adams 1980). Five types of KCNQ (KCNQ1–5) channels were previously reported to mediate IM (Lerche et al. 2000; Selyanko et al. 2000; Shapiro et al. 2000; Sogaard et al. 2001; Wang et al. 1998). Immunohistochemical co-localization of KCNQ2 and KCNQ4 channel subunit proteins was previously found in VTA neurons (Cooper et al. 2001; Kharkovets et al. 2000). We recently showed that IM regulates action potential generation to reduce the excitability by underlying fast and slow AHP components without changing the middle component of AHP (Koyama and Appel 2006a). Consistent with this finding, it was earlier shown that the KCNQ4 channel subunit is the major KCNQ subunit in DA VTA neurons and regulates the excitability of these neurons (Hansen et al. 2006). Interestingly, Moore et al. (1990) showed that ethanol reduces IM in CA1 hippocampal pyramidal neurons.
In our present study, we examined whether ethanol inhibition of IM contributed to the excitation of DA VTA neurons. For this purpose, we used acutely dissociated VTA neurons with enzyme treatment, in which both excitatory and inhibitory synaptic influences on these neurons were eliminated (Koyama et al. 2005) and nystatin-perforated patch recording to minimize intracellular dialysis and prevent the rundown of IM (Koyama and Appel 2006a). Part of this work previously appeared in abstract form (Koyama and Appel 2004).
METHODS
Preparation of dissociated neurons
Animals used in this study were treated in strict accordance with the U.S. National Institutes for Health Guide for the Care and Use of Laboratory Animals and all experimental methods were approved by the Animal Care Committee of the University of Illinois at Chicago. Each Fisher 344 rat (14–18 days old) was decapitated and the brain quickly removed. The brain was placed in the ice-cold cutting solution consisting of (in mM): 220 sucrose, 2.5 KCl, 2.4 CaCl2, 1.3 MgSO4, 1.24 NaH2PO4, 26 NaHCO3, and 11 d-glucose, which was constantly bubbled with 95% O2-5% CO2. Transverse brain slices, at a thickness of 400 μm, were made using a Vibratome. The brain slices were incubated for 30 min in artificial cerebrospinal fluid (ACSF), consisting of (in mM): 126 NaCl, 2.5 KCl, 2.4 CaCl2, 1.3 MgSO4, 1.24 NaH2PO4, 26 NaHCO3, and 11 d-glucose (osmolarity 300 mosM), which was constantly bubbled with 95% O2-5% CO2 at room temperature (23–25°C). The brain slices were then incubated in a HEPES-buffered solution (in mM: 145 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 11 d-glucose (pH adjusted to 7.4 with NaOH; osmolarity, 300 mosM) containing papain (15–18 U/ml) at 32°C for 20–25 min. After papain treatment, the brain slices were further incubated in the ACSF for 20–40 min. The VTA neurons were dissociated using a vibrating stylus apparatus dispersing cells from the brain slices as previously described (Koyama et al. 2005). Once the cell dissociation procedure was completed (4–7 min), the brain slice was removed from the culture dish and the dissociated neurons settled and adhered to the bottom of the dish within 20 min. The dissociation procedure with papain described above was earlier shown to yield acutely dissociated VTA neurons (Koyama et al. 2005) and no spontaneous synaptic potentials or synaptic currents were recorded from these neurons in the present study. DA VTA neurons were identified in current-clamp recording based on electrophysiological characteristics previously described (Koyama and Appel 2006a).
Nystatin-perforated patch recording in dissociated neurons
Electrophysiological measurement was performed using an Axo-patch-1B patch-clamp amplifier (Axon Instruments, Union City, CA). The tip resistances of the electrodes ranged from 1 to 2.5 MΩ when filled with pipette solution (in mM: 60 K-acetate, 60 KCl, 1 CaCl2, 2 MgCl2, and 40 HEPES; pH adjusted to 7.2 with KOH; final [K+]i = 131 mM; osmolality, 290 mOsm). To minimize the rundown of IM, nystatin-perforated patch recording was used as previously described (Koyama and Appel 2006a). Nystatin was dissolved in methanol at a concentration of 10 mg/ml. This nystatin stock solution was diluted with pipette solution to a final concentration of 100–200 μg/ml and the electrodes were backfilled with this solution. After cell-attached configuration had been completed, access resistance was periodically monitored and capacitative transients were cancelled. When the access resistance had reached a steady level (8–14 MΩ), the recording was started. Completion of perforated patch was distinguished from membrane rupture because the hallmark of membrane rupture was accompanied by the sudden appearance of steep capacitative transients with the reduction of access resistance to 3–7 MΩ; this can be compared with the access resistance in conventional whole cell configuration, described previously (Koyama and Appel 2006b). Pore formation with the nystatin-perforated patch technique is not accompanied by steep capacitance transients. Current- and voltage-clamp recordings were carried out with the HEPES-buffered solution constantly bubbled with 100% O2. The liquid junction potential between the pipette solution and the HEPES-buffered solution was estimated to be 5 mV (Neher 1992) and the results were corrected. Membrane currents and voltages were filtered at 1 kHz and acquired at a sampling frequency of 10 kHz. Data acquisition was performed by a DigiData 1322A interface and pClamp software version 9.0 (Axon Instruments). The dissociated VTA neurons were visualized under phase-contrast on an inverted microscope (Diaphot 300, Nikon, Tokyo, Japan). All experiments were performed at room temperature (23–25°C).
Drug application for dissociated neurons
Neurons were continuously bathed in control HEPES-buffered solution and drugs were dissolved at a final concentration in the same solution. Drug solutions were applied by a multiple channel manifold (Becton Dickinson). Each channel of the manifold was connected to a gravity-fed reservoir with tubing (ID 860 μm). The output of the manifold was connected to an outflow tube (ID 580 μm), the tip of which was placed within 200 μm of the soma of the recorded neuron. Solution flowed continuously through one manifold channel. Application of drug solutions was controlled by opening or closing valves connected to the reservoirs. The behaviorally active range of blood ethanol concentrations in the rat extends from 40 mM (sedation) to 90 mM (loss of righting reflex) (Majchrowicz and Hunt 1976); the lethal blood ethanol concentration in rats is about 200 mM (LD50 = 202 mM) (Haggard et al. 1940). Rats self-administer 44–55 mM ethanol directly into the VTA, indicating that this concentration of ethanol is reinforcing in the whole animal (Rodd-Henricks et al. 2000). In the present study, we examined ethanol concentrations in the range of 20 to 120 mM, which are pharmacologically relevant and sublethal concentrations in the rat.
Preparation of brain slices
After rapid removal of the brain, the tissue block containing the VTA was mounted in the Vibratome and submerged in the ice-cold cutting solution. Coronal sections (400 μm thick) were cut and the slice was placed on a mesh platform in the recording chamber. The slice was totally submerged in the ACSF maintained at a flow rate of 2 ml/min; the temperature in the recording chamber was kept at 35°C. The ACSF was saturated with 95% O2-5% CO2 (pH = 7.4). Equilibration time of ≥1 h was allowed after placement of the brain slice in the recording chamber before electrodes were placed in the tissue. Recording electrodes were placed in the VTA under visual control. Only those neurons that were anatomically located within the VTA and that conformed to the electrophysiological criteria for dopaminergic neurons (Mueller and Brodie 1989) were studied. These criteria include broad action potentials and regular spontaneous firing frequency at 0.5–5 Hz.
Extracellular recording in brain slices
Extracellular recording electrodes were fabricated from 1.5-mm-diameter glass tubing and were filled with 0.9% NaCl. Tip resistance of the microelectrodes ranged from 3 to 8 MΩ. The amplifier (Fintronics, Orange, CT) used in these recordings includes a window discriminator, the output of which was fed to both a rectilinear pen recorder and a computer-based data acquisition system that was used for on-line and off-line analysis of the data. The multiplexed output of the amplifier was displayed on an analog storage oscilloscope, for accurate adjustment of the window levels used to monitor single units. An IBM PC–based data acquisition system was used to calculate, display, and store the frequency of firing over 5-s and 1-min intervals.
Drug administration for brain slices
Drugs were added to the ACSF by means of a calibrated infusion pump from stock solutions 100- to 1,000-fold the desired final concentrations. The addition of drug solutions to the ACSF was performed in such a way as to permit the drug solution to mix completely with the ACSF before this mixture reached the recording chamber. The use of a calibrated, variable-speed infusion pump permits the accurate addition of several concentrations of drugs from the same stock solution. Final concentrations were calculated from the ACSF flow rate, pump infusion speed, and the concentration of drug stock solution. The small volume chamber (about 300 μl) used in this study permitted the rapid application and washout of drug solutions. Drugs typically reached equilibrium in the tissue after 2 to 3 min of application. A stock solution of 95% ethanol was used in the pump and infusion of ethanol never exceeded 1% of the flow rate of the ACSF.
Drugs and chemical agents
Dimethyl sulfoxide (DSMO), ethanol, HEPES, and nystatin were purchased from Sigma–Aldrich (St. Louis, MO). Papain was purchased from Worthington (Lakewood, NJ). 10,10-Bis(4-pyridinylmethyl)-9(10H)-anthracenone (XE991) dihydrochloride was purchased from Tocris-Cookson (Ellisville, MO).
Data analysis
Action potentials were analyzed off-line by pClamp software (version 9.0, Axon Instruments). Interspike interval (ISI) histograms were created as previously described (Cocatre-Zilgien and Delcomyn 1992). The number of bins was equal to the square root of the number of ISIs. Bin width was obtained by dividing the ISI range (maximum ISI minus minimum ISI) by the number of bins. To assess the changes of spontaneous firing with drugs, drug effect was quantitated as the mean change in firing rate (normalized as the percentage of control) for a 60-s-long interval during the peak of the drug response as previously described (Brodie et al. 1990). The formula for this normalization is
Data from cells with action potential amplitudes <50 mV were discarded. All average values are expressed as means ± SE. Statistical comparison to assess significant differences was done by one-way ANOVA as appropriate. When needed, the Student–Newman–Keuls post hoc test was used to test multiple comparisons. Correlation was evaluated by linear regression with P < 0.05 being considered significant.
RESULTS
Ethanol inhibition of M-current (IM) is concentration dependent
As illustrated in Fig. 1, IM of DA VTA neurons was recorded in nystatin-perforated patch voltage-clamp configuration, with which IM is stably recorded without rundown. IM was measured in the standard deactivation protocol (Brown and Adams 1980) with 1-s-long hyperpolarizing voltage step from a holding potential (VH) of −25 to −40 mV. Ethanol at concentrations of 40, 80, and 120 mM reduced IM amplitude as well as the negative shift of the baseline outward current in DA VTA neurons (Fig. 1, A1-A3). Figure 1B shows the measurement of the inward relaxation current caused by deactivation of IM during the voltage step. IM amplitude was 32 pA in control and ethanol (120 mM) decreased IM amplitude to 20 pA.
FIG. 1.
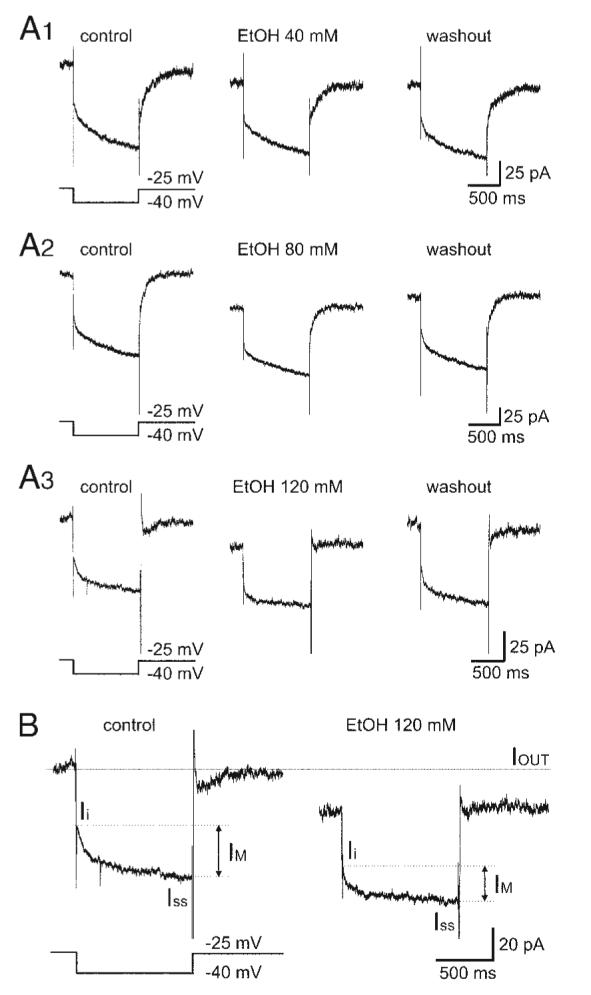
Ethanol inhibits M-current (IM) in ventral tegmental area dopamine (DA VTA) neurons. A1: IM was recorded before, during, and after application of 40 mM ethanol in a DA VTA neuron. IM was measured in the standard deactivation protocol with 1-s-long hyperpolarizing voltage step from a holding potential (VH)of −25 to −40 mV. A2: IM recorded before, during, and after application of 80 mM ethanol in a DA VTA neuron. A3: IM recorded before, during, and after application of 120 mM ethanol in a DA VTA neuron. Note the reduction in IM and the shift in the baseline outward current (IOUT) at a VH of −25 mV with ethanol. Each trace represents the average of 5 current responses. B: IM was measured as the inward relaxation current caused by deactivation of IM (arrows) during the voltage step, i.e., the difference between the instantaneous current at the beginning (Ii) and the steady-state current at the end of the voltage step (Iss) before (left) and after ethanol treatment (right). A longer dashed line represents IOUT at a VH of −25 mV before ethanol treatment.
Figure 2A shows the time course of ethanol effect on normalized IM amplitude in DA VTA neurons. Ethanol inhibition of IM was reversible and concentration dependent. Figure 2B shows the concentration–response relationship for ethanol inhibition of IM amplitude. Average maximal ethanol inhibition of IM amplitude obtained with each concentration was 6.0 ± 3.5% with 20 mM ethanol (n = 8), 7.2 ± 2.9% with 40 mM ethanol (n = 8), 15.3 ± 2.6 with 80 mM ethanol (n = 10), and 29.2 ± 2.7% with 120 mM ethanol (n = 9). From the graph in Fig. 2B, 20% inhibition of IM by ethanol was estimated to be 90 mM. Although the concentration of nystatin in the stock solution was relatively low and the concentration of methanol in the pipette solution to dissolve nystatin was relatively high, we obtained a similar ethanol effect on IM when nystatin was dissolved in DSMO (the final nystatin concentration was 100–200 μg/ml with 0.5–1% DSMO concentration in the pipette solution). Average ethanol inhibition of IM amplitude obtained with the DMSO filling solution with each concentration was 8.8 ± 1.8% with 40 mM ethanol (n = 6), 14.9 ± 1.8 with 80 mM ethanol (n = 5), and 25.5 ± 2.7% with 120 mM ethanol (n = 5). Therefore it was not likely that the relatively high concentration of methanol in the pipette solution significantly perturbed the plasma membrane when nystatin-perforated patch was completed or that the methanol interfered with the observations of ethanol effects.
FIG. 2.
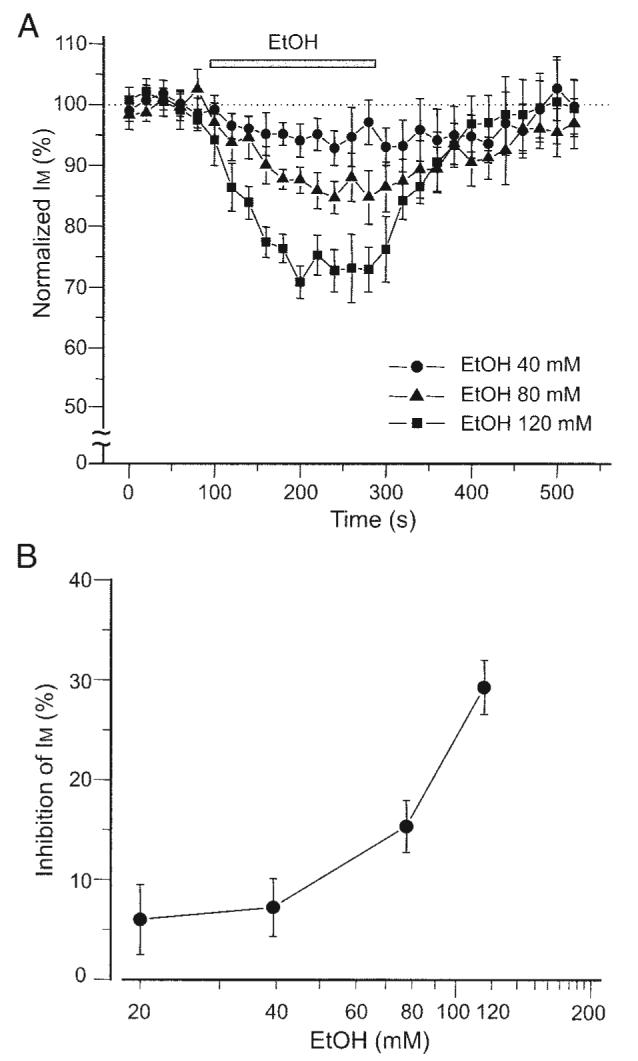
Ethanol effect is concentration dependent. A: time course of ethanol effect on IM in DA VTA neurons. IM was measured with 1-s-long hyperpolarizing voltage step from a VH of −25 to −40 mV. This hyperpolarizing voltage step was given in every 20 s. All IM amplitudes were normalized to the average IM amplitude obtained from the 5 events just before the application of ethanol. Note that ethanol inhibition of IM was concentration dependent; 40 mM (filled circles), 80 mM (filled triangles), and 120 mM (filled squares). B: concentration–response curve for ethanol inhibition of IM. Average maximal ethanol inhibition was obtained with each concentration.
Ethanol inhibition of IM is not voltage dependent
We then examined whether ethanol inhibition of IM was voltage dependent. Figure 3A shows IM induced by a series of hyperpolarizing voltage steps before, during, and after treatment with 120 mM ethanol. Ethanol inhibited IM measured with all four hyperpolarizing voltage steps in seven DA VTA neurons. Each IM in Fig. 3A was obtained by averaging the currents from the seven DA VTA neurons. Figure 3B shows the relationship between membrane voltage and normalized IM amplitude before (open circles), during (filled circles), and after (open triangles) treatment with 120 mM ethanol in the seven DA VTA neurons. The average normalized IM amplitude at −45 mV was 108.1 ± 2.1% in control, 74.2 ± 5.7% in the presence of 120 mM ethanol, and 92.3 ± 9.3% after washout of ethanol. Ethanol significantly reduced normalized IM amplitude (one-way ANOVA, F = 6.9, P < 0.01) but there was no significant difference in normalized IM amplitude between control and washout values (Student–Newman–Keuls post hoc test, P > 0.05). Figure 3C shows the relationship between membrane voltage and mean percentage inhibition of IM by 120 mM ethanol from the same seven DA VTA neurons. The average ethanol inhibition of IM obtained with each voltage was 27.3 ± 3.0% at −35 mV, 31.4 ± 4.9% at −45 mV, 31.9 ± 7.1% at −55 mV, and 31.3 ± 4.4% at −65 mV. There was no correlation between membrane voltage and IM inhibition rate by ethanol (r = −0.76, P > 0.2).
FIG. 3.

Ethanol inhibition of IM is not voltage dependent. A: average IM obtained from 7 DA VTA neurons with 4 different hyperpolarizing voltage steps before, during, and after application of 120 mM ethanol. Hyperpolarizing voltage steps were given from a VH of −25 to −65 mV in 10-mV increments. IM values measured with all 4 voltage steps were inhibited with ethanol. B: relationship between membrane voltage and normalized IM amplitude; control (open circles), 120 mM ethanol (filled circles), and washout of ethanol (open triangles). All IM amplitudes were normalized to the IM amplitude measured at −35 mV before ethanol treatment (arrow). C: relationship between voltage and average percentage inhibition by 120 mM ethanol.
Ethanol increases the firing frequency of DA VTA neurons
In nystatin-perforated patch current-clamp recording, we examined the effect of ethanol on the spontaneous firing of DA VTA neurons. Ethanol (40 mM) increased firing frequency and this ethanol effect was reversible (Fig. 4A1). Figure 4A2 shows the time course of the effect of 40 mM ethanol on spontaneous firing. The effect of 40 mM ethanol on firing frequency for 60-s-long continuous recording was analyzed by an interspike interval (ISI) histogram (see methods) (Fig. 4A3); the distribution of ISI is shifted slightly leftward. Ethanol (80 mM) increased firing frequency and this ethanol effect was also reversible (Fig. 4B1). Figure 4B2 shows the time course of the effect of 80 mM ethanol on spontaneous firing. The effect of 80 mM ethanol on firing frequency was analyzed by the ISI histogram (Fig. 4B3); the distribution of ISI is shifted leftward. As shown in Fig. 4C, ethanol increased the firing frequency of DA VTA neurons in a concentration-dependent manner. Average ethanol-induced increase in firing frequency at each concentration was 6.5 ± 1.0% by 20 mM ethanol (n = 5), 11.4 ± 3.1% by 40 mM ethanol (n = 6), and 26.8 ± 4.9% by 80 mM ethanol (n = 10). From the graph in Fig. 4C, 20% increase in spontaneous firing frequency by ethanol was estimated to be produced by 60 mM. Among 14 DA VTA neurons, 120 mM ethanol increased firing frequency by 13.9 ± 2.6% in seven neurons (50%) and prevented spontaneous firing accompanied with membrane depolarization (depolarization inhibition) in seven neurons (50%). Therefore we did not include the results by 120 mM ethanol in this study.
FIG. 4.
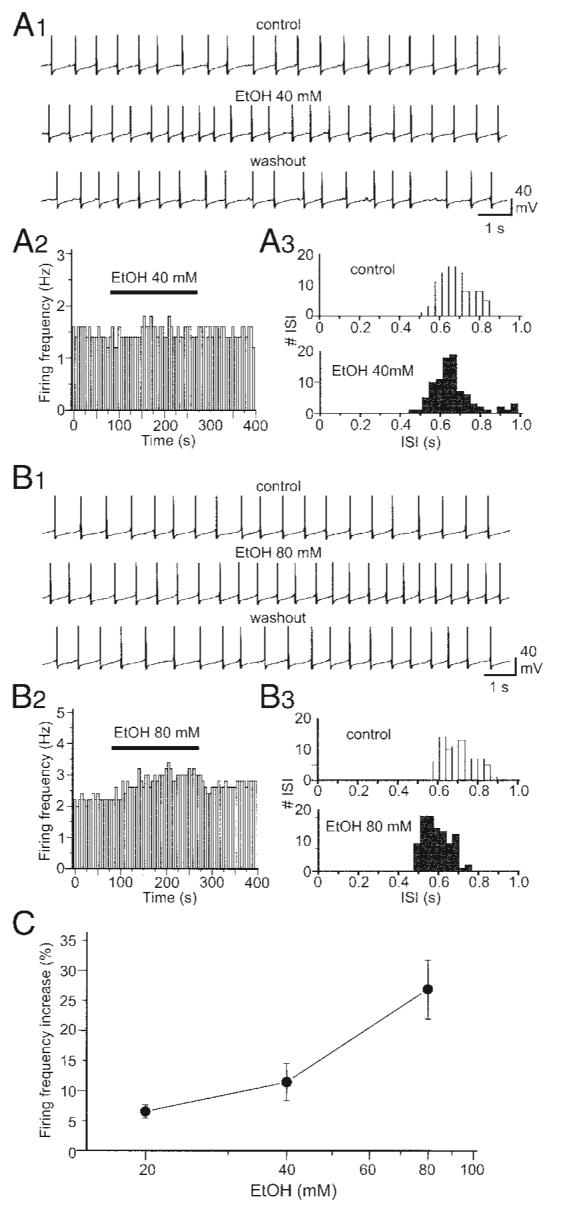
Ethanol increases the firing frequency of DA VTA neurons. A1: current-clamp recording of spontaneous firing from a typical DA VTA neuron before (top), during (middle), and after application of 40 mM ethanol (bottom). A2, left: time course of the effect of 40 mM ethanol from the same neuron in A1. Each vertical bar represents average firing frequency over a 5-s-long interval. Right: interspike interval (ISI) histogram before (white columns) and after treatment with 40 mM ethanol (striped columns) (bin width, 34 ms). B1: current-clamp recording of spontaneous firing from a typical DA VTA neuron before (top), during (middle), and after application of 80 mM ethanol (bottom). B2: time course of the effect of 80 mM ethanol from the same neuron in B1.B3: ISI histogram before (top) and after treatment with 80 mM ethanol (bottom) (bin width, 32 ms). C: concentration–response curve for ethanol-induced firing rate increase in the population of DA VTA neurons tested (20 mM, n = 5; 40 mM, n = 6; 80 mM, n = 10).
Inhibition of IM increases the firing frequency of DA VTA neurons
We next examined how IM inhibition contributes to the spontaneous firing of DA VTA neurons. For this purpose, we used a specific KCNQ channel inhibitor, XE991 (Wang et al. 1998). XE991 (1 μM) increased spontaneous firing frequency (Fig. 5A1). Figure 5A2 shows the time course of the effect of 1 μM XE991 on spontaneous firing. The effect of 1 μM XE991 on firing frequency was analyzed by the ISI histogram (Fig. 5A3); the distribution of ISI is shifted leftward. XE991 (10 μM) increased spontaneous firing frequency, an effect that was reversible (Fig. 5B1). Figure 5B2 shows the time course of the effect of 10 μM XE991 on spontaneous firing. The effect of 10 μM XE991 on firing frequency was analyzed by the ISI histogram (Fig. 5B3); the distribution of ISI is dramatically shifted leftward. As shown in Fig. 5C, XE991 increased the firing frequency of DA VTA neurons in a concentration-dependent manner. Average XE991-induced increase in firing frequency at each concentration was 8.7 ± 2.7% by 0.3 μM XE991 (n = 7), 23.6 ± 6.7% by 1 μM XE991 (n = 7), 42.0 ± 8.2% by 3 μM XE991 (n = 6), and 47.4 ± 7.4% by 10 μM XE991 (n = 8). Among 11 DA VTA neurons, 30 μM XE991 increased firing frequency by 62.1 ± 15.8% in seven neurons (64%) and completely prevented action potential generation accompanied with membrane depolarization in four neurons (36%). Therefore we did not include the results using 30 μM XE991 in this study.
FIG. 5.
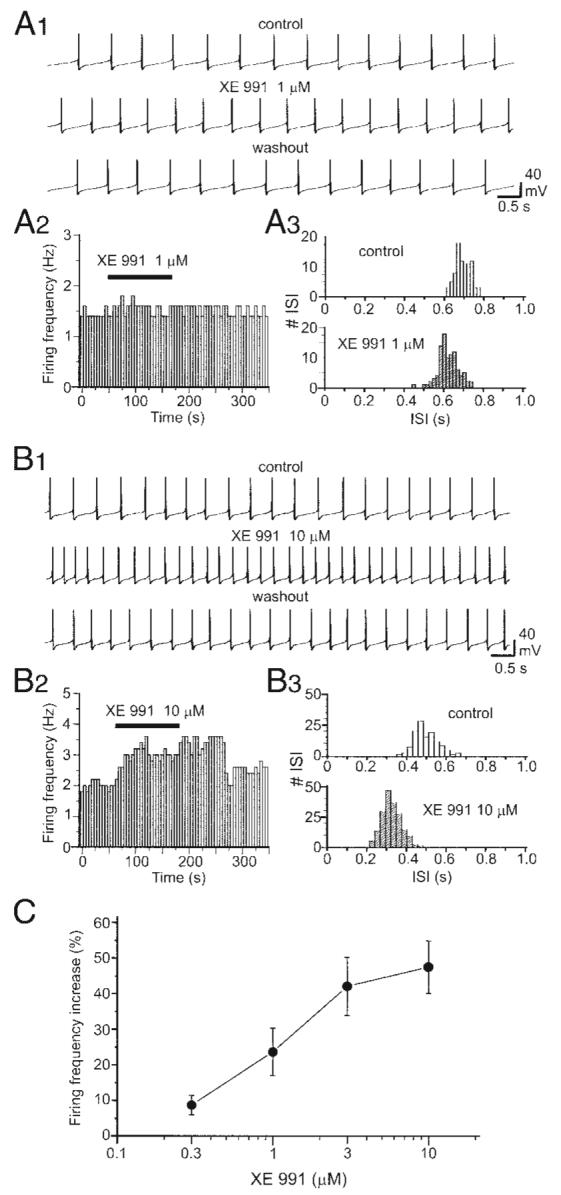
Blockade of IM increases the firing frequency of DA VTA neurons. A1: current-clamp recording of spontaneous firing from a typical DA VTA neuron before (top), during (middle), and after application of 1 μM XE991 (bottom). A2, left: time course of the effect of 1 μM XE991 from the same neuron in A1. Each vertical bar represents average firing frequency over a 5-s-long interval. Right: ISI histogram before (white columns) and after treatment with 1 μM XE991 (striped columns) (bin width, 17 ms). B1: current-clamp recording of spontaneous firing from a typical DA VTA neuron before (top), during (middle), and after application of 10 μM XE991 (bottom). B2: time course of the effect of 10 μM XE991 from the same neuron in B1.B3: ISI histogram before (top) and after treatment with 10 μM XE991 (bottom) (bin width, 35 ms). C: concentration–response curve for XE991-induced firing rate increase in the population of DA VTA neurons tested (0.3 μM, n = 7; 1 μM, n = 7; 3 μM, n = 6; 10 μM, n = 8).
Ethanol inhibition of IM and ethanol-induced increase in firing frequency
The question remained whether ethanol-induced reduction of IM could fully account for ethanol-induced excitation of DA VTA neurons. Our previous study showed that 0.3, 1, 3, and 10 μM XE991 reduced IM amplitude by 27.2 ± 3.8, 51.4 ± 2.9, 81.4 ± 1.7, and 86.6 ± 2.5% in DA VTA neurons (Koyama and Appel 2006a). By comparing the effect of XE991 on IM inhibition in the previous study and the effect of XE991 on firing frequency shown in Fig. 5C, we found a positive correlation between IM inhibition and increase in the firing frequency of DA VTA neurons produced by XE991 (Fig. 6A, open circles). The slope of the linear relationship was 0.5. In addition, we compared IM inhibition by ethanol shown in Fig. 2B and the increase in firing frequency produced by ethanol shown in Fig. 4C. In the same manner, we obtained a positive correlation between IM inhibition by ethanol and ethanol-induced increase in the firing frequency of DA VTA neurons (Fig. 6A, filled circles). The slope of this linear relationship for ethanol effects was 1.3.
FIG. 6.
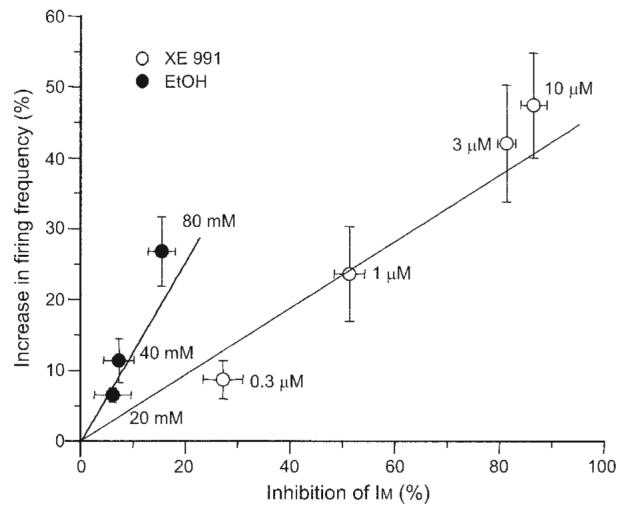
Ethanol inhibition of IM and ethanol-induced increase in firing frequency. Linear relationship between IM inhibition rate and increase in the firing frequency of DA VTA neurons; XE991 (open circles) and ethanol (filled circles).
Ethanol effect on spontaneous firing under the block of IM
Extracellular single-unit recording was used to measure the effect of ethanol (40, 80, and 120 mM) on the firing frequency of DA VTA neurons in brain slices. After washout of the last concentration of ethanol, 10 μM XE991, a concentration that blocked IM by 87% (Koyama and Appel 2006a), was applied and the same concentrations of ethanol were tested again. The percentage increase in firing frequency produced by each ethanol concentration was calculated with the formula shown in methods. Figure 7 shows the concentration–response relationship between ethanol and the percentage increase in firing frequency by ethanol before and after treatment with 10 μM XE991 from 15 DA VTA neurons. The ethanol excitation was concentration dependent (P < 0.001). Before XE991 treatment, average ethanol-induced increase in firing frequency at each concentration was 11.2 ± 1.2% by 40 mM ethanol, 20.8 ± 2.2% by 80 mM ethanol, and 30.4 ± 3.0% by 120 mM ethanol (Fig. 7, filled circles). After treatment with 10 μM XE991, average ethanol-induced increase in firing frequency at each concentration was 10.4 ± 1.6% by 40 mM ethanol, 17.7 ± 1.9% by 80 mM ethanol, and 27.9 ± 4.8% by 120 mM ethanol (Fig. 7, filled squares). Although we anticipated the partial reduction of ethanol-induced excitation in the presence of 10 μM XE991, there was no statistically significant effect of XE991 on the ethanol concentration–response curve (P > 0.05). For the 15 DA VTA neurons, 10 μM XE991 produced no significant change of firing frequency (1.3 ± 1.5%).
FIG. 7.
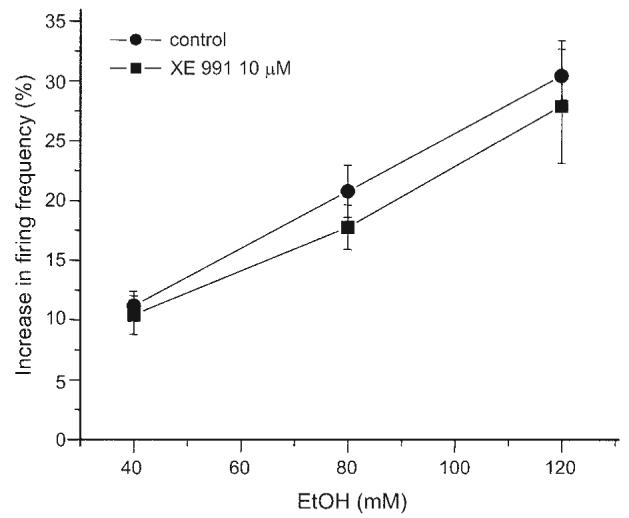
Effect of ethanol on the firing frequency of DA VTA neurons in brain slices under the block of IM. Pooled concentration–response curves for ethanol (40, 80, and 120 mM) effects on spontaneous firing rate measured in DA VTA neurons with extracellular single-unit recording in the absence and presence of 10 μM XE991 (n = 15).
DISCUSSION
In nystatin-perforated voltage-clamp recording, ethanol reduced IM amplitude in a concentration-dependent manner and ethanol inhibition of IM was reversible in DA VTA neurons. Consistent with our results, Moore et al. (1990) showed that ethanol reduces IM amplitude in CA1 hippocampal pyramidal neurons. However, ethanol reduced IM more potently in that study than in the present report. Moore et al. (1990) reported that 44 mM ethanol reduces IM by 36%. Our present study shows that 120 mM ethanol reduces IM by 30%. The difference may have arisen from the types of KCNQ channels expressed in the hippocampus and the VTA. In the hippocampal CA1 region, KCNQ2, KCNQ3, and KCNQ5 channel subunit proteins were reported to be present (Shah et al. 2002). In the VTA, on the other hand, KCNQ2 and KCNQ4 channel subunit proteins were reported to be present (Cooper et al. 2001; Kharkovets et al. 2000). Our previous study suggests that KCNQ2 and KCNQ4 channel subunits are involved in DA VTA neurons by the analysis of pharmacological properties of IM (Koyama and Appel 2006a). A recent study reported that KCNQ4 channel subunit is the major KCNQ subunit in DA VTA neurons and regulates the excitability of these neurons (Hansen et al. 2006). We also found that ethanol inhibition of IM was not voltage dependent, suggesting that the ethanol action site is different from the voltage-sensitive regions of M-channels. This is consistent with other reports indicating that IM inhibition by some drugs of abuse such as ethanol (Moore et al. 1990) and cannabinoids (Schweitzer 2000) does not appear to be voltage dependent.
In nystatin-perforated patch current-clamp recording, 20–80 mM ethanol increased the firing frequency of DA VTA neurons in a concentration-dependent manner. These results are consistent with our previous study using dissociated DA VTA neurons (Brodie et al. 1999). In the present study, we found that ethanol at a concentration of 120 mM prevented spontaneous firing by depolarization inhibition in 50% of DA VTA neurons (at room temperature). In our previous study performed at 34°C, depolarization blockade was not observed in dissociated DA VTA neurons (Brodie et al. 1999). We reported that higher concentrations of ethanol (>300 mM) produce initial increases in firing frequency followed by the complete block of firing in DA VTA neurons (depolarization inhibition) (Appel et al. 2006). There is a possibility that the ethanol-induced depolarization inhibition of spontaneous firing in DA VTA neurons may depend on the temperature of bathing solutions. However, the cellular mechanism of the depolarization inhibition by ethanol in these studies is unclear and additional studies will be needed to elucidate how ethanol produces depolarization inhibition.
Treatment with XE991 increased firing frequency in a concentration-dependent manner in DA VTA neurons. The increase in the spontaneous firing frequency of DA VTA neurons produced by XE991 correlated positively with IM inhibition. This result suggests that IM plays a role in maintaining the spontaneous firing frequency of DA VTA neurons. From the linear relationship obtained with XE991, the contribution of IM inhibition by 80 mM ethanol is estimated to cause a 6% increase in the spontaneous firing frequency, given that 80 mM ethanol inhibited IM by 15%. The ethanol-induced increase in the spontaneous firing frequency of DA VTA neurons also correlated positively with IM inhibition by ethanol. However, we found a difference in the slope values for the linear relationship of IM inhibition versus increase in spontaneous firing frequency between ethanol (1.3) and XE991 (0.5). In addition, the 20% increase in spontaneous firing frequency by ethanol was obtained at 60 mM, which was smaller than the ethanol concentration that produced 20% inhibition of IM amplitude (90 mM). Furthermore, in the pharmacological occlusion experiment using the submaximal concentration of XE991, ethanol still increased the spontaneous firing frequency of DA VTA neurons in a concentration-dependent manner. These observations suggest that if ethanol inhibition of IM underlies excitation of DA VTA neurons, other additional ethanol effects on these neurons increase the magnitude of the effect of IM reduction on firing rate. Although we anticipated the partial reduction of ethanol-induced excitation in the presence of XE991, there was no statistically significant effect of XE991 on the ethanol concentration–response curve. Because 10 μM XE991 inhibits IM amplitude by 87% (Koyama and Appel 2006a) and this XE991 effect could increase the membrane input resistance, additional ethanol-induced excitatory effects on DA VTA neurons may be amplified through the increase in the membrane input resistance and compensating for the reduction of the contribution of ethanol-induced IM inhibition to spontaneous firing frequency. It is noted that dissociated DA VTA neurons were more excited by ethanol and the block of IM than DA VTA neurons in brain slices. For example, 80 mM ethanol-induced increase in spontaneous firing frequency was 27% in dissociated neurons and 21% in neurons from brain slices. The XE991 (10 μM)–induced increase in spontaneous firing frequency was 47% in dissociated neurons and 1.3% in neurons from brain slices. Because dissociated DA VTA neurons were truncated from distal dendrites, the elimination of K+ conductance in distal dendrites presumably causes the dissociated neurons to be more excitable than the neurons in brain slices.
It is possible that ethanol may inhibit the other types of sustained K+ currents rather than IM in DA VTA neurons. We previously showed that 10 mM tetraethylammonium (TEA) blocks IM amplitude by 98% in DA VTA neurons (Koyama and Appel 2006a) and ethanol can still increase the spontaneous firing frequency of these neurons in the presence of 10 mM TEA (Appel et al. 2003). We also showed that ethanol-induced excitation of DA VTA neurons is blocked by a low concentration of quinidine (20–40 μM) (Appel et al. 2003). Thus ethanol may inhibit a quinidine-sensitive and relatively TEA insensitive sustained K+ current in addition to its inhibition of IM in DA VTA neurons. The candidates for the sustained K+ current are three different K+ channel families: Kv1 channels (Grissmer et al. 1994; Yeola et al. 1996), ether-a-go-go (eag) channels (Ludwig et al. 1994; Schonherr et al. 2002), and TASK channels (Leonoudakis et al. 1998; Meadows and Randall 2001). It is also possible that other ion channels such as Ih (Okamoto et al. 2006) could also play a role in ethanol excitation (but see Appel et al. 2003). The data of the present study taken together indicate that, although ethanol causes inhibition of IM and this results in some increase in firing frequency, another effect of ethanol is primarily responsible for the ethanol-induced increase in firing rate. Physiological reduction of this other predominantly ethanol-sensitive current (by neurotransmitter-mediated second-messenger events for example) could increase the relative importance of IM in firing of DA VTA neurons.
ACKNOWLEDGMENTS
The authors thank Dr. John McDaid and M. A. McElvain for assistance in completing the extracellular recording.
GRANTS
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grant AA-05846 to S. B. Appel.
REFERENCES
- Aguayo LG, Pancetti FC. Ethanol modulation of the gamma-aminobutyric acidA- and glycine-activated Cl-current in cultured mouse neurons. J Pharmacol Exp Ther. 1994;270:61–69. [PubMed] [Google Scholar]
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SB, Liu Z, McElvain MA, Brodie MS. Ethanol excitation of dopaminergic ventral tegmental area neurons is blocked by quinidine. J Pharmacol Exp Ther. 2003;306:437–446. doi: 10.1124/jpet.103.050963. [DOI] [PubMed] [Google Scholar]
- Appel SB, McBride WJ, Diana M, Diamond I, Bonci A, Brodie MS. Ethanol effects on dopaminergic “reward” neurons in the ventral tegmental area and the mesolimbic pathway. Alcohol Clin Exp Res. 2004;28:1768–1778. [Google Scholar]
- Appel SB, Wise L, McDaid J, Koyama S, McElvain MA, Brodie MS. The effects of long chain-length n-alcohols on the firing frequency of dopaminergic neurons of the ventral tegmental area. J Pharmacol Exp Ther. 2006;318:1137–1145. doi: 10.1124/jpet.106.105148. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Appel SB. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin Exp Res. 1998;22:236–244. [PubMed] [Google Scholar]
- Brodie MS, Liu PJ, Yu B, Appel SB. Ethanol decreases a sustained potassium current in rat dopaminergic neurons of the ventral tegmental area. Soc Neurosci Abstr. 2000;26:785. [Google Scholar]
- Brodie MS, Pesold C, Appel SB. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res. 1999;11:1848–1852. [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-z. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Cocatre-Zilgien JH, Delcomyn F. Identification of bursts in spike trains. J Neurosci Methods. 1992;41:19–30. doi: 10.1016/0165-0270(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Berger AJ. Mechanisms for the modulation of native glycine receptor channels by ethanol. J Neurophysiol. 2004;91:2685–2695. doi: 10.1152/jn.00907.2003. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G. Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res. 1985;348:201–203. doi: 10.1016/0006-8993(85)90381-6. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Haggard HW, Greenberg LA, Rakieten N. Studies on the absorption, distribution and elimination of alcohol. VI. The principles governing the concentration of alcohol in the blood and the concentration causing respiratory failure. J Pharmacol Exp Ther. 1940;69:252–265. [Google Scholar]
- Hansen HH, Ebbesen C, Mathiesen C, Weikop P, Ronn LC, Waroux O, Scuvee-Moreau J, Seutin V, Mikkelsen JD. The KCNQ opener retigabine inhibits the activity of mesencephalic dopaminergic systems of the rat. J Pharmacol Exp Ther. 2006;318:1006–1019. doi: 10.1124/jpet.106.106757. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Sibbald JR, Morrisett RA. Ethanol alters the frequency, amplitude, and decay kinetics of Sr2+-supported, asynchronous NMDAR mEPSCs in rat hippocampal slices. J Neurophysiol. 2004;91:2568–2577. doi: 10.1152/jn.00997.2003. [DOI] [PubMed] [Google Scholar]
- Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA. 2000;97:4333–4338. doi: 10.1073/pnas.97.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Appel SB. 2004 Abstract Viewer and Itinerary Planner. Society for Neuroscience; Washington, DC: 2004. Characterization of M-current in ventral tegmental area dopamine neurons and reduction by ethanol. Program No. 966.14. Online. [Google Scholar]
- Koyama S, Appel SB. Characterization of M-current in ventral tegmental area dopamine neurons. J Neurophysiol. 2006a;96:535–543. doi: 10.1152/jn.00574.2005. [DOI] [PubMed] [Google Scholar]
- Koyama S, Appel SB. A-type K+ current of dopamine and GABA neurons in the ventral tegmental area. J Neurophysiol. 2006b;96:544–554. doi: 10.1152/jn.01318.2005. [DOI] [PubMed] [Google Scholar]
- Koyama S, Kanemitsu Y, Weight FF. Spontaneous activity and properties of two types of principal neurons from the ventral tegmental area of rat. J Neurophysiol. 2005;93:3282–3293. doi: 10.1152/jn.00776.2004. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Gray AT, Winegar BD, Kindler CH, Harada M, Taylor DM, Chavez RA, Forsayeth JR, Yost CS. An open rectifier potassium channel with two pore domains in tandem cloned from rat cerebellum. J Neurosci. 1998;18:868–877. doi: 10.1523/JNEUROSCI.18-03-00868.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- Li C, Xiong K, Weight FF. Ethanol inhibition of adenosine 5′-triphosphate-activated current in freshly isolated adult rat hippocampal CA1 neurons. Neurosci Lett. 2000;295:77–80. doi: 10.1016/s0304-3940(00)01586-x. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Ludwig J, Terlau H, Wunder F, Bruggemann A, Pardo LA, Marquardt A, Stuhmer W, Pongs O. Functional expression of a rat homologue of the voltage gated ether a go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J. 1994;13:4451–4458. doi: 10.1002/j.1460-2075.1994.tb06767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majchrowicz E, Hunt WA. Temporal relationship of the induction of tolerance and physical dependence after continuous intoxication with maximum tolerable doses of ethanol in rats. Psychopharmacology (Berl) 1976;50:107–112. doi: 10.1007/BF00430477. [DOI] [PubMed] [Google Scholar]
- Meadows HJ, Randall AD. Functional characterisation of human TASK-3, an acid-sensitive two-pore domain potassium channel. Neuropharmacology. 2001;40:551–559. doi: 10.1016/s0028-3908(00)00189-1. [DOI] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Siggins GR. Ethanol diminishes a voltage-dependent K+ current, the M-current, in CA1 hippocampal pyramidal neurons in vitro. Brain Res. 1990;516:222–228. doi: 10.1016/0006-8993(90)90922-x. [DOI] [PubMed] [Google Scholar]
- Mueller AL, Brodie MS. Intracellular recording from putative dopamine-containing neurons in the ventral tegmental area of Tsai in a brain slice preparation. J Neurosci Methods. 1989;28:15–22. doi: 10.1016/0165-0270(89)90005-8. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potential in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, Neu A, Liss B, Roeper J. Ih channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci. 2002;22:1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434:117–165. doi: 10.1016/0165-0173(87)90011-7. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95:619–626. doi: 10.1152/jn.00682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peoples RW, Weight FF. Cutoff in potency implicates alcohol inhibition of N-methyl-d-aspartate receptors in alcohol intoxication. Proc Natl Acad Sci USA. 1995;92:2825–2829. doi: 10.1073/pnas.92.7.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor WR, Soldo BL, Allan AM, Dunwiddie TV. Ethanol enhances synaptically evoked GABAA receptor-mediated responses in cerebral cortical neurons in rat brain slices. Brain Res. 1992;595:220–227. doi: 10.1016/0006-8993(92)91053-h. [DOI] [PubMed] [Google Scholar]
- Rodd-Henricks ZA, McKinzie DL, Crile RS, Murphy JM, McBride WJ. Regional heterogeneity for the intracranial self-administration of ethanol within the ventral tegmental area of female Wister rats. Psychopharmacology (Berl) 2000;149:217–224. doi: 10.1007/s002139900347. [DOI] [PubMed] [Google Scholar]
- Samson HH, Tolliver GA, Schwarz-Stevens K. Oral ethanol self-administration: a behavioral pharmacological approach to CNS control mechanisms. Alcohol. 1990;7:187–191. doi: 10.1016/0741-8329(90)90003-u. [DOI] [PubMed] [Google Scholar]
- Schonherr R, Gessner G, Löber K, Heinemann SH. Functional distinction of human EAG1 and EAG2 potassium channels. FEBS Lett. 2002;514:204–208. doi: 10.1016/s0014-5793(02)02365-7. [DOI] [PubMed] [Google Scholar]
- Schweitzer P. Cannabinoids decrease the K+ M-current in hippocampal CA1 neurons. J Neurosci. 2000;20:51–58. doi: 10.1523/JNEUROSCI.20-01-00051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol. 2000;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard R, Ljungstrøm T, Pedersen KA, Olesen SP, Jensen BS. KCNQ4 channels expressed in mammalian cells: functional characteristics and pharmacology. Am J Physiol Cell Physiol. 2001;280:C859–C866. doi: 10.1152/ajpcell.2001.280.4.C859. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther. 1993;267:250–258. [PubMed] [Google Scholar]
- Ye JH, Tao L, Ren J, Schaefer R, Krnjevic K, Liu PL, Schiller DA, McArdle JJ. Ethanol potentiation of glycine-induced responses in dissociated neurons of rat ventral tegmental area. J Pharmacol Exp Ther. 2001;296:77–83. [PubMed] [Google Scholar]
- Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]