

Abstract
Little is known about the potential for engraftment of autologous hematopoietic stem cells in human adults not subjected to myeloablative conditioning regimens. Five adult patients with the p47phox deficiency form of chronic granulomatous disease received intravenous infusions of autologous CD34+ peripheral blood stem cells (PBSCs) that had been transduced ex vivo with a recombinant retrovirus encoding normal p47phox. Although marrow conditioning was not given, functionally corrected granulocytes were detectable in peripheral blood of all five patients. Peak correction occurred 3–6 weeks after infusion and ranged from 0.004 to 0.05% of total peripheral blood granulocytes. Corrected cells were detectable for as long as 6 months after infusion in some individuals. Thus, prolonged engraftment of autologous PBSCs and continued expression of the transduced gene can occur in adults without conditioning. This trial also piloted the use of animal protein-free medium and a blood-bank-compatible closed system of gas-permeable plastic containers for culture and transduction of the PBSCs. These features enhance the safety of PBSCs directed gene therapy.
Chronic granulomatous disease (CGD) is a rare inherited disorder of phagocytes associated with recurrent life-threatening infections (1, 2). CGD is caused by a defect in the phagocyte NADPH oxidase (phox) that normally generates superoxide. When normal phagocytes engulf opsonized pathogens, the oxidase becomes activated by translocation of three cytoplasmic proteins (p47phox, p67phox, and rac-2) to the cell membrane where they bind to flavocytochrome b558 (a heteromeric transmembrane protein composed of two peptides, gp91phox and p22phox) (3, 4). The genetic basis of CGD is heterogeneous (1, 5). The most common form (about two-thirds of the cases) is X chromosome-linked, resulting from mutations in the gp91phox gene. The next most common form (about one-third of the cases) is autosomal recessive resulting from mutations in the p47phox gene on chromosome 7 (2, 4). The remaining 5% of cases are due to mutations in the genes encoding p22phox (chromosome 16) or p67phox (chromosome 1).
Bone marrow transplantation can cure CGD (6, 7), indicating that the stem cells giving rise to granulocytes and monocytes are an appropriate target for gene therapy. Bone marrow transplantation in CGD has been associated with unacceptably high rates of morbidity, mortality, and graft failure, except in the case of HLA-matched sibling donors (6, 7). Specific gene therapy of autologous peripheral blood stem cells (PBSCs) would avoid these problems. The feasibility of genetic correction of CGD with retrovirus vectors has been demonstrated ex vivo by transduction of human CD34+ PBSCs from patients with each of the four forms of CGD (8–10). Furthermore, genetic correction of the gp91phox and p47phox deficiency forms of CGD has been demonstrated in vivo after stem-cell gene therapy of gene knockout CGD mice and is associated with an increased resistance to infection (11, 12).
In the CGD mouse gene therapy studies, total body radiation was used as a conditioning regimen to enhance engraftment of gene corrected stem cells. Although partial marrow ablation has been thought to be required to optimize engraftment of infused hematopoietic stem cells even in the autologous setting, a number of animal studies using syngeneic cells have suggested that infusion of large numbers of stem cells can partially overcome this barrier (13). In this clinical trial of gene therapy, we examine the potential for engraftment of transduced-gene-corrected autologous CD34+ stem cells in adult patients with the p47phox deficiency form of CGD (p47phox CGD) without marrow conditioning.
MATERIALS AND METHODS
Patients and Consent Documents.
Patients 1 to 5 have p47phox CGD as demonstrated by history of recurrent infections, by phagocytic cells that lack both oxidase activity and p47phox protein, and by p47phox gene mutation analysis (14, 15). Patients 1 to 5 are Caucasian and are, respectively, female, male, female, male, and female, and years of age at study entry were 37, 21, 18, 27, and 27. A gene-therapy phase I protocol with associated informed consent document was reviewed and approved by the National Institute of Allergy and Infectious Disease human investigation review board (Protocol 95-I-0134), by the National Institutes of Health Biosafety Committee (Approval document RD-94-XI-05), by the National Institutes of Health Recombinant DNA Advisory Committee (Protocol 9503–104), and by the U.S. Food and Drug Administration (BB IND 6100).
Protocol Clinical Procedures.
Beginning on study day 1, patients were given six daily subcutaneous injections with granulocyte colony-stimulating factor (Amgen) at 10 μg/kg to mobilize CD34+ PBSCs from the marrow (16). On both study days 5 and 6, a 10- to 15-liter apheresis stem cell collection was performed by using the CS3000 Plus blood cell separator (Baxter Healthcare, Fenwal Division, Deerfield, IL) with manufacturer recommended settings. On study days 8 and 9, the purified cultured and transduced PBSCs derived from the apheresis products were administered intravenously.
Purification of CD34+ PBSCs.
CD34+ PBSC enrichment from the apheresis product was performed by using the ISOLEX 300 SA immunomagnetic stem cell selection system (Baxter Healthcare, Immunotherapy Division, Irvine, CA). The purification procedure using this device was performed by the manufacturer’s instructions. Briefly, CD34+ PBSCs were labeled with murine anti-CD34+ mAb and the labeled cells were magnetically captured by using paramagnetic beads containing surface-bound sheep anti-mouse IgG. CD34+ cells were released from the paramagnetic beads by epitope competition by using a peptide that mimics the CD34 epitope as a releasing agent. In some cases, a few additional CD34+ cells could be released from the beads enzymatically by using chymopapain. CD34+ PBSCs were enumerated by fluorescent antibody flow cytometry analysis (16).
Closed Container System for Handling and Culturing CD34+ PBSCs.
Purified CD34+ PBSCs were handled, cultured, and transduced in a closed system of plastic containers that could be connected sterilely by using a Terumo SCD 312 sterile tubing welder (Baxter, Fenwal Division). Three types of plastic containers were used. Polyvinyl chloride (PL-146) containers (Baxter, Fenwal Division) with limited gas permeability were used to store medium. CD34+ PBSCs were cultured in gas-permeable stem cell culture (PL-2417) containers (Baxter, Immunotherapy Division) that are optimized for growth of PBSCs. Because of differences in tensile properties and construction, the early prototype PL-2417 containers available for this study were not centrifuged, but instead Life Cell gas-permeable (PL-732) containers (Baxter, Immunotherapy Division) designed for lymphocyte culture were used to centrifuge cells.
Production of Clinical Grade Retrovirus Encoding p47phox.
The ORF of human p47phox cDNA (17, 18) was inserted into the MFGS retrovirus vector (Cell Genesys, Foster City, CA) (8–10). MFGS-p47phox plasmid was transfected into the amphotropic envelope packaging line ψ-CRIP, and a vector-producing clone was selected (8–10). For production of cGMP (U.S. Food and Drug Administration current good manufacturing practice) clinical lots of retrovirus supernatant (Cell Genesys), the producer was expanded in DMEM/10% calf serum and then washed with and switched to a serum-free and animal-protein-free medium (X-VIVO 10, BioWhittaker) containing 1% human serum albumin (Baxter Healthcare, Hyland Division, Glendale, CA) for each 8-hr retrovirus supernatant harvest.
PBSC Culture and Transduction Procedures.
On the day of apheresis (culture day 0) purified CD34+ PBSCs were suspended at 0.5 to 2 × 106 cells per ml in PBSC growth medium that was serum-free and animal-protein-free (X-VIVO 10 containing 1% human serum albumin and Pixykine [PIXY321; interleukin 3/granulocyte–macrophage colony-stimulating factor fusion protein from Immunex] at 100 ng/ml and granulocyte colony-stimulating factor (Amgen) at 10 ng/ml. PBSCs were cultured overnight in a PL-2417 gas-permeable container in 7% CO2/93% air at 37°C. The next morning (culture day 1) the cells were transferred to a PL-732 container, centrifuged, and resuspended into 50% vector supernatant (titer ∼106 transducing units/ml) containing the same growth factor concentrations and at the same cell concentration as for overnight culture. Cells were spin-transduced at 1,200 × g at 32°C for 1 hr (10) and incubated 5 hr at 37°C, 7% CO2/93% air, after which cells were transferred back to PBSC growth medium in the PL-2417 container overnight. The transduction procedure was repeated on culture days 2 and 3 after which the cells were washed and resuspended in Plasmalyte (Baxter, Hyland Division) containing 1% human serum albumin for intravenous administration. Samples of cells were retained in liquid culture or plated in agarose for further analysis. Cultures of nontransduced PBSCs from the patients and PBSCs from normal volunteers (Human Investigation Review Board approved National Institutes of Health Protocol 94-I-0073) served as negative and positive controls for assays of oxidase activity.
Analysis of Transduction and Correction of Oxidase Activity.
At the end of culture day 3, PBSCs were plated in agarose to allow formation of myeloid colonies, which were evaluated for oxidase activity by using a phorbol 12-myristate 13-acetate (PMA)-stimulated nitroblue tetrazolium dye (NBT) test (8, 10). Myeloid colonies demonstrating intense staining with the formazan precipitate were scored as positive. CD34+ cells were maintained in liquid culture for 17 days and analyzed for PMA-stimulated superoxide production by using a chemiluminescence assay (8–10). SDS/PAGE and immunoblotting were used to detect production of p47phox protein in these cells (8, 10).
A flow cytometry assay of oxidant production using dihydrorhodamine 123 (DHR) loading of the cells also was performed (10, 12, 19, 20). At day 17, liquid cultures of normal CD34+ PBSCs undergoing myeloid differentiation contain 10–12% granulocytes, which are the only cells that fluoresce brightly in the flow cytometry DHR assay after PMA stimulation (10). The DHR assay also was used to detect oxidase-positive neutrophils in vivo in the peripheral blood of patients after gene therapy (19, 20). Correction of peripheral blood neutrophils also was evaluated visually by NBT staining (21).
Vector copy number in the ex vivo-transduced CD34+ PBSCs was determined by Southern blot hybridization (22), and a PCR assay was used for detection of transduced p47phox cDNA present in vivo in peripheral blood leukocyte genomic DNA (23, 24). For the PCR assay, nested oligonucleotide primers derived from p47phox cDNA sequence were designed to overlap exon junctions (14, 15) and were found empirically to amplify p47phox cDNA sequence but not genomic sequence. The outer primer pairs are AGCACTAT/GTGTACATGTTCC (bp 65–85, exons 1/2) and GACGTATGGCTCAC/CTGCATAGTTG (bp 696–672, exons 8/7). The inner primer pairs are CTACGAGTTCCAT/AAAACC (bp 140–169, exons 2/3) and CCGGTGATGT/CTGTCGCGG (bp 481–443, exons 5/4). Two detection methods were used. The inner primer pair was used to produce a labeled sequence to probe the outer primer pair PCR product by Southern blotting or was used in a second PCR to amplify a specific nested PCR sequence from the first PCR product (24).
Safety Testing of Patient Blood.
Because all five patients have a protein null phenotype of p47phox CGD, patient serum was tested for the development of antibody specific to p47phox by SDS/PAGE and immunoblot (8) detection of recombinant p47phox. Genomic DNA from patient peripheral blood cells was screened for the presence of replication competent retrovirus by using a PCR assay to detect sequence encoding amphotropic envelope (22, 24).
RESULTS
Ex Vivo Culture and Transduction of CD34+ PBSCs in Serum-Free Medium and Gas-Permeable Containers.
Patients were free of active infection at study entry, though patient 1 had recovered recently from a pneumonia. Blood studies were within normal limits except for mild anemia (all hematocrits were >26). As expected (16), granulocyte colony-stimulating factor mobilization of CD34+ cells to the peripheral blood of CGD patients was modest and varied among CGD patients. By day 5, the concentration of CD34+ cells had increased from baseline levels below two cells per μl in all patients to a level in patients 1 to 5 of 53, 27, 22, 81, and 13 CD34+ cells per μl, respectively. Despite the apheresis procedure on day 5, these counts were similar at the time of the day 6 apheresis. Ten apheresis products were collected (2 per patient) averaging 35 ± 3.7 × 109 mononuclear cells per product (mean ± SEM). After ISOLEX immunoaffinity selection, a mean of 125 ± 23.6 × 106 cells (n = 10) was recovered, at a median purity of 80% CD34+ cells resulting in a median yield of 38%.
After the 3 days of culture and transduction, each patient received two autologous products (Table 1, preparations A and B) without any symptoms or changes in vital signs. The total number of cells infused ranged from 0.1 to 4.7 × 106 cells per kg (Table 1). Transduced PBSCs averaged 91 ± 2.7% cell viability, had a colony plating efficiency of 104 ± 38.4 colonies per 1,000 cells plated, and passed safety testing for sterility, endotoxin, and replication competent retrovirus. SDS/PAGE and immunoblotting demonstrated a strong positive signal for the presence of recombinant p47phox protein in all of the transduced CGD PBSC cultures (Fig. 1, lanes B and C). Shown in Table 1 are studies performed on each product to assess the percent of cells expressing CD34 antigen on culture day 3, correction of oxidase activity by chemiluminescence and DHR assays on culture day 17, the percent of myeloid-colony-forming progenitors plated on day 3 giving rise to oxidase-positive colonies, and the vector copy number in the transduced PBSCs. The data are consistent with preservation of a primitive phenotype and a high rate of gene transfer and functional correction ex vivo. Though not shown, nontransduced PBSC cultures from the 10 apheresis products from the five patients demonstrated less than 1% of normal chemiluminescence and DHR assay and gave rise to no NBT-positive myeloid colonies. When 17-day-cultured PBSCs from the CGD patients were compared with cultured PBSCs from normal individuals, similar numbers of granulocytes are present by morphological examination in transduced and nontransduced cultures of CGD PBSCs and in the cultures of normal PBSCs, but highly fluorescent oxidase positive cells were detected in the flow cytometry DHR assay only in transduced cell cultures from patients as reported in Table 1 and from normal individuals. Though not shown, the mean fluorescence per corrected patient granulocyte in the transduced cultures was similar to that seen with granulocytes derived from cultured normal PBSCs, suggesting full restoration of oxidase activity in gene-corrected granulocytes ex vivo.
Table 1.
Evaluation of autologous PBSC after ex vivo transduction and culture
| Patient* | Prep | Cells infused† | % CD34+‡ | % of normal chemiluminescence§ | DHR assay,§ % granulocytes corrected | % NBT-positive colonies¶ | Vector copy number‖ |
|---|---|---|---|---|---|---|---|
| 1 | A | 60 | 85 | 25 | 30 | 9 | 0.05 |
| B | 200 (4.7) | 97 | 26 | 21 | 6 | 0.11 | |
| 2 | A | 12 | 84 | 26 | 90 | 29 | 0.19 |
| B | 45 (0.9) | 94 | 23 | 44 | 28 | 0.08 | |
| 3 | A | 215 | 61 | 65 | 75 | 14 | 0.13 |
| B | 66 (4.3) | 85 | 64 | 75 | 18 | 0.18 | |
| 4 | A | 77 | 92 | 27 | 34 | 9 | 0.16 |
| B | 129 (2.5) | 81 | 36 | 63 | 11 | 0.11 | |
| 5 | A | 2 | 63 | 32 | 27 | 19 | 0.13 |
| B | 2 (0.1) | 79 | 39 | 59 | 14 | 0.13 |
Each patient received by vein two preparations (Prep, A and B) of the transduced and cultured PBSCs derived from the first and second apheresis procedures, respectively.
At culture day 3 for each preparation, the number of cells shown (×10−6) were infused intravenously. Shown in the parentheses is the total number of cells (×10−6) (A plus B) infused per kg of body weight.
Measured by flow cytometry analysis at the end of culture day 3.
Assays were performed on day 17 of culture.
Cells were plated at culture day 3 and assayed 14 days later.
Measured by Southern blot of genomic DNA from the transduced and cultured PBSCs, probed with a MFGS-vector-specific 5′ long terminal repeat sequence and a cell line with known vector copy number of 1 as a reference.
Figure 1.
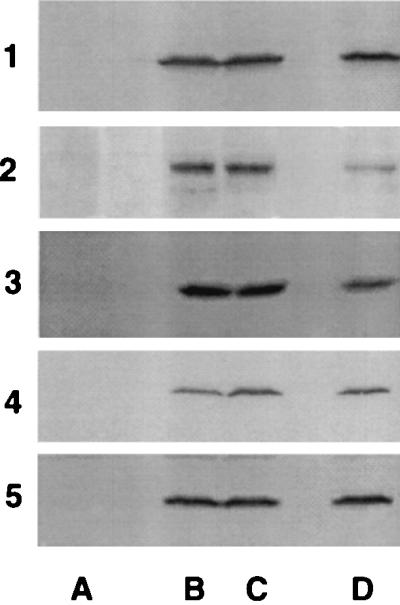
Correction of p47phox protein deficiency ex vivo. These are SDS/PAGE immunoblots demonstrating detection of p47phox protein in transduced or control CD34+ PBSCs at culture day 17. The results for studies of patients 1 to 5 are shown from the top to bottom, as indicated. Shown in lanes A are the analyses of nontransduced cultured CD34+ PBSCs from each patient and as expected no signal is detected. Shown in lanes B and C are the analyses of cultured and MFGS-p47phox-transduced CD34+ PBSCs derived from the first and second apheresis products from each patient. Shown in lanes D as a positive control in each case is an analysis of nontransduced normal control CD34+ PBSCs cultured in parallel with the patient cells.
Presence of NADPH Oxidase-Positive Neutrophils in the Peripheral Blood After Intravenous Administration of Ex Vivo Transduced Autologous CD34+ PBSCs.
The flow cytometry DHR assay was used to measure the appearance of NADPH oxidase-positive granulocytes in the peripheral blood after gene therapy. The dot plot shown in Fig. 2A demonstrates that the events generated from analysis of PMA-stimulated normal peripheral blood granulocytes cluster in a tight band at the right side of the graph, consistent with robust oxidase activation. As shown in Fig. 2B, analysis of PMA-stimulated blood granulocytes from patient 1 before gene therapy resulted in all but a single event to the left of the “positive threshold” line, characteristic of absent oxidase function. At day 24 after gene therapy, PMA-stimulated peripheral blood granulocytes from patient 1 generated almost 80 events appearing to the right of the “positive threshold” line in a tight cluster (Fig. 2C) with mean fluorescence intensity (x axis) similar to that of granulocytes from the normal control (compare Fig. 2 A and C). Although the number of corrected cells is small, the data indicate that these gene corrected granulocytes from patient 1 have acquired oxidase activity similar to normal cells. Qualitatively similar results were obtained for the other four patients. To confirm the results of the DHR assay by direct visualization of individual neutrophils, an NBT stain of PMA-stimulated peripheral blood neutrophils was performed. In the example shown in Fig. 3 with blood from patient 1 at day 26 after gene therapy, 1 in 2,000 granulocytes was oxidase-positive, consistent with the count determined by DHR assay as reported in Fig. 4.
Figure 2.

Correction of neutrophil oxidase activity in vivo. These are dot plots of the flow cytometry DHR assays of oxidant production by PMA-stimulated peripheral blood granulocytes. Shown are analyses of granulocytes. (A) Normal volunteer. (B) Patient 1 before gene therapy. (C) Patient 1 at 24 days after gene therapy. Each event (dot) represents the analysis of parameters derived from a single cell. The data shown have been gated to include only events with the forward × side scatter characteristics of granulocytes. Data are plotted to evaluate fluorescence (x axis) as a measure of oxidase activity and side scatter (y axis) as a means of distinguishing individual granulocytes (events). The “positive threshold” vertical line is set so that 95% of stimulated normal granulocytes are to the right of that line in the region defined as oxidase positive.
Figure 3.

Correction of neutrophil oxidase activity in vivo. This photomicrograph shows NBT-stained PMA-stimulated neutrophils from peripheral blood of patient 1 at day 26 after gene therapy. The cytospin preparation is counterstained red-orange with safranin to visualize segmented neutrophil nuclei. Shown in the center is a single NBT-positive neutrophil that is partly obscured by the dense blue-black precipitate of formazan, a product of NBT reduction by superoxide. This amount of precipitate is evidence of vigorous production of superoxide by this gene-therapy-corrected neutrophil. A visual count of neutrophils on this slide showed that about 1 in 2,000 cells were NBT-positive.
Figure 4.

Prolonged production of oxidase-corrected granulocytes in vivo. These bar graphs demonstrate over time the proportion of oxidase-positive neutrophils in the peripheral blood after gene therapy of the five CGD patients. For each data point, flow cytometry DHR assay was performed on peripheral blood leukocytes and the number of oxidase-positive neutrophils was determined as shown in Fig. 2. From top to bottom, the results from analyses of blood from patients 1 to 5, respectively, are shown. For all patients, the vertical axis (oxidase-positive neutrophils per 100,000 cells) is the same scale allowing direct visual comparison between patients. However, the horizonal axis is not proportional and the numbers beneath the data bars indicate the days of analyses relative to the first intravenous administration of transduced autologous CD34+ PBSCs and differ for each patient.
Each patient was followed over time for detection of corrected granulocytes in peripheral blood (Fig. 4). In each subject no oxidase-corrected granulocytes were detected for at least 2 weeks after transplantation of the autologous transduced PBSCs. After that time, an increasing number of oxidase-positive granulocytes could be detected in the peripheral blood. The peak response occurred between day 25 (patient 3) and day 53 (patient 2) with a mean of 35 days. The maximum percent of oxidase-positive granulocytes at the peak ranged from 0.004% (patient 2) to 0.051% (patient 1) with a mean of 0.019% (about 1 in 5,000 cells). The range of duration of detection of oxidase-corrected granulocytes was 51 days (patient 5) to 172 days (patient 3) with a mean of 118 days. Of note is that the kinetics of appearance of oxidase-corrected cells did not rise to a single peak followed by a smooth decay over time but instead appeared to rise and fall several times over the duration of detection of positive signal. For example, patient 3 demonstrates five (possibly six) maxima at days 25, possibly 32, 39, 53, 102, and 172.
It is of note that the greatest number of transduced PBSCs were administered to patients 1, 3, and 4 in that rank order and that these same three patients showed the longest duration of detection of corrected granulocytes. Furthermore, patients 1 and 3 had the highest peak number of such corrected granulocytes. Patients 2 and 5, in that rank order, had far fewer transduced PBSC transfused and this correlated with patients 2 and 5, in that order, having the shortest duration and patient 2 having the lowest peak number of corrected granulocytes. Patients 1 to 5 have now been followed after gene therapy for 25, 23, 22, 21, and 20 months (as of August 1997), respectively, but no oxidase-positive cells have been seen in any peripheral blood DHR assay after the last data points shown in Fig. 4. PCR detection of the transduced p47phox cDNA was performed on genomic DNA isolated from peripheral blood leukocytes as shown in Table 2 and confirms that a transient low level of gene marking of peripheral blood leukocytes occurred.
Table 2.
PCR detection of gene marking
| Pt 1
|
Pt 2
|
Pt 3
|
Pt 4
|
Pt 5
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| Day* | PCR† | Day | PCR | Day | PCR | Day | PCR | Day | PCR |
| −6 | − | −3 | − | −8 | − | −6 | − | −15 | − |
| 28 | + | 24 | +/−‡ | 3 | − | 29 | + | −3 | − |
| 33 | + | 28 | − | 26 | + | 33 | + | 29 | − |
| 124 | − | 68 | − | 55 | + | 40 | + | 34 | + |
| 147 | − | 93 | − | 70 | + | 65 | − | 61 | − |
| 241 | − | 190 | − | 92 | − | 89 | − | 82 | − |
| 313 | − | 357 | − | 119 | − | 118 | − | 117 | + |
| 362 | − | 147 | + | 132 | − | 139 | − | ||
| 175 | − | 352 | − | 335 | − | ||||
| 205 | − | ||||||||
| 232 | − | ||||||||
Pt, patient.
Number of days before or after gene therapy.
Vector p47phox cDNA sequence detected (+) or not detected (−) in blood leukocytes. All + were <0.1% of cells marked.
The +/− indicates positive signal detected by Southern blotting of the PCR product but not by nested primer detection.
Results of Safety Studies and Long-Term Clinical Follow Up.
PCR assay of amphotropic envelope sequence failed to detect evidence of replication-competent retrovirus in peripheral blood leukocytes at 1, 3, 6, and 9 months after gene therapy. Similarly, serum samples were negative for anti-p47phox antibodies at 1 and 3 months after gene therapy. All five patients are currently stable without infection. Except for patient 1, the other four patients have had no deep tissue infections during the follow-up period, and hematologic, renal, and liver function tests are normal. Patient 1 had severe Burkholderia cepacia pneumonia 3 weeks after gene therapy, from which she recovered. During her pneumonia oxidase-positive neutrophils were detected in an empyema by flow cytometry DHR assay and NBT stain, demonstrating that these gene therapy corrected cells were capable of migrating to an inflammatory focus. It is possible that host responses to this infection affected the peak level of gene corrected granulocytes seen in this individual.
DISCUSSION
Our data demonstrate the appearance of gene-corrected oxidase-positive granulocytes in the peripheral blood of each of five patients with p47phox CGD after PBSC-targeted gene therapy with vector encoding p47phox. In patient 1, we also demonstrated that the gene-corrected oxidase-positive neutrophils could migrate from the circulation to a site of infection. Moreover, the kinetics of appearance of these functionally corrected granulocytes share similar characteristics in all patients. In all patients, the first appearance of oxidase-positive granulocytes required at least 2 weeks, suggesting that engraftment, proliferation, and differentiation of the transduced PBSCs in the marrow was required. In all patients, there was an initial wave of oxidase-positive cells first peaking at 22–39 days followed by one or more waves (usually of much smaller magnitude) of oxidase-positive cells at intervals out to 6 months in some individuals. If this periodicity is real, it could be evidence of clonal succession where only a small subset of primitive progenitors in the marrow contribute to active hematopoiesis at any time (25).
Most studies of gene marking of autologous PBSCs have subjected patients to the myeloablative conditioning usually associated with cancer therapy (ref. 26; for review, see ref. 27). Less is known about engraftment potential of autologous progenitors without ablative marrow conditioning. With some disorders such as adenosine deaminase-deficient severe combined immune deficiency or Fanconi syndrome, gene-therapy-corrected lymphocytes or hematopoietic progenitors may have a selective growth advantage over uncorrected cells (28–31). In reports of the use of transduced hematopoietic blood stem cells to treat adenosine deaminase-deficient severe combined immune deficiency without myeloablation, there is evidence of prolonged engraftment (30, 31). The three newborn infants with adenosine deaminase-deficient severe combined immune deficiency treated with transduced autologous cord blood stem cells show evidence of rising levels of gene-marked lymphocytes, though marking of myeloid cells remains very low in a range similar to that seen in our present study (31). Because correction of adenosine deaminase deficiency should provide a selective growth advantage specifically to lymphocytes, this finding might have been expected. In addition, cord blood is a particularly rich source of stem cells and infants and very young children may be more receptive to engraftment of autologous cells. It remains to be seen whether permanent high-level engraftment of autologous-gene-marked hematopoietic stem cells can be achieved in nonconditioned older children or adults, where gene transfer provides no selective growth advantage.
We have demonstrated in a congenic mouse model system that low-dose nonablative radiation conditioning can increase greatly the engraftment of congenic marrow stem cells in a radiation dose-dependent manner (32). This suggests the possibility that acceptable regimens of marrow conditioning may be developed for hematopoietic-stem-cell-targeted gene therapy. Such conditioning might increase the level of gene marking from that seen in our current study to levels that could provide prolonged clinical benefit.
The DHR assay of oxidase function provides strong evidence that low-level prolonged engraftment of gene-marked hematopoietic progenitors can occur in human adults without marrow ablation or conditioning. Though the PCR confirmed this, PCR was not sensitive enough to detect marking at later time points where the flow cytometry DHR assay continued to indicate lower levels of oxidase-positive neutrophils. The PCR data demonstrate that the eventual disappearance of gene-corrected oxidase-positive granulocytes by DHR assays was not associated with continued presence in the peripheral blood of substantial numbers of leukocytes marked with a nonfunctional transduced gene. Although we cannot exclude the possibility that silencing of transcription of the transduced oxidase gene is occurring, the data are more consistent with disappearance of transduced cells. This might happen if very early progenitors rather than permanently repopulating stem cells were targeted. Several published human clinical studies of hematopoietic-stem-cell-targeted gene transfer (26, 27, 30, 31) have demonstrated low-level engraftment of retrovirus-transduced gene in blood or marrow cells by using PCR. Although these studies indicate the presence of the transduced gene, assessment of gene function in the host, as in our study and others (33), provides an important additional insight regarding the clinical potential for gene therapy. How to target the most primitive stem cells and how to prevent transcription silencing in vivo remain important issues to resolve in future studies.
An additional goal of this clinical trial was to develop and pilot the use of materials and methods that increase the safety of ex vivo gene therapy targeting hematopoietic stem cells. Animal proteins, including fetal calf serum, are widely used as required supplements to most cell culture media. Animal proteins internalized by human cells during prolonged culture may not be removed by centrifugation washing and can stimulate an immune response (34). Because gene therapy is in an early developmental stage, it is likely that any patients participating in these initial studies will be treated again in the future at a time when such treatments are more efficient. If it is at all possible to limit exposure to animal proteins, particularly fetal calf serum, in these early studies without compromising the scientific goals of the study, then such a safety feature should be incorporated into the protocol. A second important safety feature incorporated into this study was a closed system of gas-permeable flexible plastic containers for culture and transduction. The closed system reduces the contamination risk associated with pipetting cells and medium yet allows such handling to become a counter-top process. Biosafety cabinets are required at only a few steps and the system is compatible with techniques already used widely in most blood banks. We demonstrate that it is possible to incorporate these safety features without compromising PBSC viability or transduction efficiency.
The clinical potential of gene therapy is yet to be realized, and there has been considerable interest in defining both the scientific and clinical goals of human trials of gene transfer. In the case of CGD, where life-threatening infections may require many weeks or months of therapy and relapses are frequent, use of gene therapy to provide even short- to medium-term production of oxidase-positive autologous granulocytes may be clinically beneficial. This concept is supported by published studies of gene therapy in mouse models of both the X chromosome-linked (gp91phox-deficiency) and p47phox-deficiency forms of CGD that demonstrate that even transient partial correction of the oxidase defect is associated with some protection against infection challenge (11, 12). Furthermore, in human female carriers of the X chromosome-linked form of CGD, the X chromosome inactivation that occurs during embryogenesis results in phenotypic mosaicism at the cellular level in which both oxidase-positive and oxidase-negative granulocytes can be detected in the peripheral blood (21). Because this is a stochastic process, some female carriers can be found who have only 3% to 5% oxidase-positive neutrophils yet do not suffer from an increased incidence of infection. The knockout mouse studies and the clinical observations of X chromosome-linked CGD carriers suggest that even a short-term low level of gene correction in CGD could be clinically beneficial for treatment of severe prolonged infections. Until the tools are developed to achieve high-level permanent gene transfer to hematopoietic cells, our studies suggest that an achievable intermediate goal of development of gene therapy for CGD might be to augment neutrophil function in the treatment of severe infections.
Acknowledgments
Somatix Therapy Corporation, an industrial collaborator during the conduct of this study, is now a part of Cell Genesys. We thank Immunex for providing Pixykine for ex vivo culture of CD34+ PBSCs. We are grateful for the important contributions of the National Institute of Allergy and Infectious Diseases 11 East day hospital staff and the National Institutes of Health transfusion medicine apheresis staff. We thank Dr. Stephen Chanock for doing the p47phox mutation analysis of our patients. We thank Dr. Philip Murphy for critical reading of the manuscript and Dr. Douglas Kuhns for preparing Fig. 3. Finally, we thank the participating patients and the physicians who served as physician-advocates for their patients during the informed consent process.
ABBREVIATIONS
- PBSC
peripheral blood stem cell
- CGD
chronic granulomatous disease
- phox
phagocyte NADPH oxidase
- PMA
phorbol myristate acetate
- NBT
nitroblue tetrazolium dye
- DHR
dihydrorhodamine 123
References
- 1.Curnutte, J. T. (1993) Clin. Immunol. Immunopathol. 67, Suppl., 2–15. [DOI] [PubMed]
- 2.Gallin J I, Malech H L. J Am Med Assoc. 1990;263:1533–1537. [PubMed] [Google Scholar]
- 3.DeLeo F R, Quinn M T. J Leukocyte Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 4.Malech H L. Curr Opin Hematol. 1993;1:123–132. [Google Scholar]
- 5.Clark R A, Malech H L, Gallin J I, Nunoi H, Volpp B D, Pearson D W, Nauseef W M, Curnutte J T. N Engl J Med. 1989;321:647–652. doi: 10.1056/NEJM198909073211005. [DOI] [PubMed] [Google Scholar]
- 6.Calvino M C, Maldonado M S, Otheo E, Munoz A, Couselo J M, Burgaleta C. Eur J Pediatr. 1996;155:877–879. doi: 10.1007/BF02282837. [DOI] [PubMed] [Google Scholar]
- 7.Ho C M, Vowels M R, Lockwood L, Ziegler J B. Bone Marrow Transplant. 1996;18:213–215. [PubMed] [Google Scholar]
- 8.Sekhsaria S, Gallin J I, Linton G F, Mallory R M, Mulligan R C, Malech H L. Proc Natl Acad Sci USA. 1993;90:7446–7450. doi: 10.1073/pnas.90.16.7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li F, Linton G L, Sekhsaria S, Whiting-Theobald N, Katkin J P, Gallin J I, Malech H L. Blood. 1994;84:53–58. [PubMed] [Google Scholar]
- 10.Weil W M, Linton G F, Whiting-Theobald N, Vowells S J, Rafferty S P, Li F, Malech H L. Blood. 1997;89:1754–1761. [PubMed] [Google Scholar]
- 11.Bjorgvinsdottir H, Ding C, Pech N, Gifford M A, Li L L, Dinauer M C. Blood. 1997;89:41–48. [PubMed] [Google Scholar]
- 12.Mardiney M, III, Jackson S H, Spratt S K, Li F, Holland S M, Malech H L. Blood. 1997;89:2268–2275. [PubMed] [Google Scholar]
- 13.Stewart M F, Crittenden R B, Lowry P A, Pearson-White S, Quesenberry P J. Blood. 1993;10:2566–2571. [PubMed] [Google Scholar]
- 14.Casimir C M, Bu-Ghanim H N, Rodaway A R, Bentley D L, Rowe P, Segal A W. Proc Natl Acad Sci USA. 1991;88:2753–2757. doi: 10.1073/pnas.88.7.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roos D, de Boer M, Kuribayashi F, Meischl C, Weening R S, Segal A W, Ahlin A, Nernet K, Hossle J P, Bernatowska-Matuszkiewicz E, Middleton-Price H. Blood. 1996;87:1663–1681. [PubMed] [Google Scholar]
- 16.Sekhsaria S, Fleisher T A, Vowells S, Brown M, Miller J, Gordon I, Blaese R M, Dunbar C E, Leitman S, Malech H L. Blood. 1996;88:1104–1112. [PubMed] [Google Scholar]
- 17.Lomax K J, Leto T L, Nunoi H, Gallin J I, Malech H L. Science. 1989;245:409–412. doi: 10.1126/science.2547247. [DOI] [PubMed] [Google Scholar]
- 18.Volpp B D, Nauseef W M, Donelson J E, Moser D R, Clark R A. Proc Natl Acad Sci USA. 1989;86:7195–7199. doi: 10.1073/pnas.86.18.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vowells S J, Sekhsaria S, Malech H L, Shalit M, Fleisher T A. J Immunol Methods. 1994;178:89–97. doi: 10.1016/0022-1759(94)00247-t. [DOI] [PubMed] [Google Scholar]
- 20.Vowells S J, Fleisher T A, Sekhsaria S, Alling D W, Maguire T E, Malech H L. J Pediatr. 1996;128:104–107. doi: 10.1016/s0022-3476(96)70437-7. [DOI] [PubMed] [Google Scholar]
- 21.Buescher E S, Alling D W, Gallin J I. J Clin Invest. 1985;76:1581–1584. doi: 10.1172/JCI112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellem K A, O’Rourke M G, Johnson G R, Parry G, Misko I S, Schmidt C W, Parsons P G, Burrows S R, Cross S, Fell A, Li C L, Bell J R, Dubois P J, Moss D J, Good M F, Kelso A, Cohen L K, Dranoff G, Mulligan R C. Cancer Immunol Immunother. 1997;44:10–20. doi: 10.1007/s002620050349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. , Chap. 9, pp. 9.14–9.23. [Google Scholar]
- 24.Shalit M, Sekhsaria S, Li F, Mauhorter S, Mahanti S, Malech H L. J Allergy Clin Immunol. 1996;98:344–354. doi: 10.1016/s0091-6749(96)70159-8. [DOI] [PubMed] [Google Scholar]
- 25.Cook P C, Jiang S, Chertkov J L, Fan Y, Levere R D, Abraham N G. Acta Haematol. 1996;96:57–63. doi: 10.1159/000203716. [DOI] [PubMed] [Google Scholar]
- 26.Dunbar C E, Cottler-Fox M, O’Shaughnessy J A, Doren S, Carter C, Berenson R, Brown S, Moen R C, Greenblatt J, Stewart F M, Leitman S F, Wilson W H, Cowan K, Young N S, Nienhuis A W. Blood. 1995;85:3048–3057. [PubMed] [Google Scholar]
- 27.Brenner M K. J Pediatr Hematol Oncol. 1997;19:1–6. doi: 10.1097/00043426-199701000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Walsh C E, Grompe M, Vanin E, Buchwald M, Young N S, Neinhuis A W, Liu J M. Blood. 1994;84:453–459. [PubMed] [Google Scholar]
- 29.Blaese R M, Culver K W, Miller A D, Carter C S, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt J J, Rosenberg S A, Klein H, Berger M, Mullen C A, Ramsey W J, Muul L, Morgan R A, Anderson W F. Science. 1995;270:475–480. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 30.Bordignon C, Notarangelo L D, Nobili N, Ferrari G, Lasorati G, Panina P, Mazzolari E, Maggioni D, Rossi C, Servida P, Ugazio A G, Mavilio F. Science. 1995;270:470–475. doi: 10.1126/science.270.5235.470. [DOI] [PubMed] [Google Scholar]
- 31.Kohn D B, Weinberg K I, Nolta J A, Heiss L N, Lenarsky C, Crooks G M, Hanley M E, Annett G, Brooks J S, El-Khoureiy A, Lawrence K, Wells S, Moen R C, Bastian J, Williams-Herman D E, Elder M, Wara D, Bowen T, Hershfield M S, Mullen C A, Blaese R M, Parkman R. Nat Med. 1995;1:1017–1023. doi: 10.1038/nm1095-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mardiney M, III, Malech H L. Blood. 1996;87:4049–4056. [PubMed] [Google Scholar]
- 33.Mullen C A, Snitzer K, Culver K W, Morgan R A, Anderson W F, Blaese R M. Hum Gene Ther. 1996;7:1123–1129. doi: 10.1089/hum.1996.7.9-1123. [DOI] [PubMed] [Google Scholar]
- 34.Selvaggi T A, Walker R E, Fleisher T A. Blood. 1997;89:776–779. [PubMed] [Google Scholar]