

Abstract
RNA interference (RNAi) is becoming a popular method for analyzing gene function in a variety of biological processes. We have used RNAi in cultured Drosophila cells to identify trans-acting factors that regulate the alternative splicing of endogenously transcribed pre-mRNAs. We have generated a dsRNA library comprising ~70% of the Drosophila genes encoding RNA binding proteins and assessed the function of each protein in the regulation of alternative splicing. This approach not only identiWes trans-acting factors regulating specific alternative splicing events, but also can provide insight into the alternative splicing regulatory networks of Drosophila. Here, we describe this RNAi approach to identify alternative splicing regulatory proteins in detail.
Keywords: Alternative splicing, RNA interference, Drosophila
1. Introduction
Alternative splicing is a powerful mechanism by which a single gene can give rise to multiple mRNA isoforms, and therefore multiple proteins. Because different protein isoforms can have distinct biological functions, alternative splicing plays an important role in regulating gene expression [4,1]. Alternative splicing also plays a central role in expanding the size and diversity of the proteome. In fact, it is estimated that as many as 74% of human genes encode alternatively spliced transcripts [5]. Despite the prevalence of alternative splicing, we have a very limited knowledge about the underlying regulatory mechanisms. For example, only a handful of splicing regulators has been identified to date and the endogenous regulatory targets for most of these regulators are not known. Given the sheer number of alternatively spliced exons that exists in metazoan organisms, it is almost certain that numerous splicing regulatory factors have yet to be discovered.
To begin identifying proteins involved in regulating alternative splicing, we have performed an RNA interference (RNAi) screen in cultured Drosophila melanogaster cells. D. melanogaster is an excellent model organism to study alternative splicing for several reasons. First, the components of the spliceosome are highly conserved between human and the fruitfly [8]. Second, it is easy to perform both genetic and biochemical experiments using the animals and extracts from cultured cells or embryos, respectively. Finally, it is relatively easy and inexpensive to perform RNAi in Drosophila cells. In contrast to mammalian cells, where expensive synthetic siRNAs or vectors that express shRNAs are used, RNAi in cultured Drosophila cells can be performed by simply adding long (~400–500 bps) dsRNAs that can be enzymatically synthesized in the lab to the culture media [10]. The simplicity of this system has allowed several genome-wide RNAi screens to be performed in Drosophila culture cells and have led to the identification factors involved in biological processes ranging from cell viability to cell morphology [6,7,2,3]. In this chapter, we will describe in detail the RNAi approach we use to identify and analyze RNA binding proteins involved in controlling alternative splicing.
2. Methods
An overview of the RNAi screen to identify RNA binding proteins involved in alternative splicing is schematically depicted in Fig. 1. First, we constructed a library of dsRNAs corresponding to genes predicted to encode RNA binding proteins. This was done by cloning the target genes, transcribing the two single-stranded RNAs for each gene, and annealing the RNAs to form dsRNA. Next, these dsRNAs were administered to the cultured Drosophila cells. After incubating the dsRNAs with the cells for 4 days, total cellular RNA was extracted and used to analyze the alternative splicing of endogenous pre-mRNAs by RT-PCR. Below, we will describe each of these steps in detail.
Fig. 1.
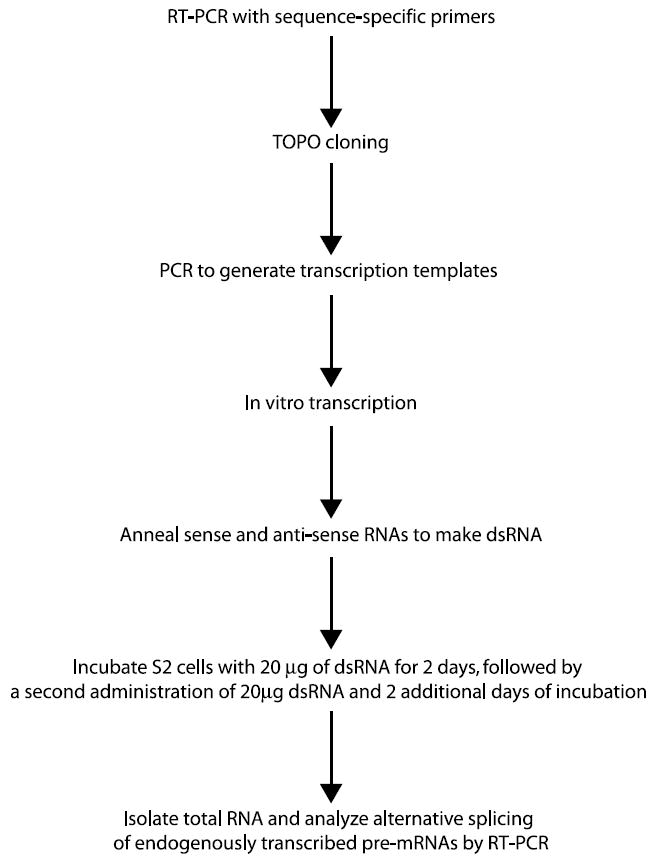
An outline of how to use RNAi to identify trans-acting factors involved in regulating alternative splicing.
2.1. Construction of dsRNA library
2.1.1. Cloning of cDNA fragments of the target genes
To generate a dsRNA library for the majority of the RNA binding proteins in D. melanogaster , we searched the InterPro database and identified 320 genes that are annotated as encoding proteins containing known RNA binding motifs (http://www.ebi.ac.uk/proteome). Gene-specific primers were designed for each of the RNA binding protein genes using primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) based on sequence information available in Flybase (http://www.flybase.org). These primer pairs were designed to amplify cDNA fragments 250–450 bp in length that did not share any regions of 19 consecutive nucleotides of identity or greater to other genes in the genome. These primer pairs were then used to amplify the cDNA fragments by reverse transcription-polymerase chain reaction (RT-PCR) using total RNA isolated from S2 cells. These PCR products were individually cloned into the pCRII-TOPO vector (Invitrogen) which contains a T7 and SP6 promoter Xanking the cloning site. Finally, each clone was sequenced to verify its identity.
2.1.1.1. Reverse transcription
Reverse transcription was performed in a 20 μl reaction mixture containing 1 μl (10 U/μl) RNase inhibitor (Invitrogen), 265ng Random hexamer (Invitrogen), 4 μl 5 × First strand buffer (supplied with Superscript II), 10 mM DTT, 0.5 mM dNTPs, 1 μl (100 U/μl) Superscript II (Invitrogen), and 5 μg total Drosophila RNA isolated using Trizol (Invitrogen) as directed by the manufacturer.
2.1.1.2. Polymerase chain reaction
PCR was carried out in a 50 μl reaction containing 5 μl 10× PCR buffer-(MgCl2), 1.5 mM MgCl2, 0.2 mM dNTPs, 0.2 μM primers, 0.5 μl Taq polymerase (5 units/μl), 2 μl of RT reaction mixture. The reactions were incubated at 94 °C for 2 min, and then cycled 35 times at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, followed by a final 3 min incubation at 72 °C.
2.1.1.2.1. Cloning into pCRII-TOPO
0.25 μl PCR products were immediately incubated with 0.25 μl pCRII-TOPO vector (Invitrogen) in a 2 μl reaction containing 100 mM NaCl at room temperature for 5 min. The whole reaction mixture was transformed into One Shot cells (Invitrogen). DNA from individual colonies was sequenced to confirm the identity of the clones.
2.1.2. Preparation of double-stranded RNA
Each vector was individually used as a PCR template with the M13 forward and reverse primers to generate dsRNA transcription templates. The PCR products were then separately transcribed with T7 and SP6 RNA polymerase (AmpliScribe Kits from Epicentre), followed by DNase I treatment to remove the template DNA. We chose to use two different RNA polymerases to separately transcribe the top and bottom RNA strands, rather than a single polymerase to simultaneously transcribe both strands, to ensure that the RNA we were adding to the cells was indeed dsRNA. Typically, RNA yields of the T7 and SP6 polymerase in vitro transcription are 15 and 10 mg/ml, respectively. Double-stranded RNA was generated by incubating 20 μg of each RNA strand in a 50 μl reaction containing 100 mM NaCl, 20 mM Tris, pH 8.0, and 1 mM EDTA at 68 °C for 10 min and 37 °C for 30 min. The integrity and efficiency of each of these steps was monitored by agarose gel electrophoresis (Fig. 2).
Fig. 2.
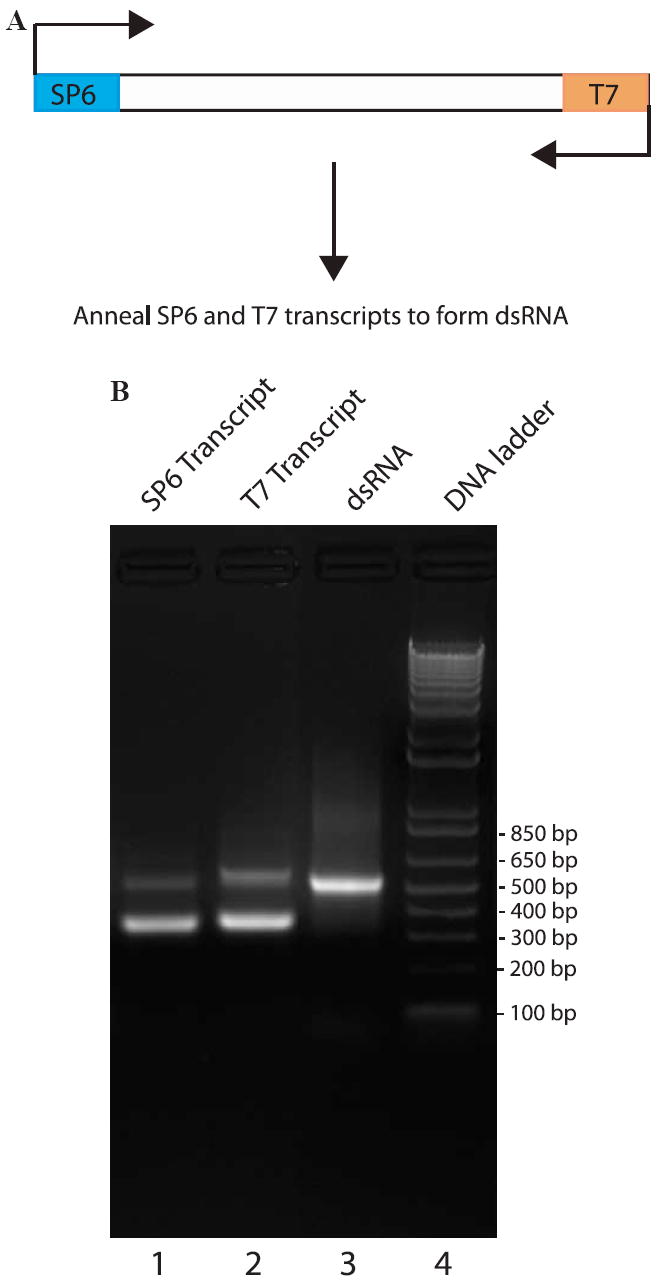
Analysis of RNA annealing efficiency and the integrity of dsRNA. (A) A PCR product Xanked by T7 and SP6 RNA polymerase promoters is transcribed with each RNA polymerase to generate single-stranded RNA. These RNA strands are then annealed together to generate double-stranded RNA. (B) Analysis of RNA by agarose gel electrophoresis. The SP6 and T7 transcripts separately synthesized from a vector were run on a 1.5% agarose gel electrophoresis (lanes 1 and 2). These two RNAs were annealed by incubating them together at 78 °C for 10 min and slowly cooling the reaction at 37 °C for 30 min. The annealed dsRNA has a clearly distinct mobility in an agarose gel (lane 3) than either single-stranded RNA. A DNA marker was loaded in lane 4.
2.1.2.1. PCR amplification of the in vitro transcription templates
The PCRs were carried out in 20 μl reactions containing 1 × PCR buffer lacking MgCl2 (supplied with Taq DNA polymerase), 1.5 mM MgCl2, 0.5 mM dNTPs, 0.5 mM M13 forward primer, 0.5 mM M13 reverse primer, 0.5 U Taq DNA polymerase, and 5 ng of the cDNA clone vector. The reactions were incubated at 94 °C for 2 min, and then cycled 35 times at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, followed by a final 3 min incubation at 72 °C.
2.1.2.2. In vitro transcription reactions
To transcribe the RNA for RNAi, we use the AmpliScribe kits from Epicentre. Each RNA strand is separately transcribed in a 20 μl reaction with either T7 or SP6 RNA polymerase. Each reaction contains 1 × AmpliScribe T7 or SP6 buffer (supplied in kit), 7.5 mM each of ATP, CTP, GTP, and UTP, 2 μl of AmpliScribe T7 or SP6 enzyme solution, 10 mM DTT, and 1 μg of the Template DNA (PCR product). The transcription reactions are then incubated at 37 °C for 3 h.
2.1.2.3. Annealing the single-stranded RNAs to make dsRNA
The dsRNA is next formed by annealing the two complementary single-stranded RNAs synthesized in the previous section. The annealing reactions are set up in a 50 μl volume and contain 20 μg of each single-stranded RNA, 10 mM NaCl, 2 mM Tris–HCl, pH 8.0, and 0.1 mM EDTA. The annealing reactions are then incubation at 78 °C for 10 min followed by a slow cooling to room temperature over approximately 30 min.
2.2. RNA interference screen
To perform the RNAi screen to identify new splicing regulators, we utilize Drosophila Schneider S2 cells adapted for growth at 27°C in serum-free medium (Drosophila-SFM (Invitrogen, catalog # 10797-017)). Because serum can interfere with the ability of the dsRNA uptake into the cells, the use of serum-free medium eliminates the need to remove serum-containing media before administration of the dsRNA. The Drosophila-SFM is supplemented with 1 × penicillin–streptomycin (Invitrogen) and 2mM l-glutamine (Invitrogen). The Drosophila Schneider S2 cells can also be purchased from Invitrogen (catalog # R690-07). We maintain stocks of S2 cells in 100ml plastic Erlenmeyer flasks at 27°C in a shaking incubator at a cell density between 1 ×106 and 1 ×107 cells/ml.
2.2.1. Administration of the dsRNAs to the S2 cells
To perform RNAi, 20 μg of each dsRNA is added to ~1 × 106 cells Schneider (S2) cells in six-well culture dishes. The dsRNA is added directly to the culture medium. After 2 days, a second dose of 20 μg of dsRNA is added to each well and the cells incubated for two additional days. We have found that incubating the cells with dsRNA two times over the course of four days give a fairly robust knockdown of the targeted gene. After a total of four days, total RNA is isolated using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. The alternative splicing of any gene of choice is then analyzed by RT-PCR. Typically, one of the two primers is 32P-end labeled and the PCR products are subjected to denaturing PAGE. The ratio between the different alternative isoforms is then determined by phosphorimager analyzer. To monitor the level of depletion, a primer pair that can amplify a region of the targeted mRNA outside of the dsRNA region can be used for RT-PCR amplification. When antibodies are available for the target protein, the level of depletion can also be checked by Western blotting.
3. Discussion
RNAi is a powerful method that allows one to rapidly assess the function of hundreds or thousands of proteins at a time. The RNAi screen we have conducted, which consisted of 250, or roughly 70%, of the Drosophila RNA binding proteins, led to the identification of 47 proteins that are involved in controlling the alternative splicing of 19 alternative exons from three different pre-mRNAs [9]. This type of screen can quickly lead to the identification of candidate splicing regulators of any pre-mRNA of interest. In addition to analyzing the splicing of pre-mRNAs that are endogenously transcribed in the S2 cells, we have recently extended this assay to analyze the splicing of pre-mRNAs transcribed from transfected mini-gene plasmids (S. Olson and B.R.G., unpublished data). This approach should prove to be a powerful method to identify proteins that regulate the splicing of specific pre-mRNAs and to elucidate their functions.
RNAi oVers a new approach to analyze the function of proteins involved in pre-mRNA splicing. Typically, the functions of such proteins have been determined by depletion and add-back experiments. However, these assays can sometimes be problematic because of potential co-depletion of unknown proteins involved in pre-mRNA splicing. More importantly, these assays require knowing the identity of the splicing regulator and having the necessary reagents (antibodies). An additional approach that is frequently taken to identify proteins that regulate the splicing of specific genes is to Wrst identify important regulatory RNA sequences by mutagenesis and subsequently, the protein that binds to these sequences by UV-crosslinking. Though frequently successful, these approaches can be time consuming and challenging. In contrast, the RNAi screening approach described above is rapid and requires neither the knowledge about the identity and location of splicing regulatory elements nor the development of a mini-gene or in vitro splicing system. Coupling the RNAi approach with the more traditional biochemical and tissue culture systems should lead to a rapid increase in our understanding of the mechanisms involved in controlling alternative splicing.
Acknowledgments
Work in our laboratory is supported by Grants GM62516 and GM67842 from the NIH to B.R.G.
References
- 1.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Consortium HF, Paro R, Perrimon N. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- 3.DasGupta R, Kaykas A, Moon RT, Perrimon N. Functional genomic analysis of the Wnt-wingless signaling pathway. Science. 2005;308:826–833. doi: 10.1126/science.1109374. [DOI] [PubMed] [Google Scholar]
- 4.Graveley BR. Sex, agility, and the regulation of alternative splicing. Cell. 2002;109:409–412. doi: 10.1016/s0092-8674(02)00750-x. [DOI] [PubMed] [Google Scholar]
- 5.Johnson J, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Schoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 6.Kiger A, Baum B, Jones S, Jones M, Coulson A, Echeverri C, Perrimon N. A functional genomic analysis of cell morphology using RNA interference. J Biol. 2003;2:27. doi: 10.1186/1475-4924-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lum L, Yao S, Mozer B, Rovescalli A, Von Kessler D, Nirenberg M, Beachy PA. Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science. 2003;299:2039–2045. doi: 10.1126/science.1081403. [DOI] [PubMed] [Google Scholar]
- 8.Mount SM, Salz HK. Pre-messenger RNA processing factors in the Drosophila genome. J Cell Biol. 2000;150:F37–F44. doi: 10.1083/jcb.150.2.f37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Worby CA, Simonson-Leff N, Dixon JE. RNA interference of gene expression (RNAi) in cultured Drosophila cells. Sci STKE. 2001;1:PL1. doi: 10.1126/stke.2001.95.pl1. [DOI] [PubMed] [Google Scholar]