

Abstract
Nontypeable Haemophilus influenzae (NTHi) is strongly associated with exacerbations of chronic obstructive pulmonary disease, which often coincide with viral respiratory infections. TLR2 contributes importantly to innate immunity to NTHi, but whether this pathway is affected by simultaneous antiviral responses is unknown. To analyze potential interactions, resident murine and human alveolar macrophages (AMφ) were exposed, in the presence or absence of the appropriate rIFN-β, to synthetic lipopeptides corresponding to the triacylated N-terminal fragments of three outer membrane proteins (OMP) (PCP, P4, and P6) that are highly conserved among different NTHi strains. Synthetic OMP elicited strong release of IL-6, the principal inducer of airway mucin genes, and induced CCL5 and CXCL10 from murine AMφ only when IFN-β was also present. Surprisingly, combined stimulation by OMPs and IFN-β also markedly enhanced TNF-α release by murine AMφ. Stimulation with PCP plus IFN-β induced IFN-regulatory factor 1 expression and sustained STAT1 activation, but did not alter the activation of MAPKs or NF-κB. AMφ derived from STAT1-deficient mice did not demonstrate increased production of TNF-α in response to PCP plus IFN-β. Analysis of wild-type and STAT1-deficient AMφ using real-time PCR showed that increased TNF-α production depended on transcriptional up-regulation, but not on mRNA stabilization. The synergistic effect of synthetic OMP and IFN-β was conserved between murine AMφ and human AMφ for IL-6, but not for TNF-α. Thus, IFN-β, which is produced by virally infected respiratory epithelial cells, converts normally innocuous NTHi OMP into potent inflammatory stimulants, but does so via different mechanisms in mice and humans.
Nontypeable Haemophilus influenzae (NTHi)3 almost universally colonizes the human upper respiratory tract (1). This commensal Gram-negative bacterium generally exists in harmony with the host, but can induce otitis media in children under age 3 and lower respiratory tract infections in the elderly or those with chronic lung disease (2, 3). NTHi is also increasingly recognized to play a key role in the pathogenesis of chronic obstructive pulmonary disease (COPD), a debilitating and potentially fatal condition that is rapidly increasing in prevalence worldwide. NTHi is the most commonly recovered bacteria during acute exacerbations of chronic bronchitis (AECB) (4, 5), episodes of increased cough, sputum production, and breathlessness that are a major cause of hospitalization and functional decline in COPD. Hence, understanding the immune response to NTHi is a goal of considerable public health importance.
NTHi succeeds as a colonizer and pathogen largely because it has multiple means by which it evades the adaptive immune response. NTHi comprises heterogeneous populations of distinct strains with extreme interstrain antigenic variation. Rapid succession of NTHI strains is one mechanism for persistence in the lower airways in COPD. Individual strains of NTHi can also vary the antigenicity of their outer membrane lipoproteins (OMP) under immunological pressure (2). NTHi secrete any of three IgA1 proteases; individual NTHi strains can alter their IgA protease, so that it not only cleaves IgA1 at a different site but also changes its antigenic properties (2). The microorganism can survive both extracellularly in biofilms (6) and within respiratory epithelial cells and macrophages (Mφ) (7, 8), in each site remaining relatively safe from host defenses. These factors have so far thwarted attempts to develop effective vaccines against NTHi.
Nevertheless, NTHi can activate innate immune defenses, whose stereotypic pathogen recognition receptors are less susceptible to such antigenic trickery. Although NTHi appears to evade TLR4 stimulation in part by incorporating host-derived sialic acid into its lipo-oligosaccharide (9), crude extract of NTHi organisms or whole lipoprotein P6 induce IL-8 and TNF-α production by human mononuclear phagocytes purified from peripheral blood, and activate the NF-κB and p38 MAPK pathways in human epithelial cells via TLR2 (10, 11). The capacity of innate immune receptors to recognize NTHi led us to ask whether alveolar Mφ (AMφ), the principal innate immune effector of the lower respiratory tract, might contribute to the inflammatory response induced by NTHi during AECB.
Based on the known association between viral respiratory infections and AECB (3, 12–14), we postulated that any such AMφ contribution to NTHi-induced inflammation would depend on stimulation with type I IFN (IFN-α/IFN-β). In addition to their well-established role in containing viral infections, type I IFNs have more recently been shown to be centrally involved in both innate and adaptive immunity to Gram-negative bacteria (reviewed in Refs. 15 and 16). IFN-βnull mice are resistant to LPS-induced lethality through a mechanism involving the IFN-α/IFN-βR′-associated kinase Tyk2 and the transcription factor STAT1 (17). Importantly, we have recently shown that murine AMφ cannot activate STAT1 in response to stimulation via TLR4 or TLR3, due to a block in the autocrine secretion of IFN-β seen in other Mφ subtypes (18). Nevertheless, murine AMφ respond vigorously to exogenous IFN-β or IFN-γ (18). Collectively, these facts imply that production of type I IFNs by virally infected respiratory epithelial cells might overcome the apparent ability of NTHi to evade immune detection.
The goal of this study was to determine whether IFN-γ modulates the AMφ response to NTHi-derived pathogen-associated molecular patterns. Triacylated lipopeptides, such as NTHI OMP, are known to stimulate TLR2 in combination with TLR1 (19, 20). For this purpose, we stimulated resident murine and human AMφ in vitro using three different synthetic tripalmitoylated peptides derived from the invariant N terminus of three OMP, PCP, P4, and P6, that are common to all strains of NTHi (2, 11). As a positive control, we used the well-known TLR2 agonist (20) Escherichia coli-derived lipopeptide Pam3CysSK4 (LP), which we have previously shown induces substantial TNF-α production by both murine AMφ and peritoneal Mα (PMα) (18). As outcomes, we assayed cytokines and chemokines theorized to be responsible for the cardinal symptoms of AECB, exaggerated mucous production, and inflammatory cell recruitment (21–23). Our results indicate that type I IFN combines with OMP-mediated stimulation via TLR2 to elicit a strong inflammatory response by resident murine and human AMφ. We also provide data showing that IFN-β contributes to transcriptional increased induction of TNF-α via a novel STAT1-dependent mechanism in murine AMφ but not human AMφ.
Materials and Methods
Mice
We purchased the following specific pathogen-free female mice from the indicated vendors: C57BL/6 mice from Charles River Laboratories; B6.129-Tlr2tm1Kir/j (TLR2−/−) mice from The Jackson Laboratory; and 129S6/SvEv-Stattm1Rds (STAT1−/−) and control 129S6/SvEv mice from Taconic Farms. Resident AMφ and PMφ were harvested as previously described (18). This study followed a protocol approved by the Animal Care Committee of the local institutional review board.
Synthetic bacterial lipoprotein analogs, other reagents, and Mφ cell lines
The following synthetic bacterial lipoprotein analogs were purchased from EMC Microcollections: Pam3Cys-Ala-Asn-Thr-Asp-Ile-Phe-Ser-Gly-Asp-Val-Tyr-Ser-Ala-Ser-Gln (abbreviated as PCP); Pam3Cys-Gly-Ser-His-Gln-Met-Lys-Ser-Glu-Gly-His-Ala-Asn-Met-Gln-Leu (abbreviated as P4); Pam3Cys-Ser-Ser-Ser-Asn-Asn-Asp-Ala-Ala-Gly-Asn-Gly-Ala-Ala-Gln-Thr (abbreviated as P6); and Pam3Cys-Ser-Lys-Lys-Lys-Lys-OH (also known as Pam3CysSK4) (abbreviated as LP). LPS (E. coli 0111:B4, Sigma-Aldrich) was repurified as described previously (18). Recombinant murine and human IFN-β were purchased from R&D Systems. The murine MH-S Mφ cell line and the human Mφ cell line THP-1 were purchased from American Type Culture Collection and grown in complete medium (RPMI 1640 containing 10% heat-inactivated FBS, 20 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin/streptomycin (all from Invitrogen Life Technologies)) in a 5% CO2 environment at 37°C. Before use in experiments, THP-1 were differentiated using 80 nM vitamin D3 (Calbiochem) to induce expression of CD14, which contributes to TLR2-mediated responses (24, 25).
Human subjects
Human AMφ were isolated from five male subjects undergoing clinically indicated fiberoptic bronchoscopy as part of the diagnostic evaluation of solitary pulmonary nodules. Subjects (age 71.2 ± 10.0 years) (mean ± SD) all had COPD (forced expiratory volume, 1 second 49.3 ± 16.7% predicted) and had a smoking history of >20 packs per year, although only one actively smoked. No subjects had pulmonary abnormalities thought to be due to infectious, interstitial, or systemic disease. The study was approved by the institutional review board of the Ann Arbor Veterans Affairs Healthcare System, and all the subjects gave their signed consent. Bronchoalveolar lavage (BAL) was collected from the radiographically normal lung contralateral to the nodule, using 30-ml aliquots of normal saline to 180-ml total.
Isolation of human Mφ
BAL cells were centrifuged (500 × g for 10 min) and washed twice with HBSS. Cell viability was assessed by trypan blue exclusion; BAL macrophages were isolated by plastic adherence. PBMC were obtained from the same donors and monocytes were isolated by plastic adherence as previously described (26). This method of purification results in >95% pure Mφ populations, as determined by morphological and surface marker analysis. After removal of nonadherent cells, Mφ were grown before stimulation in complete medium in a 5% CO2 environment at 37°C.
Mφ stimulation
Murine or human Mφ or Mφ cell lines were seeded at 8 × 105 cells/well in 24-well tissue culture plates (Costar) for Western blot analysis, or at 5 × 104 cells/well in 96-well plates (Costar) for ELISA and real-time PCR and incubated for the indicated times in complete medium alone, or complete medium containing 100 ng/ml of each of the three NTHI OMPs (PCP, P4, and P6), or 100 ng/ml LP, without or with the addition of 1000 U/ml rIFN-β of the appropriate species. In some experiments, Mβ were also cultured in medium containing 100 ng/ml OMP-free LPS.
Cytokine/chemokine ELISAs
The supernatants concentrations of TNF-α, IL-6, CCL5 (RANTES), and CXCL10 (IFN-γ-inducible protein 10 (IP-10)) were determined by ELISA, using Duo Set Development Systems (R&D Systems).
Cell extracts, Western blot analysis, and gel documentation
Cell extract protein analysis and gel documentation was conducted as previously described (18). Primary Abs anti-STAT1, anti-phospho-STAT1 Y701, anti-p38, and anti-p-p38 were all from Cell Signaling Technology. The anti-IFN-regulatory factor-1 (IRF-1) Ab was from Santa Cruz Bio-technology. Densitometric analysis of films was performed using Gel Expert hardware and software (Nucleo Tech).
Real-time PCR and comparative quantitation analysis of murine mRNAs
For real-time analysis, total RNA was prepared from control and stimulated cells using the Absolutely RNA RT-PCR miniprep kit (Stratagene). DNase-treated total RNA was converted to cDNA and subsequently to specific PCR products using Brilliant SYBR Green QRT-PCR master mix kit, 1 step (Stratagene). cDNA conversion, amplification, and data analysis were performed using a Mx3000P Real-Time PCR System computerized cycler from Stratagene. We used the following primers, designed using software available at 〈http://biotools.idtdna.com/Primerquest/〉 (IDT), and synthesized and HPLC purified by Invitrogen Life Technologies: TNF-α sense, AGCCGATGGGTTGTACCTTGTCTA; TNF-α antisense, TGAGATAGCAAATCGGCTGACGGT; IL-6 sense, ATCCAGTTGC CTTCTTGGGACTGA; IL-6 antisense TAAGCCTCCGACTTGT GAAGTGGT; GAPDH sense, TATGTCGTGGAGTCTACTGGT; GAPDH antisense, GAGTTGTCATATTTCTCGTGG. Primers were used at 75 nM each in 25-μl reactions; cycle parameters were: 40 s at 55°C for the reverse transcription step, followed by a denaturation step at 95°C for 10 min and 36 cycles, each consisting of 30 s denaturation at 95°C, 1 min annealing at 60°C and 30 s at 72°C polymerization. Fluorescence data were collected and analyzed as previously described (18). For use in mRNA stability analysis, actinomycin D was obtained from EMD Biosciences.
Statistical analysis
Data were expressed as the mean ± SD of replicates in single experiments, or mean ± SEM. Samples were compared by two-tailed Student t test analyses. Statistical calculations were performed using Statview version 5.0 and Super ANOVA version 1.11 software (SAS Institute) on a Macintosh PowerPC G4 computer. Significant differences were defined as p <0.05.
Results
IFN-β and NTHi-derived OMPs synergize to up-regulate inflammatory cytokine and chemokine production by resident murine AMφ
To verify that Mφ stimulation by the synthetic NTHi OMP analogs PCP, P4, and P6 depended strictly on TLR2, we first tested each at doses up to 100 ng/ml on AMφ from TLR2−/− mice. As a known TLR2 agonist, we used E. coli-derived LP (20), which we have previously shown induces substantial TNF-α production by both murine AMφ and PMφ(18). In these experimental conditions, neither the three NTHi OMP nor LP showed any ability to induce TNF-α, whereas OMP-free LPS, a pure TLR4 stimulus, evoked a strong TNF-α production (Fig. 1), as anticipated (18). PMφ from TLR2−/− mice also showed a similar response (data not shown). Thus, the stimuli used throughout the remainder of this study depend strictly on TLR2.
FIGURE 1.
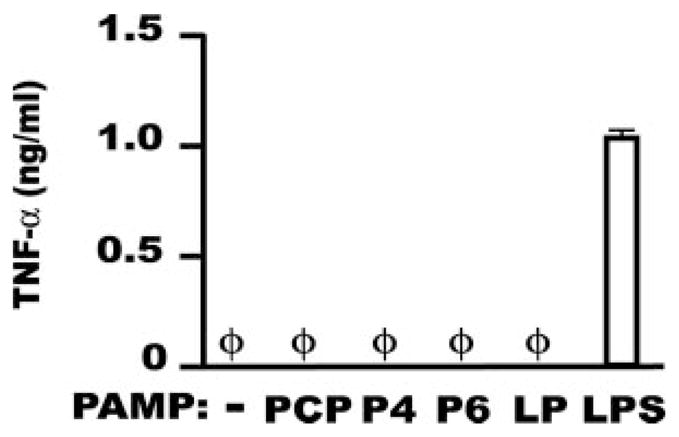
Murine AMφ from TLR2−/− mice do not respond to the NTHi OMP PCP, P4, and P6. Resident AMφ from TLR2−/− mice were incubated for 6 h in the presence of medium alone or medium containing 100 ng/ml of each of the three NTHI OMPs (PCP, P4, and P6). As a control for TLR2 stimulation, we used the synthetic lipopeptide Pam3CysSK4 (referred to in this and subsequent figures as LP). The sources and structures of the synthetic lipopeptides used in this work are described in detail in Materials and Methods. To test for cell responsiveness, 100 ng/ml OMP-free LPS was used as positive control for TLR4 stimulation. Supernatants were collected and assayed by ELISA for TNF-α. φ, None detected. Data are mean ± SD of triplicate wells in a single experiment.
Next, to investigate whether IFN-β modulates the AMφ response to NTHi-derived TLR2 OMPs, we stimulated resident murine AMφ from wild-type C57BL/6 mice with each of the three synthetic NTHi OMPs or the positive control LP, in the presence or absence of murine IFN-β. ELISA analysis of the AMφ supernatants disclosed two major findings. First, AMφ production of IL-6 in response to the NTHi OMPs depended almost entirely on combined stimulation with IFN-β (Fig. 2A). Treatment with IFN-β alone showed no effect. Second, addition of IFN-β strongly enhanced TNF-α release by AMφ, relative to each of the three synthetic OMPs alone (Fig. 2B). Treatment with IFN-β alone did not induce TNF-α production. These results did not depend on the strain of mice, because similar findings were obtained using the AMφ cell line MH-S, of BALB/c origin (data not shown), and another inbred strain (see below).
FIGURE 2.
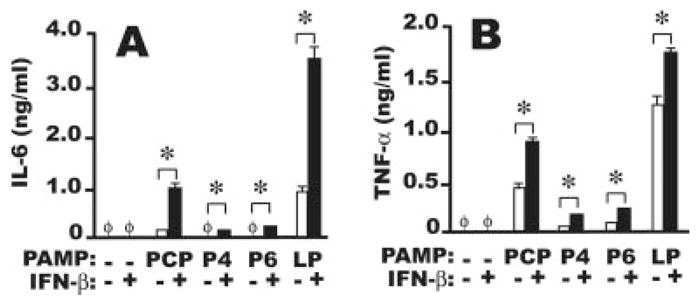
Murine AMφ respond strongly to NTHi OMP only in the presence of IFN-β. Resident AMφ from normal C57BL/6 mice were incubated for 6 h in the presence of medium alone, 1000 U/ml IFN-β, 100 ng/ml of each of four OMPs (PCP, P4, P6, and positive control LP) (□), or 100 ng/ml of each OMPs plus 1000 U/ml IFN-β( ■). Supernatants were collected and assayed by ELISA for (A) IL-6 and (B) TNF-α. φ, None detected. Data are mean ± SD of triplicate wells.*, p <0.05, unpaired Student t test. One of three independent experiments with similar results is shown.
It remained possible that the apparent inability of NTHi OMPs to induce IL-6 without costimulation by IFN-β might result from an inadequate period of AMφ stimulation. To exclude such a kinetic explanation, we next stimulated murine AMφ overnight and assayed production of IL-6, as well as the chemokines CCL5 (RANTES) and CXCL10 (IP-10), which are thought to play pathogenetic roles in AECB (22, 23). Enhancement of IL-6 release by the simultaneous treatment with the different OMPs and IFN-β was even more pronounced, although IL-6 was just detectable in wells stimulated with the OMPs alone (Fig. 3A). Combinatorial stimulation also abundantly induced CCL5 and CXCL10, which were not seen on stimulation with the OMPs or LP alone (Fig. 3, B and C). Using the same combinatorial stimulations, comparable observations were made with resident PMφ (data not shown). Hence, exogenous type I IFN can increase murine AMφ production of inflammatory mediators central to the pathogenesis of AECB in humans on stimulation with otherwise ineffective concentrations of NTHi-derived products.
FIGURE 3.
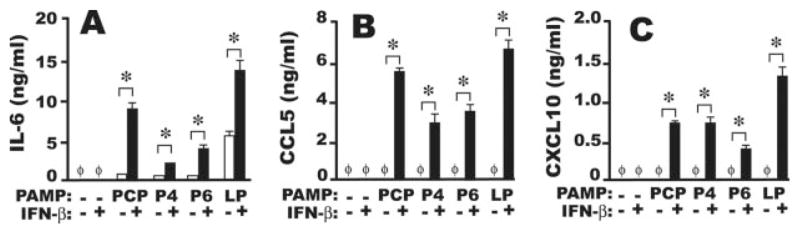
Overnight stimulation of murine AMφ with NTHi OMP plus IFN-β increases IL-6 production and induces the chemokines RANTES and IP-10. Resident AMφ from normal C57BL/6 mice were incubated for 18 h in the presence of medium alone, 1000 U/ml IFN-β, 100 ng/ml of each of four OMPs (PCP, P4, P6, and positive control LP) (□), or 100 ng/ml of each OMP plus 1000 U/ml IFN-β (■). Supernatants were collected and assayed by ELISA for (A) IL-6, (B) CCL5, and (C) CXCL10.φ, None detected. Data are mean ± SD of triplicate wells.*, p <0.05, unpaired Student t test. One of two independent experiments with similar results is shown.
Sustained induction of IRF-1 and of pY701 STAT1 by combined TLR2 and IFN-β stimulation
Increased IL-6 and induction of CCL5 and CXCL10 by IFN-β results from the presence in the murine and human promoters of these proinflammatory genes of specific sequences that bind such IFN-regulated transcription factors as IFN-inducible transcription factor IRF-1, IRF-3, or the IFN-stimulated gene factor 3 (ISGF3) complex (27–32). Significantly, we found that stimulation of AMφ by the synthetic OMP PCP plus IFN-β induced rapid synthesis of IRF-1 (Fig. 4, A and B, top). We also found that the combined stimulation induced phosphorylation of STAT1 on Y701 (Fig. 4, A and B, second from top), indicative of ISGF3 activity (18). AMφ abundantly expressed IRF-3 protein at steady state (data not shown).
FIGURE 4.

Combined stimulation of murine AMφ with the NTHi OMP PCP and IFN-β induces long-term activation of STAT1 and increases expression of IRF-1 without affecting activation of MAPKs. Resident AMφ from normal C57BL/6 mice were incubated for the indicated times in the presence of medium alone (lane C′), 1000 U/ml IFN-β (lanes IFN-β), PCP 100 ng/ml or a combination of both (lanes PCP+IFN-β). Cells were then harvested and processed for Western blot analysis as specified in Materials and Methods. The membranes were probed with (from the top) anti-IRF-1, anti-pY701 STAT1 anti-STAT1, anti p-p38, and anti-p38. Note that the pY701 STAT1 film in B has been deliberately overexposed relative to the other rows to emphasize the absence of STAT1 phosphorylation by PCP alone. C–F, Densitometric quantitative analysis of the band intensities of (C and E) IRF-1 and (D and F) pY701 STAT1 in the films shown in A (C and D) and B (E and F), respectively. As a control for loading, band intensities were normalized relative to the corresponding p38 bands. AU, arbitrary units. IFN-β, light gray bars; PCP plus IFN-β, ■. Similar results were obtained in an additional independent experiment of equivalent design.
However, the synergistic effect of combined TLR2 plus IFN-β stimulation on TNF-α production (Fig. 2B) was unanticipated from the literature. Accordingly, we next analyzed known activation pathways resulting from the triggering of either TLR2 or IFN-β receptors to gain an understanding of potential mechanisms. For these experiments, we used only the PCP-derived lipopeptide, which had shown the greatest effects on resident AMφ (Figs. 2 and 3). As expected (15, 33), we found activation or strong induction of the transcription factors IRF-1 (Fig. 4, A and B, top), STAT1 (Fig. 4, A and B, second from top), and STAT2 (data not shown) only when AMφ were stimulated with IFN-β. Addition of IFN-β to AMφ stimulated with PCP did not alter the pattern of activation of p38 (Fig. 4B, fourth from top) or of JNK, ERK MAPKs, or NF-κB p65 (data not shown). Treatment of AMφ with IFN-β alone for up to 4 h did not activate JNK or NF-κB p65 (data not shown), and had very minimal effect on induction of phospho-p38 (Fig. 4A, fourth from top) and phospho-ERK (data not shown).
Interestingly, the presence of PCP during IFN-β stimulation increased IRF-1 protein (Fig. 4A, top, C, and E) and markedly sustained phosphorylation of STAT1 at Y701 through 4 h, relative to stimulation by IFN-β alone (Fig. 4A, second from top, and D). As anticipated, TLR2 stimulation alone by PCP did not induce STAT1 phosphorylation (Fig. 4B, second from top).
Based on these results, and on the observation that TLR2 stimulation likewise does not induce activation of the IRF-3 complex (34, 35), we speculated that the augmented TNF-α production we found on combined stimulation by IFN-β and NTHi OMP (Fig. 2B) might reflect a transcriptional mechanism by which signals emanating from the IFN-βR and signals from the TLR2 pathway might converge on target inflammatory genes.
A specific transcriptional role for IFN-β in the increased production of TNF-α by murine AMφ
To address this possibility, murine resident AMφ were treated for 3 or 6 h with IFN-β, PCP, or both simultaneously, and the levels of TNF-α mRNA were determined by real-time PCR. Confirming our hypothesis, the combination rapidly and strongly increased TNF-α mRNA concentrations above those seen on treatment with PCP alone (Fig. 5A). As expected, IFN-β treatment alone had minimal effect on TNF-α mRNA induction.
FIGURE 5.
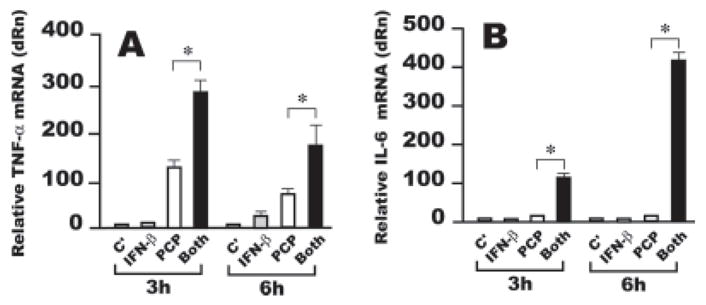
Increased induction of TNF-α by combined stimulation of AMφ with the NTHi OMP PCP and IFN-β is transcriptionally regulated. Resident AMφ from normal C57BL/6 mice were cultured for 3 or 6 h in the presence of medium alone (C′) (light gray bars), 1000 U/ml IFN-β (IFN-β ) (gray bars), 100 ng/ml PCP (□), or a combination of the same doses of PCP and IFN-β (Both) (■). Total cellular RNA was collected and processed for real-time PCR; samples were analyzed for (A) TNF-α or (B) IL-6 mRNA expression. The relative quantities of specific mRNA from treated AMφ are plotted compared with the calibrator mRNA (TNF-α or IL-6 mRNA in control AMφ, which is defined as a value of 1). dRn is baseline-corrected, reference dye-normalized fluorescence.*, p <0.05, unpaired Student t test. Data are mean ± SEM of triplicate wells in each of two independent experiments with similar results.
We also analyzed the effect of combined treatment with PCP and IFN-β on induction of IL-6 mRNA, because increased mRNA levels of that cytokine have been found on combined treatment of HeLa cells with IFN-β and the TLR3 stimulant dsRNA (36). Strong and persistent induction of IL-6 was also seen only when both stimuli were given (Fig. 5B). Similar results were obtained using the murine AMφ cell line MH-S (data not shown). These results indicate that IFN-β, when it is present during TLR2 stimulation, regulates the transcription of both TNF-α and IL-6 in murine AMφ.
A well-known mechanism to control TNF-α mRNA expression relies on its destabilization due to A φ U-rich elements in the 3′ untranslated region of the gene (37). To test whether the observed increase in TNF-α mRNA amounts could be ascribed to increased mRNA stability, we used the transcriptional inhibitor actinomycin D (10 μg/ml) and using real-time PCR, analyzed mRNA turnover in murine AMφ treated with PCP or PCP plus IFN-β. We found that TNF-α mRNA underwent rapid turnover regardless of IFN-β, i.e., with either treatment, >90% of the specific TNF-α mRNA was lost after incubation of the stimulated AMφ with the drug (Fig. 6). Thus, increased mRNA stability does not appear to be responsible for increased production of TNF-α by IFN-β in murine Mφ.
FIGURE 6.
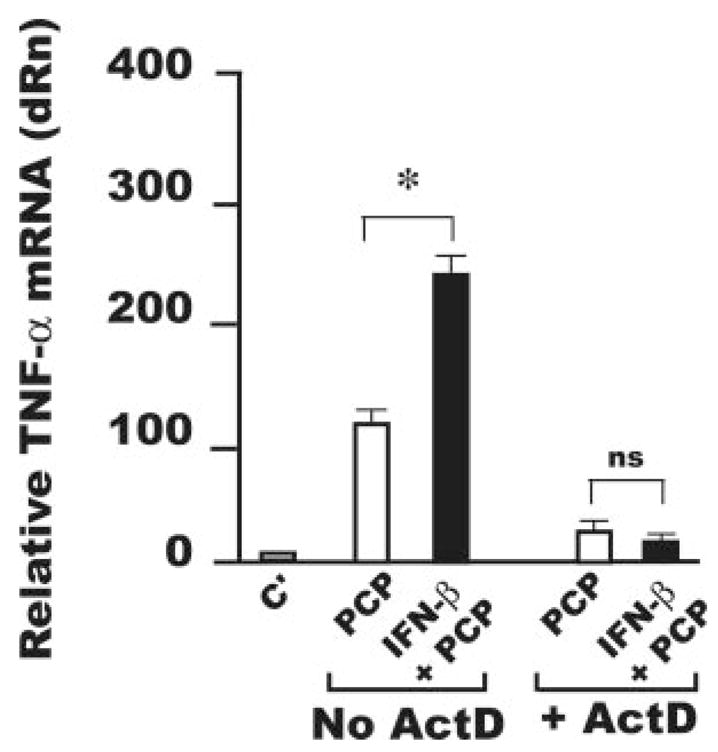
Increased mRNA stability is not responsible for increased TNF-α production by AMφ stimulated with the NTHi OMP PCP and IFN-β. Resident AMφ from normal C57BL/6 mice were cultured for 3 h in the presence of medium alone (C′) (light gray bars), 100 ng/ml PCP (□), or a combination of PCP and IFN-β (1000 U/ml) (■). After 3 h, the RNA polymerase inhibitor actinomycin D (10 μg/ml, ActD) was added to a fraction of the samples and all the samples were incubated for 3 additional hours. Total cellular RNA was collected and processed for real-time PCR and samples were analyzed for TNF-α mRNA expression. The relative quantities of specific mRNA from treated AMφ are plotted compared with the calibrator mRNA (TNF-α mRNA in control AMφ, which is defined as a value of 1). dRn is baseline-corrected, reference dye-normalized fluorescence.*, p <0.05, unpaired Student t test. Data are mean ± SEM of triplicate wells in each of two independent experiments with similar results.
Increased murine TNF-α mRNA is dependent on the transcription factor STAT1
All our previous results pointed to a direct transcriptional role for IFN-β in the observed increase in TNF-α mRNA by IFN-β cotreatment. Because IFN-β treatment can elicit both STAT1-dependent and STAT1-independent mechanisms (15, 33), we next analyzed TNF-α protein production in supernatants of AMφ of wild-type or STAT1-gene targeted mice stimulated by PCP or PCP plus IFN-β. Increased production of TNF-α was detected only in AMφ of wild-type mice (Fig. 7A). As anticipated from our results and Ref. 38, IL-6 up-regulation was also STAT1 dependent (Fig. 7B).
FIGURE 7.
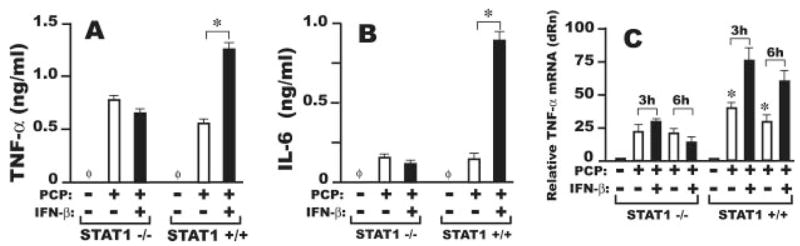
Increased induction of TNF-α by combined stimulation of murine AMφ with the NTHi OMP PCP and IFN-β is STAT1 dependent. Resident AMφ from STAT1−/− or STAT1−/−. mice were cultured in the presence of medium alone (C′), 100 ng/ml PCP (□) or a combination of 100 ng/ml PCP and 1000 U/ml IFN-β (■). Supernatants were harvested at 6 h and analyzed by ELISA for (A) TNF-α or (B) IL-6 concentration. (C) In parallel cultures, total RNA was prepared at 3 or 6 h after stimulation and analyzed for TNF-α mRNA expression by real-time PCR.*, p <0.05, unpaired Student t test. Data are mean ±SD of triplicate wells in each of two independent experiments with similar results.
Real-time PCR analysis of TNF-α mRNA levels of resident AMφ from both strains of mice after 3 or 6 h of incubation with PCP or PCP plus IFN-α showed that STAT1, directly or indirectly, is indispensable for the up-regulated TNF-α mRNA levels observed on combined TLR2 and IFN-β stimulation (Fig. 7C).
Combined stimulation of human IFN-β and NTHi-derived OMP up-regulates IL-6 but not TNF-α production by human AMφ
Because NTHi is a human pathogen, we wanted to test whether the findings obtained using murine Mφ could be extended to human Mφ. To this purpose, we examined cytokine production by primary resident human AMφ, which we exposed to human IFN-β, NTHi OMP analogs or both. The TLR2 agonist LP was used as a positive control, as it has been shown to stimulate human Mφ (39). We assessed both TNF-α and IL-6 production, because published data indicated that in humans as in mice, IL-6 gene expression involves both NF-κB activation (27, 28) and STAT1-dependent IRF-1 synthesis (38).
Adherence-purified human AMφ responded to PCP and P6 (but not to P4) by releasing substantial amounts of IL-6 only in the presence of human IFN-β (Fig. 8A). However, AMφ production of TNF-α was not significantly up-regulated by costimulation in any of the donors (Fig. 8B). Similar responses were seen using Mφ isolated from PBMC of two of the same donors and using the vitamin D3-differentiated THP-1 Mφ cell line (data not shown). We considered the possibility that chronic lower respiratory infections might blunt the responsiveness of COPD patients to exogenous IFN-β. The increased IL-6 production on simultaneous treatment with IFN-β and PCP or P6 (Fig. 8A) argues that any such effect is incomplete, but to further investigate this possibility, we tested for basal STAT1 phosphorylation and for spontaneous IFN-β release in two additional subjects with COPD, and found none (data not shown). Thus, although the NTHi-derived lipopeptides PCP and P6 acquire strong IL-6-inducing activity when used to stimulate Mφ in the presence of human IFN-β, the STAT1-dependent increase in TNF-α production they induce in mice appears not to be conserved in humans.
FIGURE 8.
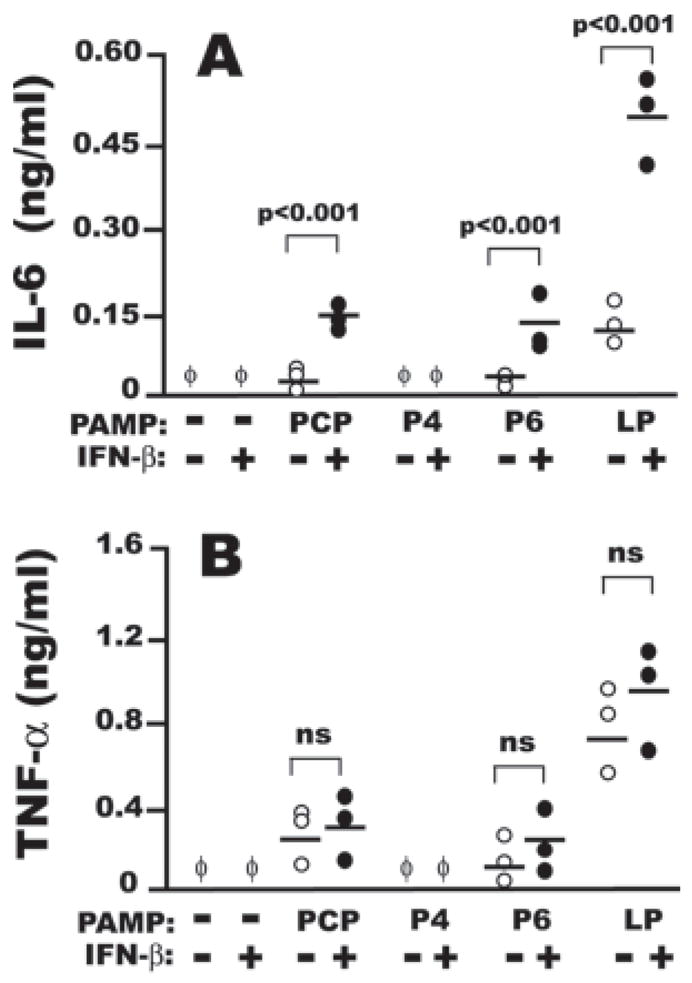
Human AMφ respond synergistically to NTHi lipopeptides and recombinant human IFN-β. Resident AMφ from three donors were incubated for 6 h in the presence of medium alone, 1000 U/ml IFN-β, 100 ng/ml of each of the indicated synthetic OMPs (○) or a combination of one OMP and IFN-β(●). The supernatants were collected and processed for ELISA for IL-6 (A) and TNF-α (B).φ, None detected. The three independent experiments performed are shown together and assessed for statistical significance by paired Student t test. The mean of all the donors is also shown in each experimental group.
This disparity prompted us to analyze the 5′ region of the IL-6 and TNF-α genes in the two species. The human IL-6 promoter contains sequences that bind IRF-1, activity of which (in addition to NF-κB) is essential for IL-6 production (28). Using TESS promoter analysis software (〈www.cbil.upenn.edu/tess/〉), we found corresponding IRF-1-binding sites in the murine IL-6 promoter (GenBank accession no. M20572). Analysis of the murine TNF-α promoter (GenBank accession no. AB062426) revealed two potential IFN-stimulated response elements (ISRE) that match the consensus sequence AGTTTCNNTTCNC/T (33). These elements are located 122 and 499 bp upstream from the ATG translational start site. In agreement with our ELISA data in the human system (Fig. 8B), we found no such ISRE consensus sequences in the promoter of the human TNF-α gene (GenBank accession no. AB088112).
Discussion
These results demonstrate that costimulation by type I IFN causes resident murine and human AMφ to mount a strong inflammatory response to NTHi OMPs that are themselves relatively innocuous. Using TLR2-specific synthetic tripalmitoylated lipopeptides that correspond to the invariant portion of three highly conserved NTHi OMPs (2), we found that IFN-β markedly increases a cytokine (IL-6) and induces two chemokines (CCL5, CXCL10) that drive mucus production and inflammatory cell recruitment, respectively. This observation is noteworthy because TLR2 is unique, among the nine TLRs common to mice and humans, for being incapable of autocrine/paracrine STAT1 activation via IFN-β production (34, 35). We now show that direct activation of STAT1 by IFN-β during TLR2 signaling permits induction of cytokines and chemokines which are typically induced by TLR3 or TLR4 stimulation alone. We also found that TLR2-induced TNF-α production is up-regulated by a STAT1-dependent transcriptional mechanism in murine AMφ, but not in human AMφ. By showing how IFN-β modulates the host response to components of a Gram-negative pathogen, these data emphasize that the importance of type I IFN to innate immunity is not limited to its antiviral properties (15, 16), and provide a partial explanation for the association of viral respiratory tract infections with AECB.
Mechanistically, the effects we found on addition of IFN-β to TLR2 stimuli (synergistic increase in IL-6, induction of CCL5 and CXCL10, and disparity in the effect on TNF-α between mice and humans) are all explained by the presence or absence of ISRE in the promoters of these proinflammatory genes. Both the murine and human IL-6 genes contain IRF-1-binding sites. CCL5 and CXCL10 production in both species depends strictly on the IFN-regulated factors IRF-3 and IRF-1, respectively (30, 31). By contrast, the human TNF-α promoter lacks the ISRE elements found in the mouse. Interestingly, the TNF family member TRAIL is induced by IFN-β via STAT1 activation in human cells (40), suggesting that it is the human TNF-α promoter that has diverged from a primordial mechanism retained in the mouse. Because IRF-binding sites in the murine TNF-α promoter match the ISRE inner core, this region could bind either STAT or IRF proteins (41). Indeed, previous chromatin immunoprecipitation data identified phospho-STAT1 binding in the murine TNF-α gene (42). Clearly, the precise mechanisms by which STAT1 up-regulates TNF-α transcription in the murine system, and their significance, require further investigation.
It should not be surprising that the finding that TNF-α production by TLR2-stimulated resident murine Mφ can be magnified by a STAT1-dependent transcriptional mechanism has gone undetected. LPS stimulation of murine PMφ (the stimulus and cell type most commonly used to study TNF-α production) itself induces IFN-β via an autocrine/paracrine loop (43). Our findings provide a potential explanation for the resistance of IFN-β−/− mice to LPS-induced lethality (17) and for similarities between IFN-β−/− mice and TNF-α−/− mice (44), that led Deonarain et al. (44) to hypothesize a critical immunoregulatory interrelationship between IFN-β and TNF-α. We now extend their data using a TLR2-dependent stimulus and resident murine AMφ, a cell type we have shown cannot produce IFN-β upon LPS stimulation (18).
The AMφ products that we measured have considerable potential to contribute to lung pathology. IL-6 is the only cytokine that directly up-regulates expression by primary human tracheobron-chial epithelial cells of the principal airway mucin genes, MUC5B and MUC5AC (45). CCL5 and CXCL10 are potent recruitment signals for cell types bearing their respective receptors, CCR5 (T cells, monocytes) and CXCR3 (T, B, NK, NKT, and mast cells) (46). As a non-Glu-Leu-Arg-containing CXC chemokine, CXCL10 also has angiogenic and profibrotic properties that might contribute to the marked small airway thickening and peribronchial fibrosis seen in advanced COPD (47).
COPD is increasingly recognized as an inflammatory disease (47). Although the roles of bacterial infections in AECB and in COPD progression remain controversial (48, 49), several recent findings imply that NTHi contribute importantly to both processes. NTHi is the bacteria most commonly isolated from expectorated sputum of both stable COPD patients and during AECB (4, 5), and the increased frequency of its isolation during AECB, relative to the stable state (5), is consistent with an etiological role. In a study using bronchoscopic protected brush sampling, isolation of NTHi increased between the stable and exacerbated state (50). Moreover, molecular evidence of NTHi persistence despite negative sputum cultures (51) implies that NTHi colonization of the lower tracheobronchial tree in COPD has likely been underestimated. Hence, our results suggest that production of type I IFN by virally infected airway cells already colonized by NTHi could be a common trigger for the inflammation characteristic of AECB.
A potential limitation of our human data is that AMφ were obtained from subjects with a history of COPD, although none had suffered recent exacerbation. Hence, it is possible that the responsiveness of their AMφ to IFN-β was altered, relative to normal subjects or healthy smokers, by previous lower airway infections. Testing this interesting possibility will require considerably greater study.
The two strain-conserved OMP that we showed elicit a strong response in human Mφ, PCP and P6, are attractive candidate immunogens (2, 11). Indeed, other TLR2 stimulants are currently being evaluated in such settings (52), and TLR2 ligands have been shown to exert an adjuvant role in human Th1 responses (53). Coadministration of IFN-β as an adjuvant may increase immunization efficiency, as type I IFN powerfully stimulates development and activity of monocyte-derived dendritic cells (54, 55).
In summary, we show that IFN-β, which is produced by virally infected respiratory epithelial cells, converts normally innocuous NTHi OMPs into potent stimulants for AMφ production of inflammatory mediators, but does so via different mechanisms in mice and humans. By activating STAT1, exogenous IFN-β compensates for the inability of TLR2-initiated signals to be amplified by the autocrine/paracrine loop observed during TLR3 and TLR4 activation.
Acknowledgments
We thank Dr. David Fox for critically reviewing the manuscript; Joetta Steinke, R.R.T., and Mary Christensen, R.R.T., for patient recruitment; Bonnie Schweitzer, R.N., for assistance in the bronchoscopies; all the members of the Ann Arbor Veterans Affairs Research Enhancement Award Program for helpful suggestions and discussions; and Joyce O’Brien for secretarial support.
Footnotes
This work was supported by Merit Review funding and a Research Enhancement Award Program grant from the Department of Veterans Affairs and by RO1 HL056309 and HL082480 from the U.S. Public Health Service.
Abbreviations used in this paper: NTHi, nontypeable Haemophilus influenzae; COPD, chronic obstructive pulmonary disease; AECB, acute exacerbations of chronic bronchitis; OMP, outer membrane lipoprotein; Mφ, macrophage; AMφ, alveolar Mφ; LP, lipopeptide Pam3CysSK4; PMφ, peritoneal Mφ; BAL, bronchoalveolar lavage; IRF, IFN-regulatory factor; ISRE, IFN-stimulated response element; IP-10, IFN-γ-inducible protein 10.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Kuklinska D, Kilian M. Relative proportions of Haemophilus species in the throat of healthy children and adults. Eur J Clin Microbiol. 1984;3:249–252. doi: 10.1007/BF02014895. [DOI] [PubMed] [Google Scholar]
- 2.Foxwell AR, Kyd JM, Cripps AW. Nontypeable Haemophilus influenzae: pathogenesis and prevention. Microbiol Mol Biol Rev. 1998;62:294–308. doi: 10.1128/mmbr.62.2.294-308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bandi V, Jakubowycz M, Kinyon C, Mason EO, Atmar RL, Greenberg SB, Murphy TF. Infectious exacerbations of chronic obstructive pulmonary disease associated with respiratory viruses and non-type-able. Haemophilus influenzae. FEMS Immunol Med Microbiol. 2003;37:69–75. doi: 10.1016/S0928-8244(03)00100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson R. Evidence of bacterial infection in acute exacerbations of chronic bronchitis. Semin Respir Infect. 2000;15:208–215. doi: 10.1053/srin.2000.18070. [DOI] [PubMed] [Google Scholar]
- 5.Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465– 471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 6.Murphy TF, Kirkham C. Biofilm formation by nontypeable Haemophilus influenzae: strain variability, outer membrane antigen expression and role of pili. BMC Microbiol. 2002;2:7. doi: 10.1186/1471-2180-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.St Geme JW, III, Falkow S. Haemophilus influenzae adheres to and enters cultured human epithelial cells. Infect Immun. 1990;58:4036–4044. doi: 10.1128/iai.58.12.4036-4044.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forsgren J, Samuelson A, Ahlin A, Jonasson J, Rynnel-Dagoo B, Lindberg A. Haemophilus influenzae resides and multiplies intracellularly in human adenoid tissue as demonstrated by in situ hybridization and bacterial viability assay. Infect Immun. 1994;62:673– 679. doi: 10.1128/iai.62.2.673-679.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouchet V, Hood DW, Li J, Brisson JR, Randle GA, Martin A, Li Z, Goldstein R, Schweda EK, Pelton SI, et al. Host-derived sialic acid is incorporated into Haemophilus influenzae lipopolysaccharide and is a major virulence factor in experimental otitis media. Proc Natl Acad Sci USA. 2003;100:8898–8903. doi: 10.1073/pnas.1432026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuto T, Xu H, Wang B, Han J, Kai H, Gu XX, Murphy TF, Lim DJ, Li JD. Activation of NF-κB by nontypeable Haemophilus influenzae is mediated by Toll-like receptor 2-TAK1-dependent NIK-IKK α/β-IκBα and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci USA. 2001;98:8774–8779. doi: 10.1073/pnas.151236098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berenson CS, Murphy TF, Wrona CT, Sethi S. Outer membrane protein P6 of nontypeable Haemophilus influenzae is a potent and selective inducer of human macrophage proinflammatory cytokines. Infect Immun. 2005;73:2728–2735. doi: 10.1128/IAI.73.5.2728-2735.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seemungal T, Harper-Owen R, Bhowmik A, Moric I, Sanderson G, Message S, Maccallum P, Meade TW, Jeffries DJ, Johnston SL, Wedzicha JA. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1618–1623. doi: 10.1164/ajrccm.164.9.2105011. [DOI] [PubMed] [Google Scholar]
- 13.Rohde G, Wiethege A, Borg I, Kauth M, Bauer TT, Gillissen A, Bufe A, Schultze-Werninghaus G. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax. 2003;58:37– 42. doi: 10.1136/thorax.58.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:115–120. doi: 10.1513/pats.2306030. [DOI] [PubMed] [Google Scholar]
- 15.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675– 687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 16.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (α/β) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 17.Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. 2003;4:471– 477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- 18.Punturieri A, Alviani RS, Polak T, Copper P, Sonstein J, Curtis JL. Specific engagement of TLR4 or TLR3 does not lead to IFN-β-mediated innate signal amplification and STAT1 phosphorylation in resident murine alveolar macrophages. J Immunol. 2004;173:1033–1042. doi: 10.4049/jimmunol.173.2.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Modlin RL. Activation of Toll-like receptors by microbial lipoproteins: role in host defense. J Allergy Clin Immunol. 2001;108:S104–S106. doi: 10.1067/mai.2001.118299. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 21.Nelson S, Summer WR, Mason CM. The role of the inflammatory response in chronic bronchitis: therapeutic implications. Semin Respir Infect. 2000;15:24–31. doi: 10.1053/srin.2000.0150024. [DOI] [PubMed] [Google Scholar]
- 22.Hogg JC. Chronic obstructive pulmonary disease: an overview of pathology and pathogenesis. Novartis Found Symp. 2001;234:4–19. doi: 10.1002/0470868678.ch2. discussion 19 –26. [DOI] [PubMed] [Google Scholar]
- 23.Barnes PJ. New concepts in chronic obstructive pulmonary disease. Annu Rev Med. 2003;54:113–129. doi: 10.1146/annurev.med.54.101601.152209. [DOI] [PubMed] [Google Scholar]
- 24.Muta T, Takeshige K. Essential roles of CD14 and lipopolysaccharide-binding protein for activation of Toll-like receptor (TLR)2 as well as TLR4 reconstitution of TLR2- and TLR4-activation by distinguishable ligands in LPS preparations. Eur J Biochem. 2001;268:4580–4589. doi: 10.1046/j.1432-1327.2001.02385.x. [DOI] [PubMed] [Google Scholar]
- 25.Manukyan M, Triantafilou K, Triantafilou M, Mackie A, Nilsen N, Espevik T, Wiesmuller KH, Ulmer AJ, Heine H. Binding of lipopeptide to CD14 induces physical proximity of CD14, TLR2 and TLR1. Eur J Immunol. 2005;35:911–921. doi: 10.1002/eji.200425336. [DOI] [PubMed] [Google Scholar]
- 26.Punturieri A, Filippov S, Allen E, Caras I, Murray R, Reddy V, Weiss SJ. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J Exp Med. 2000;192:789–799. doi: 10.1084/jem.192.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanabe O, Akira S, Kamiya T, Wong GG, Hirano T, Kishimoto T. Genomic structure of the murine IL-6 gene: high degree conservation of potential regulatory sequences between mouse and human. J Immunol. 1988;141:3875–3881. [PubMed] [Google Scholar]
- 28.Sanceau J, Kaisho T, Hirano T, Wietzerbin J. Triggering of the human interleukin-6 gene by interferon-γ and tumor necrosis factor-α in monocytic cells involves cooperation between interferon regulatory factor-1, NF κB, and Sp1 transcription factors. J Biol Chem. 1995;270:27920–27931. doi: 10.1074/jbc.270.46.27920. [DOI] [PubMed] [Google Scholar]
- 29.Genin P, Algarte M, Roof P, Lin R, Hiscott J. Regulation of RANTES chemokine gene expression requires cooperativity between NF-κB and IFN-regulatory factor transcription factors. J Immunol. 2000;164:5352–5361. doi: 10.4049/jimmunol.164.10.5352. [DOI] [PubMed] [Google Scholar]
- 30.Lin R, Heylbroeck C, Genin P, Pitha PM, Hiscott J. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol Cell Biol. 1999;19:959–966. doi: 10.1128/mcb.19.2.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohmori Y, Hamilton TA. Cooperative interaction between interferon (IFN) stimulus response element and κB sequence motifs controls IFN γ-and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J Biol Chem. 1993;268:6677– 6688. [PubMed] [Google Scholar]
- 32.Majumder S, Zhou LZ, Chaturvedi P, Babcock G, Aras S, Ransohoff RM. p48/STAT-1α -containing complexes play a predominant role in induction of IFN-γ-inducible protein, 10 kDa (IP-10) by IFN-γ alone or in synergy with TNF-α. J Immunol. 1998;161:4736–4744. [PubMed] [Google Scholar]
- 33.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 35.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 36.Harcourt JL, Offermann MK. Interferon-α synergistically enhances induction of interleukin-6 by double stranded RNA in HeLa cells. Eur J Biochem. 2000;267:2768–2777. doi: 10.1046/j.1432-1327.2000.01300.x. [DOI] [PubMed] [Google Scholar]
- 37.Sariban E, Imamura K, Luebbers R, Kufe D. Transcriptional and posttranscriptional regulation of tumor necrosis factor gene expression in human monocytes. J Clin Invest. 1988;81:1506–1510. doi: 10.1172/JCI113482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Leung S, Qureshi S, Darnell JE, Jr, Stark GR. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-α. J Biol Chem. 1996;271:5790–5794. doi: 10.1074/jbc.271.10.5790. [DOI] [PubMed] [Google Scholar]
- 39.Parker LC, Whyte MK, Vogel SN, Dower SK, Sabroe I. Toll-like receptor (TLR)2 and TLR4 agonists regulate CCR expression in human monocytic cells. J Immunol. 2004;172:4977– 4986. doi: 10.4049/jimmunol.172.8.4977. [DOI] [PubMed] [Google Scholar]
- 40.Choi EA, Lei H, Maron DJ, Wilson JM, Barsoum J, Fraker DL, El-Deiry WS, Spitz FR. Stat1-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand and the cell-surface death signaling pathway by interferon β in human cancer cells. Cancer Res. 2003;63:5299–5307. [PubMed] [Google Scholar]
- 41.Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22:145–152. doi: 10.1089/107999002753452755. [DOI] [PubMed] [Google Scholar]
- 42.Takagi K, Takagi M, Kanangat S, Warrington KJ, Shigemitsu H, Postlethwaite AE. Modulation of TNF-α gene expression by IFN-γ and pamidronate in murine macrophages: regulation by STAT1-dependent pathways. J Immunol. 2005;174:1801–1810. doi: 10.4049/jimmunol.174.4.1801. [DOI] [PubMed] [Google Scholar]
- 43.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-α -induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 44.Deonarain R, Verma A, Porter AC, Gewert DR, Platanias LC, Fish EN. Critical roles for IFN-β in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor α. Proc Natl Acad Sci USA. 2003;100:13453–13458. doi: 10.1073/pnas.2230460100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 46.Brightling CE, Ammit AJ, Kaur D, Black JL, Wardlaw AJ, Hughes JM, Bradding P. The CXCL10/CXCR3 axis mediates human lung mast cell migration to asthmatic airway smooth muscle. Am J Respir Crit Care Med. 2005;171:1103–1108. doi: 10.1164/rccm.200409-1220OC. [DOI] [PubMed] [Google Scholar]
- 47.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 48.Sethi S. Bacteria in exacerbations of chronic obstructive pulmonary disease: phenomenon or epiphenomenon? Proc Am Thorac Soc. 2004;1:109–114. doi: 10.1513/pats.2306029. [DOI] [PubMed] [Google Scholar]
- 49.Rosell A, Monso E, Soler N, Torres F, Angrill J, Riise G, Zalacain R, Morera J, Torres A. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med. 2005;165:891– 897. doi: 10.1001/archinte.165.8.891. [DOI] [PubMed] [Google Scholar]
- 50.Monso E, Ruiz J, Rosell A, Manterola J, Fiz J, Morera J, Ausina V. Bacterial infection in chronic obstructive pulmonary disease: a study of stable and exacerbated outpatients using the protected specimen brush. Am J Respir Crit Care Med. 1995;152:1316–1320. doi: 10.1164/ajrccm.152.4.7551388. [DOI] [PubMed] [Google Scholar]
- 51.Murphy TF, Brauer AL, Schiffmacher AT, Sethi S. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:266–272. doi: 10.1164/rccm.200403-354OC. [DOI] [PubMed] [Google Scholar]
- 52.Muller SD, Muller MR, Huber M, Esche Uv U, Kirschning CJ, Wagner H, Bessler WG, Mittenbuhler K. Triacyl-lipopentapeptide adjuvants: TLR2-dependent activation of macrophages and modulation of receptor-mediated cell activation by altering acyl-moieties. Int Immunopharmacol. 2004;4:1287–1300. doi: 10.1016/j.intimp.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 53.Sieling PA, Chung W, Duong BT, Godowski PJ, Modlin RL. Toll-like receptor 2 ligands as adjuvants for human Th1 responses. J Immunol. 2003;170:194–200. doi: 10.4049/jimmunol.170.1.194. [DOI] [PubMed] [Google Scholar]
- 54.Santini SM, Lapenta C, Logozzi M, Parlato S, Spada M, Di Pucchio T, Belardelli F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med. 2000;191:1777–1788. doi: 10.1084/jem.191.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santini SM, Lapenta C, Belardelli F. Type I interferons as regulators of the differentiation/activation of human dendritic cells: methods for the evaluation of IFN-induced effects. Methods Mol Med. 2005;116:167–181. doi: 10.1385/1-59259-939-7:167. [DOI] [PubMed] [Google Scholar]