

Abstract
The axon guidance cue netrin is importantly involved in neuronal development. DCC (deleted in colorectal cancer) is a functional receptor for netrin and mediates axon outgrowth and the steering response. Here we show that different regions of the intracellular domain of DCC directly interacted with the tyrosine kinases Src and focal adhesion kinase (FAK). Netrin activated both FAK and Src and stimulated tyrosine phosphorylation of DCC. Inhibition of Src family kinases reduced DCC tyrosine phosphorylation and blocked both axon attraction and outgrowth of neurons in response to netrin. Mutation of the tyrosine phosphorylation residue in DCC abolished its function of mediating netrin-induced axon attraction. On the basis of our observations, we suggest a model in which DCC functions as a kinase-coupled receptor, and FAK and Src act immediately downstream of DCC in netrin signaling.
During embryonic development, neurons are guided to specific targets by extracellular cues in their environment. Axon growth cones sense various chemoattractive and chemorepulsive signals and translate these signals, via intracellular signal transduction pathways, into cellular movements that ultimately steer them to their correct targets. Several axon guidance molecules have been discovered and characterized, including a soluble family of proteins called netrins1–3. Netrins can stimulate axon growth in addition to eliciting both attractive and repulsive responses4.
Genetic studies in Caenorhabditis elegans indicate that UNC-40 and UNC-5 are functional receptors for UNC-6, a netrin homolog5–7. The mammalian homolog of UNC-40 is DCC, originally identified as a tumor suppressor gene8. DCC mediates both the axon growth and the chemoattractive function of netrin6,7,9. In addition, DCC mediates growth cone repulsion when in a complex with the UNC-5 receptor5,10–13. The UNC-40 and UNC-5 receptor complex is required for the dorsoventral repulsion of both neurons and gonads in C. elegans. The biological functions of netrin and its receptors in axon growth and growth cone guidance have been well studied; the intracellular signal transduction pathways downstream of the netrin receptors, however, are only partially understood.
Tyrosine phosphorylation may be involved downstream of guidance receptors, as phosphorylation of both DCC and UNC-5 has been observed14. Furthermore, coexpression of Src, a tyrosine kinase, increases UNC-5 tyrosine phosphorylation, indicating a possible role of Src in netrin signaling. FAK is activated by integrins and other extracellular signals. FAK activation stimulates multiple cellular signal transduction events leading to cell adhesion, motility and cell morphological changes15–17. In response to integrin stimulation, FAK binds to Src and stimulates Src activity. Both the scaffold functions and kinase activity of FAK contribute to integrin-induced signal transduction. Src, in turn, phosphorylates and activates FAK. Therefore, FAK and Src act synergistically to signal downstream targets. Interestingly, the Src family tyrosine kinases (SFKs) are highly enriched in the nervous system and may be involved in the signal transduction of axon guidance receptors18,19.
In this report, we show that netrin activates FAK and Src and stimulates tyrosine phosphorylation of DCC. The carboxy (C)-terminal domain of FAK interacts with the C-terminal P3 domain of DCC, and this FAK-DCC interaction is required for netrin to stimulate tyrosine phosphorylation and activation of FAK. Furthermore, netrin-stimulated tyrosine phosphorylation of DCC and FAK requires SFKs. We also found that DCC interacts with Src and that inhibition of SFKs by PP2 blocks netrin-stimulated axon growth of commissural neurons from embryonic rat spinal explants. Alteration of Src function using inhibitors or by overexpression of Src mutants blocked the netrin-induced axon attraction of Xenopus laevis spinal neurons. In addition, the importance of DCC tyrosine phosphorylation in netrin signaling was also demonstrated by the axon attraction assay, indicating that DCC is a key downstream substrate of FAK-Src in the DCC receptor complex. Our data demonstrate that FAK and SFKs act immediately downstream of the netrin receptor DCC and are importantly involved in netrin-induced signal transduction.
Results
Netrin stimulates tyrosine phosphorylation of DCC and FAK
It has been reported that netrin induces tyrosine phosphorylation of DCC in transfected HEK293 cells14. It is not known, however, whether the endogenous DCC is tyrosine phosphorylated or which kinase is responsible for DCC tyrosine phosphorylation. To determine whether netrin stimulates tyrosine phosphorylation of DCC in vivo, we tested dorsal spinal neurons from chick embryos. We observed that netrin increased tyrosine phosphorylation of both DCC and FAK (Fig. 1a). To confirm that these effects were due to netrin and not to contaminating components in the netrin preparation, we repeated the test using immunoaffinity-purified netrin and found a similar level of DCC and FAK tyrosine phosphorylation (data not shown). Furthermore, preincubation of the cultured neurons with an extracellular antibody to DCC (anti-DCC) blocked the netrin-stimulated FAK tyrosine phosphorylation by approximately 70% (Fig. 1b). These observations demonstrate that netrin induces tyrosine phosphorylation of endogenous DCC and FAK.
Figure 1.
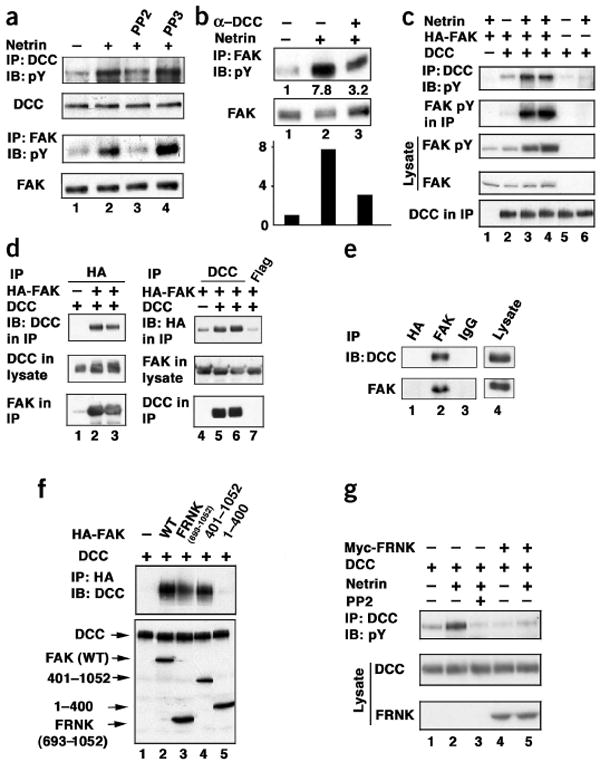
Netrin-stimulated tyrosine phosphorylation of DCC and FAK. (a) Netrin stimulates tyrosine phosphorylation of DCC and FAK in embryonic spinal neurons. IB, immunoblot; anti-pY, anti-phosphotyrosine antibody. (b) DCC extracellular domain antibody blocks netrin-induced FAK tyrosine phosphorylation. Bar graph shows quantitative data. α-DCC, anti-DCC. (c) Netrin stimulates tyrosine phosphorylation of DCC and FAK in transfected HEK293 cells. HEK293 cells were transfected with the indicated expression vectors. After netrin stimulation, DCC was immunoprecipitated and subjected to the indicated IB analysis. Lanes 3 and 4 are duplicates. (d) Co-IP of FAK with DCC. Left, results of IP with anti-HA followed by anti-DCC IB. Right, the reciprocal IP done. Anti-Flag IP was included as a negative control. (e) DCC and FAK interact at endogenous levels. Endogenous FAK was immunoprecipitated from the membrane fraction isolated from mouse brain lysate. The presence of endogenous DCC was determined by IB. Anti-HA and anti-Myc IgGs were used as negative controls. Lane 4 reflects a shorter exposure than lanes 1–3. (f) The C-terminal domain (FRNK) of FAK interacts with DCC. HEK 293 cells were transfected with the indicated plasmids. The various FAK deletions were immunoprecipitated by anti-HA and the presence of DCC was determined by IB. Expression levels of DCC and different FAK deletions in cell lysates are shown by IB. (g) FRNK inhibits netrin-induced DCC tyrosine phosphorylation. HEK293 cells were transfected with DCC with or without FRNK. Cells were stimulated with netrin for 20 min and tyrosine phosphorylation of DCC was determined by IB. The anti-phosphotyrosine western blot in this panel was exposed longer than that in c to visualize DCC tyrosine phosphorylation induced by netrin.
FAK and DCC associate with each other
To examine the function of FAK in DCC tyrosine phosphorylation, we cotransfected DCC and FAK into HEK293 cells. FAK coexpression increased DCC tyrosine phosphorylation (Fig. 1c). Importantly, tyrosine phosphorylation of both DCC and FAK were enhanced by netrin. We also observed that netrin stimulated DCC tyrosine phosphorylation in a dosage- and time-dependent manner, with an effective concentration similar to that reported to induce axon outgrowth2 (see Supplementary Fig. 1 online). Notably, our phosphotyrosine western blot detected a DCC-coprecipitated band with a molecular weight similar to FAK (Fig. 1c), indicating that DCC may associate with FAK. We directly tested the interaction between DCC and FAK by coimmunoprecipitation (co-IP) of overexpressed proteins in HEK293 cells. We found that DCC was specifically immunoprecipitated by FAK (Fig. 1d). Reciprocal co-IP also confirmed that FAK was recovered in anti-DCC immunoprecipitates (Fig. 1d). We estimated that approximately 2–5% of the transfected DCC was coimmunoprecipitated with FAK. The interaction between DCC and FAK was confirmed by in vitro pulldown experiments (Supplementary Fig. 1). To confirm the interaction between DCC and FAK under physiological conditions, we carried out co-IP studies from mouse brain tissue. DCC was detected in the anti-FAK immunoprecipitates, but not in the negative control (Fig. 1e). These data demonstrate that DCC and FAK can form a complex under physiological conditions.
We examined which region of FAK is responsible for the interaction with DCC. Deletion experiments indicated that the C-terminal domain of FAK is necessary and sufficient to mediate interaction with DCC (Fig. 1f). A naturally existing alternatively spliced form of FAK called FRNK (FAK-related nonkinase) corresponds to this C-terminal domain (amino acids 693–1052) of FAK and functions in a dominant-negative manner20. The interaction between DCC and FRNK was also observed in a yeast two-hybrid assay (data not shown), suggesting that they directly interact. The above observations indicate that FRNK may have an inhibitory role in netrin signaling. We tested whether FRNK could block DCC tyrosine phosphorylation induced by netrin. Our data show that netrin-stimulated DCC tyrosine phosphorylation was inhibited by FRNK coexpression (Fig. 1g), further supporting the notion that endogenous FAK binding is required for netrin-stimulated tyrosine phosphorylation of DCC.
Physical and functional interaction between DCC and Src
Previous studies have established a positive relationship between the activations of FAK and Src16,17. Hence, we tested the effect of Src using the SFK-specific inhibitor PP2 (ref. 21). PP2, but not its inactive analog PP3, inhibited netrin-stimulated tyrosine phosphorylation of DCC and FAK (Fig. 1a). To confirm the involvement of Src in DCC signaling, we coexpressed DCC and Src in HEK293 cells. We observed that Src significantly stimulated DCC tyrosine phosphorylation (Fig. 2a). DCC tyrosine phosphorylation was efficiently inhibited by a concentration of PP2 as low as 0.2 μM (Supplementary Fig. 1), supporting a specific role of endogenous SFK in netrin signaling. In addition, when only a small amount of Src was coexpressed with DCC, tyrosine phosphorylation of DCC could be further stimulated by netrin (Fig. 2b). Interestingly, netrin also increased phosphorylation of Tyr418 in Src (Fig. 2b), which is a site that is required for Src activation, and is thus used as an indicator19. These data suggest that Src stimulates DCC phosphorylation and is also a potential downstream effector of DCC.
Figure 2.
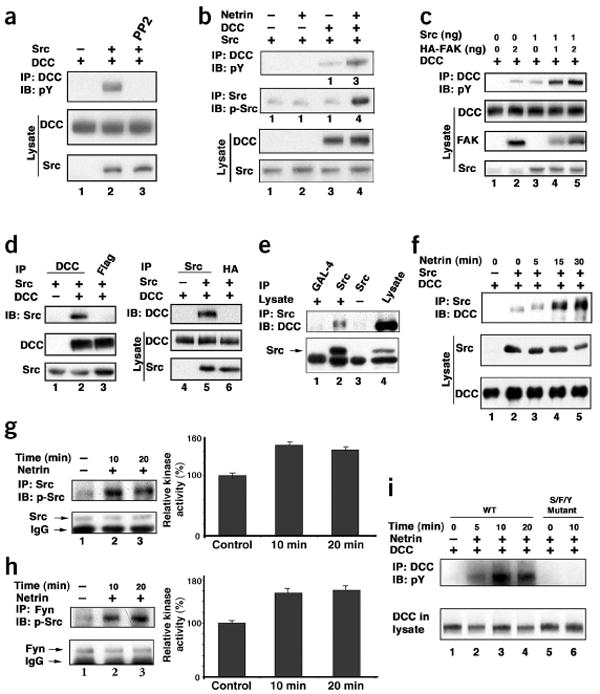
Src interacts with DCC and is activated by netrin. (a) Src activates DCC tyrosine phosphorylation. HEK293 cells were transfected with DCC (500 ng) and Src (50 ng) and treated with PP2 as indicated. Tyrosine phosphorylation of DCC was determined by anti-DCC IP followed by anti-PY20 IB. (b) Netrin stimulates Src. Cells were transfected with DCC (250 ng) and Src (10 ng) and stimulated with netrin. Tyrosine phosphorylation of DCC and Src were determined using anti-pY20 and Src-Y418, respectively. (c) Synergistic tyrosine phosphorylation of DCC by Src and FAK. DCC was cotransfected with various amounts of FAK and Src as indicated. Tyrosine phosphorylation of DCC was determined. (d) Co-IP of Src with DCC. HEK293 cells were transfected as indicated and subjected to IP with either anti-DCC (left) or anti-Src (right). Co-IP of Src (left) or DCC (right) was determined by IB. (e) Interaction of endogenous Src and DCC. Endogenous Src was immunoprecipitated. The presence of endogenous DCC in the Src IP was determined by IB. IP with anti-GAL-4 and anti-Src incubated with buffer only were used as negative controls. (f) Netrin enhances the interaction between DCC and Src in transfected HEK293 cells. Src was immunoprecipitated by Src antibody and the IP was probed with DCC antibody for DCC. (g) Netrin stimulates Src tyrosine phosphorylation and kinase activity in the E12 mouse brain cortex neurons. Left, Src tyrosine phosphorylation; right, quantification of kinase activity after 10 min and 20 min of netrin application. (h) Netrin stimulates Fyn tyrosine phosphorylation and kinase activity. Experiments are similar to those in g. (i) Netrin did not induce DCC tyrosine phosphorylation in Src/Fyn/Yes (S/F/Y) triple-knockout MEF cells. DCC and UNC-5 were cotransfected into wild-type (WT) and Src/Fyn/Yes triple knockout MEFs, and then netrin-induced DCC tyrosine phosphorylation was determined.
We examined whether FAK and Src cooperate to stimulate DCC phosphorylation. Cotransfection of both FAK and Src resulted in synergistic enhancement in DCC tyrosine phosphorylation (Fig. 2c). These results indicate that both FAK and Src positively contribute to DCC tyrosine phosphorylation. Multiple tyrosine residues in FAK have been mapped as targets of phosphorylation15–17. Netrin induced the most dramatic increase in tyrosine phosphorylation on residues Tyr576 and Tyr577 (Supplementary Fig. 1). Src has been implicated in the phosphorylation of these two residues which are important for FAK activation22. PP2 significantly inhibited tyrosine phosphorylation of Tyr576 and Tyr577, but had less effect on other tyrosine residues (Supplementary Fig. 1), further supporting a positive role of Src in netrin-induced FAK activation.
To test whether Src interacts with DCC, we used IP with cotransfected DCC and Src. DCC was present in the Src immunoprecipitates, and Src was present in the DCC immunoprecipitates (Fig. 2d). A Src-specific antibody coimmunoprecipitated with DCC from a mouse brain lysate, indicating an interaction between the two proteins in vivo (Fig. 2e). We further examined a possibility of direct interactions among DCC, FAK and Src. The immunoprecipitated FAK and Src interacted with immunopurified DCC in vitro (Supplementary Fig. 1), indicating that DCC interacts directly with FAK and Src.
The effect of netrin stimulation on the interaction between DCC and Src was examined. Our results show that netrin stimulation enhanced the interaction between DCC and Src (Fig. 2f). Similar experiments were done for FAK, and we found that netrin stimulation had no significant effect on the interaction between DCC and FAK (Supplementary Fig. 1). These results indicate that DCC uses different mechanisms to interact with FAK and Src.
To determine the effect of netrin on Src activation in neurons, we immunoprecipitated Src from mouse embryonic cortical neurons. We found that netrin stimulated Src tyrosine phosphorylation and kinase activity (Fig. 2g). Similarly, Fyn, a member of the SFKs23, was activated by netrin (Fig. 2h). We also found that Fyn interacts with DCC (Supplementary Fig. 1). To determine the functional importance of SFKs in netrin stimulation, we examined the Src/Fyn/Yes triple-knockout mouse embryonic fibroblasts (MEF)24. Netrin did not stimulate tyrosine phosphorylation of transfected DCC (Fig. 2i). Our results demonstrate that SFKs are required for netrin to induce DCC tyrosine phosphorylation.
Tyr1420 is the major tyrosine phosphorylation site in DCC
The intracellular domain of DCC contains four tyrosine residues (Fig. 3a). Our results indicate that mutation of Tyr1420 almost completely abolishes tyrosine phosphorylation of DCC, whereas individual mutation of the other three tyrosine residues has a minor effect on DCC tyrosine phosphorylation (Fig. 3a,b). We further confirmed Tyr1420 as the primary FAK-dependent phosphorylation site by creating a mutant DCC in which three tyrosine residues were replaced by phenylalanine, leaving one tyrosine residue. As expected, replacement of all four tyrosine residues by phenylalanine in the intracellular domain of DCC (DCC-4Y/F) completely abolished DCC tyrosine phosphorylation. Similar experiments were done with Src, which demonstrated that Tyr1420 was the major tyrosine phosphorylation site in DCC induced by coexpression of Src with DCC (Fig. 3c).
Figure 3.
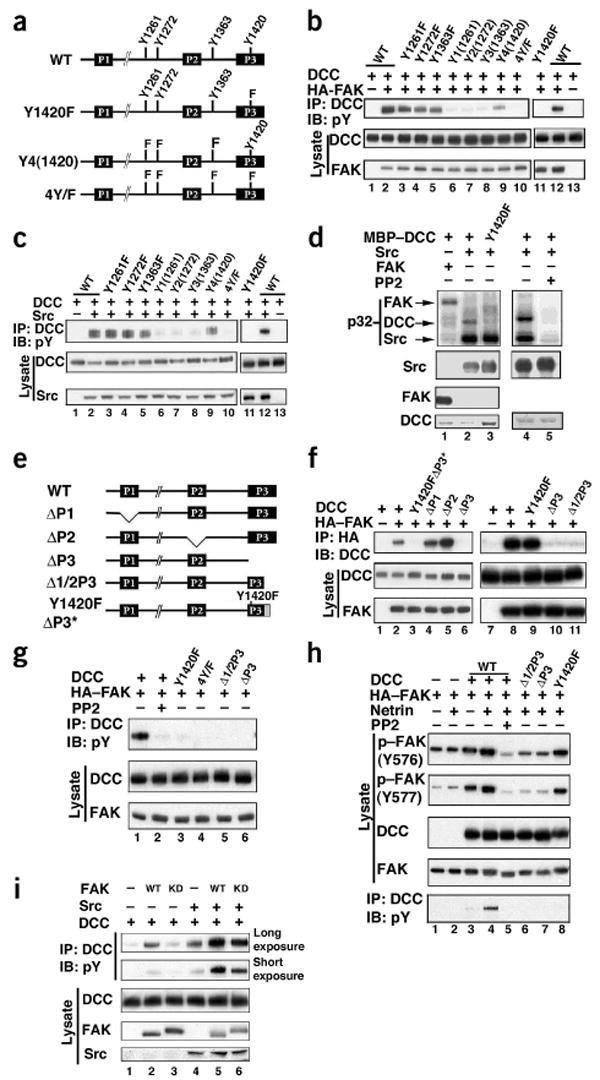
Tyr1420 in DCC is phosphorylated and the P3 domain in DCC interacts with FAK. (a) Schematic of DCC intracellular domain, showing the conserved P1, P2 and P3 domains along with four tyrosine residues. Also shown are some of the constructs used in Figure 3b–h. (b) FAK enhanced DCC Tyr1420 phosphorylation. HEK293 cells were transfected with the indicated plasmids. Y1, Y2, Y3 and Y4 denote mutations in the tyrosines of the intracellular domain of DCC, except for the one indicated in parenthesis. (c) As in b, except with Src coexpressed instead of FAK. (d) In vitro phosphorylation of DCC by Src and not FAK. Immunoprecipitated Src and HA-FAK were incubated with MBP-DCC. Above, 32P incorporation. Below, total amounts of Src, FAK and MBP-DCC used in the kinase reactions. (e) Intracellular domain of DCC. DCC-ΔP1, ΔP2, ΔP3 and Δ1/2P3 correspond to deletions of residues 1147–1171, 1335–1356, 1412–1447 and 1426–1447, respectively. Shaded box denotes mutation of 22 C-terminal residues in DCC-1420Y/FΔP3*. (f) The C-terminal half of the P3 domain of DCC is required for FAK binding. Experiments were similar to those shown in b. (g) FAK binding is essential for tyrosine phosphorylation of DCC by FAK. DCC and various mutants were coexpressed with HA-FAK in HEK293 cells. Tyrosine phosphorylation (top) and total amount of protein (middle) of immunoprecipitated DCC are shown. (h) The P3 domain of DCC is essential for FAK activation. HEK293 cells were transfected as indicated. The netrin-DCC induced phosphorylation status of FAK at Tyr576 and Tyr577 was determined using phospho-specific antibody. (i) Kinase-inactive FAK cooperates with Src to stimulate DCC phosphorylation. DCC was cotransfected in HEK293 cells with 2 ng of wild-type FAK (WT), kinase-dead FAK (KD) and Src as indicated. Phosphorylation of immunoprecipitated DCC was detected (two different exposures are presented).
To determine whether FAK or Src can directly phosphorylate DCC, we carried out in vitro phosphorylation experiments using purified recombinant DCC as a substrate. We found that Src could directly phosphorylate DCC, whereas FAK could not (Fig. 3d). The ability of Src to phosphorylate DCC was inhibited by PP2, and DCC-Y1420F (DCC in which tyrosine residue 1420 is replaced by phenylalanine) was not phosphorylated by Src. Hence, these data demonstrate that tyrosine phosphorylation of Tyr1420 in DCC results from direct phosphorylation by Src but not FAK.
Different regions of DCC interact with FAK and Src
The intracellular region of DCC contains three motifs (P1, P2 and P3), which are conserved across species (Fig. 3e)7,12. Deletion of P1 (residues 1147–1171, ΔP1) did not affect FAK binding (Fig. 3f). Notably, deletion of the P2 domain (residues 1335–1356, ΔP2) reproducibly enhanced the ability of DCC to bind FAK, suggesting that the P2 domain may have an inhibitory effect on FAK binding. In contrast, deletion of the P3 domain (residues 1412–1447, ΔP3) abolished FAK binding (Fig. 3f) and mutation of the tyrosine phosphorylation site (DCC-Y1420F) had no effect on the interaction with FAK (Fig. 3e,f). We also constructed a Y1420FΔP3* mutant, in which the C-terminal 22 residues of DCC (1426–1447) are replaced by 10 unrelated residues. The mutant was not able to interact with FAK (Fig. 3e,f). The importance of the C-terminal half of the P3 domain in FAK interaction was confirmed by the Δ1/2P3 mutant (deletion of the 22 C-terminal amino acids), which also did not interact with FAK. As expected, PP2 treatment (100 min) did not inhibit the interaction between DCC and FAK (Supplementary Fig. 1). These results demonstrate that the C-terminal half of the P3 domain, but not tyrosine phosphorylation of DCC, is important for FAK interaction.
The importance of FAK binding in DCC tyrosine phosphorylation was examined with the Δ1/2P3 and ΔP3 mutants. FAK binding was essential for DCC tyrosine phosphorylation in response to FAK coexpression, although FAK was not directly responsible for DCC phosphorylation (Fig. 3d,g and see model in Supplementary Fig. 2). We tested whether the interaction between DCC and FAK is important for netrin and DCC to activate FAK. We observed that netrin treatment of DCC-Δ1/2P3 and ΔP3 mutants showed a markedly decreased ability to stimulate tyrosine phosphorylation of FAK. In contrast, mutation of the tyrosine phosphorylation sites in DCC had no significant effect on the ability of DCC to stimulate FAK tyrosine phosphorylation (Fig. 3h). These data suggest that the ability to interact with FAK is important for DCC to activate FAK.
The fact that FAK is required for DCC tyrosine phosphorylation, whereas it does not directly phosphorylate DCC, indicates that FAK may also function as a scaffold protein. We examined the effect of kinase-inactive mutant FAK-KD on DCC tyrosine phosphorylation. Expression of FAK-KD induced a minor increase of DCC phosphorylation, as compared to the wild-type FAK (Fig. 3i). Coexpression of FAK-KD and Src stimulated DCC phosphorylation, although this combination was less effective than the coexpression of wild-type FAK and Src. These observations indicate that both kinase-dependent and kinase-independent functions of FAK contribute to DCC phosphorylation. Kinase-independent scaffolding functions of FAK have been reported previously25,26. The above data support the view that FAK may also function as a scaffold by recruiting Src to the DCC complex (Supplementary Fig. 2).
DCC tyrosine phosphorylation is not required for Src binding because DCC-4Y/F was found to bind Src (Fig. 4a). Deletion of the P1, P2 or P3 domain did not affect the interaction with Src (Fig. 4b, data not shown for P1 and P2 deletions). This would suggest that Src and FAK bind different regions of DCC. We found that application of the inhibitor PP2 (20–100 min) weakened the interaction between Src and DCC (Fig. 4b), indicating that Src kinase activity promotes its interaction with DCC. However, our observations that DCC-4Y/F still binds Src (Fig. 4a) suggest that tyrosine phosphorylation of a protein other than DCC is probably required for the full interaction between DCC and Src.
Figure 4.
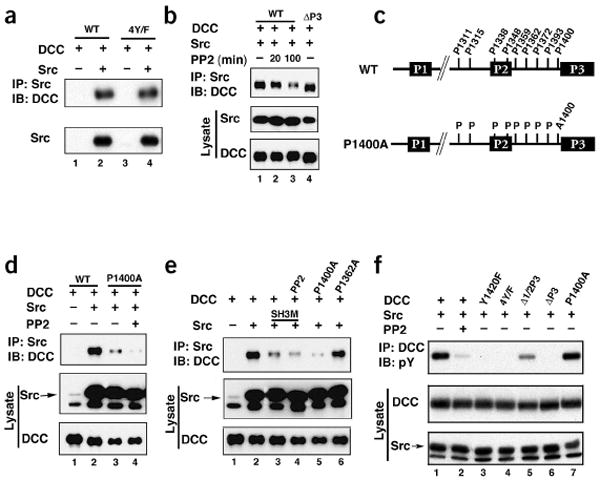
The 1400PXXP motif in DCC and the SH3 domain in Src are required for DCC and Src interaction. (a) Mutation of all DCC intracellular tyrosine residues did not affect Src-DCC interaction. HEK293 cells were transfected as indicated. Src was immunoprecipitated and bound DCC or DCC-4Y/F was determined by immunoblotting (IB). (b) Src kinase activity is required for optimal Src-DCC interaction. HEK293 cells were transfected as indicated. Cells were treated with PP2 for 20 min or 100 min followed by Src IP. Bound DCC was determined by IB. (c) Diagram of all nine putative PXXP motifs in the DCC intracellular domain. The amino acid number denotes the first proline in the PXXP motif and the P1400A mutant is also shown. (d) DCC-P1400A is compromised in Src binding. HEK293 cells were transfected with indicated plasmids. Src was immunoprecipitated and the presence of DCC in IP was determined by immunoblot. (e) The Src SH3 domain is important for the interaction with DCC. DCC-P1362A was included as a control. (f) Tyrosine phosphorylation of DCC mutants stimulated by Src. Experiments are similar to those shown in Figure 3g.
The fact that Src binds to DCC-4Y/F suggests that the SH3 domain of Src may interact with DCC. The intracellular domain of DCC contains nine PXXP motifs as putative SH3 binding sites (Fig. 4c). Deletion of residues 1309–1318, which contains the first two PXXP motifs, and 1335–1356, which contains the next two PXXP motifs, showed no significant effect on the interaction between DCC and Src (data not shown). Point mutations of PXXP motifs at Pro1359, Pro1362, Pro1372, Pro1393 and Pro1400 were constructed and tested. We observed that mutation of Pro1400 disrupted the interaction with Src (Fig. 4d,e), whereas mutation of other proline residues did not affect the interaction (Fig. 4e and data not shown). These results demonstrate that the PXXP motif, beginning at residue 1400, is important for Src binding and that the Src SH3 domain is likely to be involved. We created a Src SH3-domain mutant (Src-SH3M) by mutating the conserved Trp127, and this mutant showed reduced interaction with DCC (Fig. 4e). Notably, PP2 was able to further inhibit the interaction between Src and DCC-P1400A (a mutant in which proline residue 1400 has been replaced by alanine) and between Src-SH3M and DCC (Fig. 4d,e), which suggests that the DCC interaction with the Src SH3 domain is not the only factor contributing to the interaction. It is possible that the Src SH2 domain might interact with another phosphotyrosine-containing protein, possibly FAK, to enhance the Src interaction with DCC (Supplementary Fig. 2).
We next determined the ability of Src to phosphorylate DCC mutants. Elimination of FAK binding in DCC decreased Src-induced DCC phosphorylation (Fig. 4f). Furthermore, Src could still increase phosphorylation of DCC-P1400A, which is defective in Src interaction. These observations are consistent with a model that FAK recruits Src to the DCC complex and Src phosphorylates DCC (Supplementary Fig. 2). The kinase-inactive FAK mutant, FAK-KD, consistently cooperated with Src to stimulate DCC phosphorylation (Fig. 3i).
Inhibition of netrin-stimulated axon growth by PP2
To determine the function of Src in netrin-stimulated axon growth, we tested the effect of the Src kinase inhibitor PP2 on the axon outgrowth of dorsal spinal explants isolated from E12 rats in response to netrin stimulation. Consistent with previous studies1,2,27, there was little axon outgrowth from these explants in the absence of netrin-1 after 20 h in culture (Fig. 5a). Netrin-1 dramatically stimulated commissural axon outgrowth from the explants (Fig. 5a). Immunohistochemical analysis with an antibody to TAG-1, a marker for commissural neurons, confirmed the commissural nature of these axons (data not shown). Commissural axon outgrowth induced by netrin-1 was inhibited by PP2, but not by PP3 (Fig. 5a,b). In contrast, PP2 had no significant inhibition on the spontaneous axon outgrowth (Fig. 5c). These results support the view that tyrosine phosphorylation and activity of the SFKs are important for netrin-stimulated axon growth.
Figure 5.
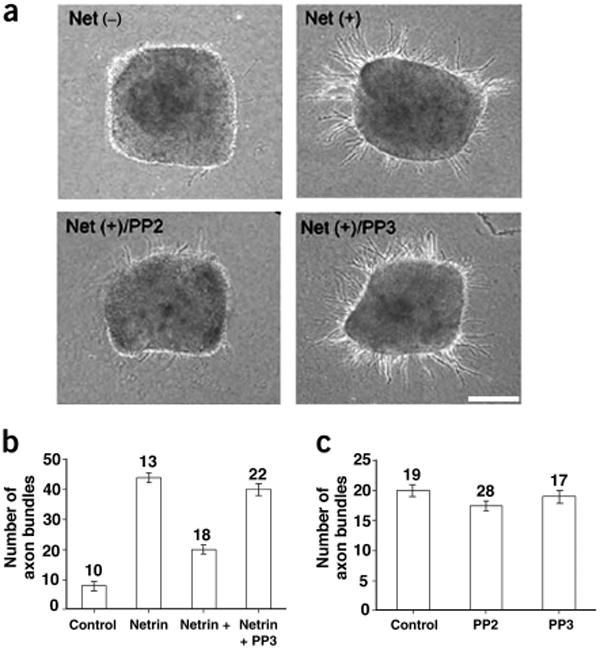
Src activity is required for netrin-induced axon growth. (a) PP2 inhibits netrin-stimulated axon growth. Explants from the dorsal half of E12 spinal cords were cultured in collagen gels for 20 h. Netrin-1-induced outgrowth of axon bundles was inhibited by PP2 but not PP3. Scale bar, 100 μm. (b) PP2 inhibits netrin-stimulated axon growth. The netrin (20 h) and PP2 (0.2 μM) treatments are indicated (netrin versus control, P < 0.0001; netrin versus netrin + PP2, P < 0.0001; netrin versus netrin + PP3, P < 0.57. (c) PP2 did not inhibit spontaneous axon growth. The explants were cultured for 40 h. PP2 compared with control, P < 0.33; PP3 compared with control, P < 0.88. Numbers denote explants counted.
Function of Src and DCC phosphorylation in netrin response
A gradient of netrin induces chemoattraction of X. laevis spinal neuron growth cones via the DCC receptor12,28. We tested the effect of various PP2 concentrations on the netrin-induced outgrowth and attraction of the X. laevis spinal neuron growth cones. As expected, netrin induced both axon growth and attraction (Fig. 6). We found that incubation with the Src inhibitor PP2 at a concentration higher than 0.2 μM inhibited axon growth (data not shown), further supporting the importance of SFK in netrin-stimulated axon growth. However, 0.1 μM PP2 only partially reduced axon growth (Fig. 6c), which enabled us to test the effect of PP2 on axon turning. We observed that 0.1 μM PP2 almost completely blocked netrin-induced axon turning, whereas 0.1 μM PP3 had no effect (Fig. 6a–c). These data are consistent with a role for SFKs in netrin-induced growth cone attraction.
Figure 6.
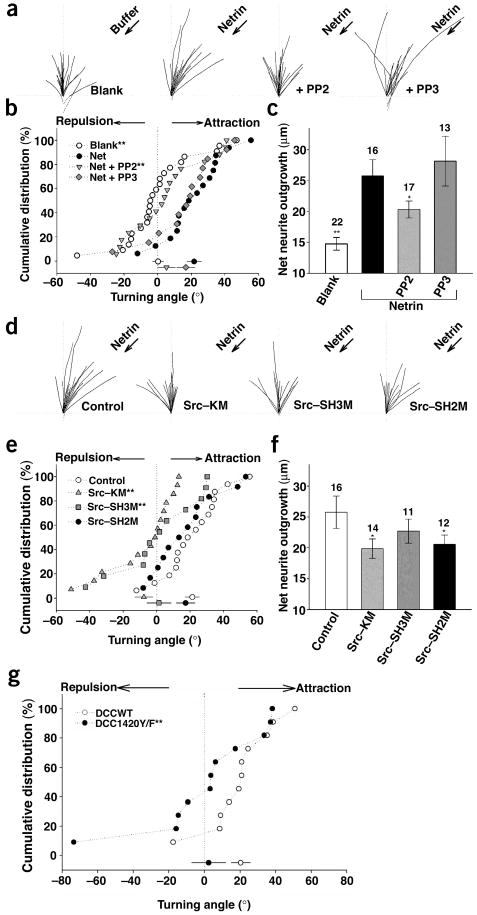
Src function or DCC tyrosine phosphorylation is required for DCC-mediated axon attraction. (a) Superimposed neurite trajectories of DCC-expressing neurons in the presence of 0.1 μM PP2 or PP3 during 1-h exposure to a netrin gradient (2.5 μg/ml in the pipette, arrows) for all the neurons examined. Arrows, presence of netrin gradient or buffer control. ‘PP2’ and ‘PP3,’ presence of 0.1 μM PP2 and PP3, respectively, in the presence of a netrin gradient. (b) Cumulative distribution of turning angles. Significant differences from the control groups are marked (*P < 0.05, **P < 0.001; Kolmogorov-Smirnov test). (c) Average neurite extension during the 1-h assay for experiments shown in a. Numbers denote the total number of growth cones examined. Error bars, s.e.m. (d) Traced neurite trajectories of Src-overexpressing mutants in DCC-expressing neurons exposed to a netrin gradient similar to that in a. The netrin gradient (arrow) was present in all panels. ‘Control’ indicates no Src cotransfection. Src-KM, Src-SH3M and Src-SH2M are the kinase-inactive, SH3-domain and SH2-domain mutants, respectively. (e) Distribution of turning angles for the data in d. (f) Average neurite extension during the 1-h assay for experiments shown in d. (g) Expression of DCC-Y1420F blocks netrin-induced attraction. X. laevis spinal neurons with ectopic expression of DCC or DCC-Y1420F mutant were analyzed by in vitro axon turning experiments in the presence of a netrin gradient. Graph show the cumulative distribution of turning angles for all neurons examined. Turning responses of growth cones observed in wild-type DCC (DCCWT; open circle, n = 11) and DCC mutant (DCC-Y1420F; closed circle, n = 11) overexpressing neurons in response to a netrin-1 gradient (2.5 μg/ml) are shown. Significant differences from the control groups are marked (**P < 0.001 compared with WT; Kolmogorov-Smirnov test).
We further tested different Src mutants in the axon turning assay. Expressing the kinase-inactive Src-KM blocked both netrin-induced axon attraction and growth (Fig. 6d–f), supporting the importance of Src in netrin signaling. The SH3 domain mutant (Src-SH3M) also abolished the netrin-induced attraction, but it had a smaller effect on netrin-induced axon growth (Fig. 6d–f). In contrast, expression of the SH2 domain mutant (Src-SH2M) showed a more dramatic inhibition of axon growth but had little effect on axon turning (Fig. 6d–f). The differential effects of Src-SH2M and Src-SH3M on netrin response indicate that our results probably are not due to a nonspecific toxic effect of the Src mutants. These results suggest that SH2 and SH3 domains of Src may participate preferentially in different signaling pathways downstream of DCC, one in axon outgrowth and the other in axon attraction.
To determine the functional significance of DCC tyrosine phosphorylation in netrin signaling, we tested the phosphorylation-defective DCC-Y1420F mutant. We found that netrin-induced attraction was significantly diminished in X. laevis spinal neurons expressing DCC-Y1420F but not wild-type DCC (Fig. 6g). Because the X. laevis spinal neurons without ectopic DCC expression showed an attractive response to netrin, our data suggest that DCC-Y1420F functions as a dominant-negative receptor and blocks signaling of the endogenous DCC. Our observations demonstrate that tyrosine phosphorylation of DCC is important for netrin signaling, and DCC also serves as a key downstream substrate of the FAK-Src complex.
Discussion
In this report, we have demonstrated that netrin stimulation results in the activation of FAK and Src tyrosine kinases, as well as tyrosine phosphorylation of DCC, in dorsal spinal neurons, cortical neurons and transfected HEK293 cells. Experiments with an axon outgrowth assay using rat spinal neurons and in vitro axon turning of X. laevis spinal neurons suggest that SFK is centrally involved in DCC-mediated netrin signaling. SFK activity is required for both axon growth and attraction in response to netrin stimulation. We propose that DCC functions as a tyrosine kinase–coupled receptor by directly interacting and activating FAK and Src. This conclusion is supported by independent and complementary studies29,30. Interestingly, another recent report shows that clr-1, a protein tyrosine phosphatase, is involved in UNC-6/netrin–mediated axon attraction in C. elegans31. Similarly to high eukaryotes, the UNC-40/DCC receptor is required for axon attraction induced by UNC-6/netrin in C. elegans. This study suggests that clr-1 probably dephosphorylates a key protein in the netrin signaling pathway, possibly UNC-40/DCC. These findings are consistent with our conclusions that DCC functions as a tyrosine kinase–coupled receptor and that tyrosine phosphorylation of DCC is critical for netrin signaling.
Signal transduction via tyrosine kinase–coupled receptors has been widely used by many transmembrane receptors, such as cytokines and T-cell receptors. Our study supports a model by which DCC may also use a similar mechanism (Supplementary Fig. 2). Three motifs in the intracellular domain, known as P1, P2 and P3, are conserved among DCC family members from different species7,12. The P1 domain has been implicated in the interaction with UNC-5 and is important for the functional complex of DCC with UNC-5 (ref. 12). The DCC/UNC-5 receptor complex is important for netrin to induce growth cone repulsion. The P2 domain, which is rich in prolines, may play a role in protein-protein interactions. The P3 domain has been shown to interact with the Slit receptor, Robo32. The association between Robo and DCC may be important for Slit to silence the netrin-induced growth cone attraction. We demonstrated that FAK and Src interact with DCC under physiological conditions, via different mechanisms and through different regions of DCC. FAK interacts via its C-terminal domain, with the C-terminal half of the P3 domain of DCC (Supplementary Fig. 2). The SH3 domain of Src interacts with a single PXXP motif, beginning at residue Pro1400, of DCC. Furthermore, the interaction between Src and DCC is stimulated by netrin and partially inhibited by PP2, indicating a role of tyrosine phosphorylation in the interaction between Src and DCC. In contrast, the interaction between FAK and DCC is not stimulated by netrin nor inhibited by PP2.
Our results suggest a model in which different regions of DCC are responsible for interacting with FAK and Src (Supplementary Fig. 2). We propose that netrin binding to DCC activates the associated FAK. FAK autophosphorylates on Tyr397 and recruits Src (or enhances the interaction between Src and DCC). Thus, FAK also shows a scaffolding function, likely by recruiting Src to the receptor complex. FAK and Src in the receptor complex positively stimulate each other. The active FAK and Src then phosphorylate target molecules, including the DCC receptor, which in turn induce downstream signaling events.
The importance of SFK in netrin signaling is demonstrated by our axon turning experiments using the Src inhibitor PP2 and dominant-negative Src mutants. Furthermore, the importance of DCC tyrosine phosphorylation is supported by our observation that the DCC-Y1420F mutant interferes with netrin-induced axon attraction. Therefore, in addition to functioning as an upstream receptor, DCC also serves as a critical downstream substrate of FAK and Src in netrin signaling. This mechanism of action is similar to that used by cytokine receptors, where phosphorylation of the receptor by Jak family kinases is important for cytokine signaling. However, DCC tyrosine phosphorylation is not required for the activation of Src and FAK. Tyrosine-phosphorylated DCC may create binding sites for SH2-containing downstream signaling molecules. Future studies will be needed to identify substrates phosphorylated by Src and FAK, as well as proteins that interact with phospho-DCC—these studies will provide further insights into the mechanism of DCC signaling in response to netrin stimulation.
Several proteins have been reported to participate in netrin signaling, including PI3K33, Rho family GTPases34–36 and ERK MAP kinase37,38. How DCC receptor activation leads to modulation of these downstream molecules is currently unknown. Both FAK and Src are known to regulate PI3K, Rho family small GTPases and ERK16,25. Therefore, our study provides a possible mechanism by which DCC could modulate these downstream signaling molecules.
Netrin can stimulate both axon growth and turning initiated by the same netrin receptor, DCC. However, the intracellular signaling pathways for axon growth and turning may diverge after the receptor-proximal events. For example, the nuclear factor of activated T cells (NFAT) is involved in netrin-induced axon growth but not the turning response39. In contrast, the Slit receptor Robo silences netrin-induced turning but not the outgrowth response32. We observed that expression of the kinase-inactive Src mutant, Src-KM, blocked both axon outgrowth and turning in response to netrin (Fig. 6d–f). These data suggest that Src activation is a receptor-proximal event and important for both axon outgrowth and the turning responses. Notably, mutations in the SH2 and SH3 domains preferentially inhibited netrin-induced axon outgrowth and turning, respectively. These observations suggest that Src-SH2M and Src-SH3M may selectively block downstream signaling pathways leading to axon outgrowth or turning. Future experiments are required to test whether Src-SH2M blocks the NFAT pathway and whether Src-SH3M mimics the effect of Robo silencing.
Methods
Plasmids and cell culture
The neuronal isoform of c-Src (GenBank accession number 625219) was constructed by PCR from a human brain cDNA library, and subcloned into pcDNA3 (Invitrogen). FAK and related plasmids were described previously40. Src SH3 (127/128WW/RR), SH2 (178R/A) and kinase dead (298K/M) mutants, as well as DCC-ΔP1 (Δ1147–1170), ΔP2 (Δ1335–1356), ΔP3 (Δ1412–1447), Y1420F, P1400A and other DCC expression constructs, were constructed by mutagenesis (Stratagene). pGEX-KG-DCC, pEBG3X-HV-DCC, MBP-DCC/pMAL-p2 and AP (alkaline phosphatase)-netrin/pcDNA3 were constructed by PCR and subcloned into the indicated vectors. HEK293 cells stably expressing chick myc-netrin were a generous gift from M. Tessier-Lavigne (Stanford University).
Antibodies
We used the following antibodies in our experiments: anti-DCC (PharMingen and Oncogene), anti-Fyn, pan-anti-Src, anti-Lyn, anti-Yes, anti-FAK (Santa Cruz Biotechnology), anti-Src (Oncogene, Santa Cruz Biotechnology and Upstate Biotechnology), anti-phospho-Src (Bioscience and Cell Signaling), anti-hemagglutinin (HA) (Covance), anti-phospho-Tyr (PY20 from Transduction Laboratories and 4G10 from Upstate Biotechnology) and an anti-phospho-FAK kit (Biosource).
X. laevis embryo microinjection and primary spinal neuron cultures
Capped mRNAs were synthesized using mMESSAGE mMACHINE (Ambion) as suggested by the manufacturer. The transcription products were purified using QIAquick PCR Purification Kit (QIAGEN). In vitro fertilization was carried out as described12. Src or DCC mutant mRNAs were injected into two blastomeres of X. laevis embryos at the four-cell stage. For each blastomere, we injected 4–8 μl of mRNA solution containing either 0.15 μg/μl or 0.25 μg/μl of green fluorescent protein (GFP) mRNA. GFP was used as an indicator for expression of the sample mRNA Cultures of spinal neurons were prepared as described previously41.
Immunoprecipitation and in vitro binding
HEK293 cells were transfected using the Lipofectamine (Invitrogen) method. For immunoprecipitation (IP), generally, cells were lysed 48 h after transfection in mild lysis buffer (MLB: 20 mM Tris-Cl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin and 5 μg/ml leupeptin) and subjected to IP with 1 μg of antibody. Two hours later, protein A/G–agarose beads (Santa Cruz Biotechnology) were added for a further 1.5 h and then washed four times prior to addition of 1× SDS sample buffer. For phosphotyrosine immunoblot experiments, PLC buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 10 mM NaPPi, 100 mM NaF, 1 mM vanadate and protease inhibitors) was used in cell lysis. To inhibit SFK activity, the inhibitor PP2 (Calbiochem) was added to serum-starved cells at a concentration of 0.2–5 μM in 1% DMSO for 1 h. Tyrosine phosphorylation was determined by immunoblotting using PY20 antibody.
Three fresh mouse brains were Dounce-homogenized in 12 ml of PBS containing protease inhibitors. Lysates were centrifuged three consecutive times at 15,000g to remove the insoluble fraction and then ultracentrifuged at 100,000g for 40 min. The soluble fraction was removed and the pellet fraction was washed once with PBS. The pellet was resuspended in PLC buffer and then centrifuged at 15,000g for 5 min to remove insoluble debris. The solubilized membrane fraction was then divided into two groups and subjected to IP with, in the case of the Src IP, 5 μg of mouse antibodies specific to Src B12 or GAL4 RK5C1. In the case of the FAK IP, solubilized membranes were divided into three groups and immunoprecipitated with 5 μg of the antibodies specific to FAK C20, HA probe or c-Myc. IPs were subjected to immunoblotting (IB) with anti-DCC (PharMingen), which recognizes the intracellular domain of DCC.
HEK293 cells were transfected independently with expression constructs for HA-FAK and Src. At 24 h after transfection, cells were lysed in MLB with protease inhibitors and cleared by centrifugation at 15,000g. The lysates were then incubated for 1 h with GST or GST-DCC intracellular domain (expressed and purified from Escherichia coli or HEK293 cells) precoupled to glutathione-agarose beads. Beads were then washed several times and protein was eluted with 1× SDS sample buffer. Bound HA-FAK or Src was determined by immunoblotting with HA-specific (Babco) or Src-specific antibody.
In vitro kinase assay
The kinase assay was done essentially as described previously42. HA-FAK, Src and Fyn were obtained by immunoprecipitation from expression of transiently transfected HEK293 cells, primary neuron cultures from mouse embryonic brain cortex and dorsal spinal cord of chick embryos. MBP-DCC protein purified from E. coli and Cdc2 purchased from Upstate Biotechnology (cat.# 17-111) were used as substrates. The phosphorylation levels of DCC by FAK, Src and Src and Fyn kinase activities were determined by phosphoimaging and scintillation counting. The nitrocellulose membrane was stained by Coomassie blue to determine MBP-DCC levels. The levels of FAK, Src and Fyn were determined by immunoblotting.
Neuronal cultures and analysis of axon outgrowth
For the spinal cords, small rectangular explants were made from the dorsal half of rat embryonic day 12 (E12) and mouse E13 spinal cords. Explants were transferred into a drop of rat tail collagen, and another drop of collagen was added on the explants. After polymerization, gels were covered with conditioned media of control HEK293 cells or netrin-1-secreting HEK293 cells in the absence or presence of pharmacological reagents. Overnight cultures were used to observe the effect of netrin-1 on axon outgrowth. The number and length of axon bundles from the spinal cord explants were measured with NIH Image software (National Institutes of Health).
Dorsal spinal cords were dissected from E7 chick embryos. The isolated spinal cords were triturated to generate dissociated neuronal cells, which were plated on laminin-coated plates in neuron culture medium as described2. For primary cortical neuronal culture, the cortex was dissected out from E12 mouse embryonic brain in HBSS buffer and was triturated mechanically in L15 medium supplemented with 0.5% B27, 1% N2 and 30 mM glucose. The dissociated neurons were cultured in six-well plates coated with poly-l-lysine and laminin. The cells were cultured overnight and then stimulated with netrin. Use of laboratory animals was approved by the University of Michigan Committee on the Use and Care of Animals (Protocol #8719).
Growth cone turning assay
A microscopic gradient of netrin-1 protein was produced as previously described12. Microscopic images of neurites were recorded with a CCD camera (Hitachi KP-M2U) attached to a phase contrast microscope (Olympus CK-40) and analyzed using NIH Image 1.62. The final turning angle, defined by the angle between the original direction of neurite extension and a straight line connecting the positions of the growth cone at the onset and the end of the 1-h period, was measured. Only those growth cones with net extension of at least 7 μm over the 1-h period were included for analysis.
Acknowledgments
We thank B. Kruger for critical reading of the manuscript, T. Zhu for technical assistance, and W. Xiong for communicating unpublished information. This work is supported by grants from the National Institutes of Health (K.-L.G.), a Rackham Graduate Fellowship (H.V.) and the Pharmocological Sciences Training Program (GMO7767 to J.A.).
Footnotes
Competing Interests Statement: The authors declare that they have no competing financial interests.
Note: Supplementary information is available on the Nature Neuroscience website.
References
- 1.Serafini T, et al. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 1994;78:409–424. doi: 10.1016/0092-8674(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994;78:425–435. doi: 10.1016/0092-8674(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 3.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 4.Culotti JG, Merz DC. DCC and netrins. Curr Opin Cell Biol. 1998;10:609–613. doi: 10.1016/s0955-0674(98)80036-7. [DOI] [PubMed] [Google Scholar]
- 5.Chan SS, et al. UNC-40, a C. elegans homolog of DCC (Deleted in Colorectal Cancer), is required in motile cells responding to UNC-6 netrin cues. Cell. 1996;87:187–195. doi: 10.1016/s0092-8674(00)81337-9. [DOI] [PubMed] [Google Scholar]
- 6.Keino-Masu K, et al. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell. 1996;87:175–185. doi: 10.1016/s0092-8674(00)81336-7. [DOI] [PubMed] [Google Scholar]
- 7.Kolodziej PA, et al. frazzled encodes a Drosophila member of the DCC immunoglobulin subfamily and is required for CNS and motor axon guidance. Cell. 1996;87:197–204. doi: 10.1016/s0092-8674(00)81338-0. [DOI] [PubMed] [Google Scholar]
- 8.Fearon ER, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247:49–56. doi: 10.1126/science.2294591. [DOI] [PubMed] [Google Scholar]
- 9.Fazeli A, et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- 10.Leung-Hagesteijn C, et al. UNC-5, a transmembrane protein with immunoglobulin and thrombospondin type 1 domains, guides cell and pioneer axon migrations in C. elegans. Cell. 1992;71:289–299. doi: 10.1016/0092-8674(92)90357-i. [DOI] [PubMed] [Google Scholar]
- 11.Hamelin M, Zhou Y, Su MW, Scott IM, Culotti JG. Expression of the UNC-5 guidance receptor in the touch neurons of C. elegans steers their axons dorsally. Nature. 1993;364:327–330. doi: 10.1038/364327a0. [DOI] [PubMed] [Google Scholar]
- 12.Hong K, et al. A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell. 1999;97:927–941. doi: 10.1016/s0092-8674(00)80804-1. [DOI] [PubMed] [Google Scholar]
- 13.Leonardo ED, et al. Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature. 1997;386:833–838. doi: 10.1038/386833a0. [DOI] [PubMed] [Google Scholar]
- 14.Tong J, et al. Netrin stimulates tyrosine phosphorylation of the UNC-5 family of netrin receptors and induces Shp2 binding to the RCM cytodomain. J Biol Chem. 2001;276:40917–40925. doi: 10.1074/jbc.M103872200. [DOI] [PubMed] [Google Scholar]
- 15.Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- 16.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta. 2001;1540:1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 17.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 18.Lowell CA, Soriano P. Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 1996;10:1845–1857. doi: 10.1101/gad.10.15.1845. [DOI] [PubMed] [Google Scholar]
- 19.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 20.Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a noncatalytic domain of the focal adhesion–associated protein tyrosine kinase pp125FAK. Mol Cell Biol. 1993;13:785–791. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanke JH, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 22.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sperber BR, et al. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bockholt SM, Burridge K. An examination of focal adhesion formation and tyrosine phosphorylation in fibroblasts isolated from src-, fyn-, and yes- mice. Cell Adhes Commun. 1995;3:91–100. doi: 10.3109/15419069509081279. [DOI] [PubMed] [Google Scholar]
- 25.Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. 1999;4:D102–D113. doi: 10.2741/cary. [DOI] [PubMed] [Google Scholar]
- 26.Richardson A, Malik RK, Hildebrand JD, Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol Cell Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tessier-Lavigne M, Placzek M, Lumsden AG, Dodd J, Jessell TM. Chemotropic guidance of developing axons in the mammalian central nervous system. Nature. 1988;336:775–778. doi: 10.1038/336775a0. [DOI] [PubMed] [Google Scholar]
- 28.Ming GL, et al. cAMP-dependent growth cone guidance by netrin-1. Neuron. 1997;19:1225–1235. doi: 10.1016/s0896-6273(00)80414-6. [DOI] [PubMed] [Google Scholar]
- 29.Liu GBH, et al. Netrin requires the focal adhesion kinase and the Src family kinases to induce axon outgrowth and to attract axons. Nat Neurosci. 2004;7:1222–1232. doi: 10.1038/nn1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ren XGM, et al. Focal adhesion kinase in netrin-1 signaling. Nat Neurosci. 2004;7:1204–1212. doi: 10.1038/nn1330. [DOI] [PubMed] [Google Scholar]
- 31.Chang C, Yu TW, Bargmann CI, Tessier-Lavigne M. Inhibition of netrin-mediated axon attraction by a receptor protein tyrosine phosphatase. Science. 2004;305:103–106. doi: 10.1126/science.1096983. [DOI] [PubMed] [Google Scholar]
- 32.Stein E, Tessier-Lavigne M. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291:1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- 33.Ming G, et al. Phospholipase C-γ and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron. 1999;23:139–148. doi: 10.1016/s0896-6273(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 34.Li X, et al. The adaptor protein Nck-1 couples the netrin-1 receptor DCC (Deleted in Colorectal Cancer) to the activation of the small GTPase Rac1 through an atypical mechanism. J Biol Chem. 2002;277:37788–37797. doi: 10.1074/jbc.M205428200. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Saint-Cyr-Proulx E, Aktories K, Lamarche-Vane N. Rac1 and Cdc42 but not RhoA or Rho kinase activities are required for neurite outgrowth induced by the Netrin-1 receptor DCC (deleted in colorectal cancer) in N1E–115 neuroblastoma cells. J Biol Chem. 2002;277:15207–15214. doi: 10.1074/jbc.M109913200. [DOI] [PubMed] [Google Scholar]
- 36.Shekarabi M, Kennedy TE. The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol Cell Neurosci. 2002;19:1–17. doi: 10.1006/mcne.2001.1075. [DOI] [PubMed] [Google Scholar]
- 37.Forcet C, et al. Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature. 2002;417:443–447. doi: 10.1038/nature748. [DOI] [PubMed] [Google Scholar]
- 38.Ming GL, et al. Adaptation in the chemotactic guidance of nerve growth cones. Nature. 2002;417:411–418. doi: 10.1038/nature745. [DOI] [PubMed] [Google Scholar]
- 39.Graef IA, et al. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 40.Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol. 2003;23:8030–8041. doi: 10.1128/MCB.23.22.8030-8041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabti N, Poo MM. Culturing spinal cord neurons and muscle cells from Xenopus embryos. In: Banker G, Goslin K, editors. Culturing Nerve Cells. MIT Press; Cambridge, Massachusetts, USA: 1991. pp. 137–154. [Google Scholar]
- 42.Schaller MD, et al. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]