

Abstract
The zinc finger transcription factor Krüppel-like factor 4 (KLF4) is frequently down-regulated in colorectal cancer. Previous studies showed that the expression of KLF4 was activated by the colorectal cancer tumor suppressor adeno-matous polyposis coli (APC) and that KLF4 repressed the Wnt/β-catenin pathway. Here, we examined whether KLF4 plays a role in modulating intestinal tumorigenesis by comparing the tumor burdens in mice heterozygous for the ApcMin allele (ApcMin/+) and those heterozygous for both the ApcMin and Klf4 alleles (Klf4+/−/ApcMin/+). Between 10 and 20 weeks of age, Klf4+/−/ApcMin/+ mice developed, on average, 59% more intestinal adenomas than ApcMin/+ mice (P < 0.0001). Immunohistochemical staining showed that Klf4 protein levels were lower in the normal-appearing intestinal tissues of Klf4+/−/ApcMin/+ mice compared with wild-type, Klf4+/−, or ApcMin/+ mice. In contrast, the levels of β-catenin and cyclin D1 were higher in the normal-appearing intestinal tissues of Klf4+/−/ApcMin/+ mice compared with the other three genotypes. Klf4 levels were further decreased in adenomas from both ApcMin/+ and Klf4+/−/ApcMin/+ mice compared with their corresponding normal-appearing tissues. Reverse transcription-PCR showed an inverse correlation between adenoma size and Klf4 mRNA levels in both Klf4+/−/ApcMin/+ and ApcMin/+ mice. There was also a progressive loss of heterozygosity of the wild-type Apc allele in adenomas with increasing size from Klf4+/−/ApcMin/+ and ApcMin/+ mice. Results from this study show that KLF4 plays an important role in promoting the development of intestinal adenomas in the presence of ApcMin mutation.
Introduction
Colorectal cancer is a major cause of cancer mortality in the United States. More than 80% of colorectal cancers contain mutations in the adenomatous polyposis coli (APC) tumor suppressor gene (1). APC is present in the normal intestinal mucosa with an increasing gradient of expression in mature epithelial cells located in the upper crypt region (2). Overexpression of APC leads to cell cycle arrest at the G1-S and G2-M boundaries (3–6). In addition, APC antagonizes the pro-proliferative Wnt pathway by negatively regulating the steady-state level of intracellular β-catenin (7–9). When APC is inactivated by mutation, Wnt signaling is unimpeded, resulting in the nuclear accumulation of β-catenin and subsequent activation of downstream target genes such as cyclin D1 and c-MYC that promote cell proliferation (10, 11).
The nuclear transcription factor Krüppel-like factor 4 (KLF4; also known as gut-enriched Krüppel-like factor or GKLF), is a member of the C2H2-zinc finger–containing proteins exhibiting homology to the Drosophila melanogaster segmentation gene product, Krüppel (12–16). KLF4 is highly expressed in the terminally differentiated, postmitotic intestinal epithelial cells and is an inhibitor of cell proliferation (17, 18). We previously showed that KLF4 was transcrip-tionally activated by p53 following DNA damage (19) and caused cell cycle arrest at both the G1-S and G2-M boundaries (20, 21). These results indicate that KLF4 is an important factor in mediating the checkpoint functions of p53 following DNA damage. In the intestine, the KLF4 promoter is regulated by APC in a CDX2-dependent manner; CDX2 is an intestine-specific transcription factor that controls intestinal development (22). Conversely, KLF4 has been shown to regulate colonic cell growth by inhibiting β-catenin activity (23, 24). Accordingly, studies have shown a potentially causal relationship between KLF4 and several kinds of human cancers. For example, the expression of KLF4 is often reduced in tumors of the gastrointestinal tract (25–29). In addition, loss of heterozygosity (LOH) and promoter hypermethylation are thought to be possible reasons for the reduced expression of KLF4 in a subset of colorectal cancers (25). However, whether KLF4 plays an in vivo role in the development of intestinal tumors has not been established.
The ApcMin/+ mice are an excellent model for studying intestinal tumorigenesis (30). The mutant mice, multiple intestinal neoplasia (Min; ref. 30), carry a truncating mutation at codon 850 of the murine Apc gene (31). In a C57BL/6J background, ApcMin/+ mice develop, on average, 30 adenomatous polyps in the intestines, with a predominant distribution in the small intestine (30). All intestinal adenomas are established by 100 days of age or sooner and new tumors do not arise continuously over the remaining life span of the animals (32). In the current study, we investigated the in vivo role of KLF4 in intestinal tumorigenesis in the setting of the ApcMin mutation.
Materials and Methods
Mice
Founder C57BL/6J male mice heterozygous for the ApcMin allele (ApcMin/+) were purchased from The Jackson Laboratory. Founder C57BL/6J male mice heterozygous for the Klf4 allele (Klf4+/−) were previously established (33). A colony of each genotype was established by mating the founder male with wild-type C57BL/6J female mice. Allele-specific PCR assays were used to identify the ApcMin (34) and the Klf4 (33) mutations. ApcMin/+ males were subsequently mated with Klf4+/− females to obtain mice wild-type for both alleles, heterozygous for the Klf4 allele (Klf4+/−), heterozygous for the ApcMin allele (ApcMin/+), or heterozygous for both the Klf4 and ApcMin alleles (Klf4+/−/ApcMin/+).
Tissue harvesting and tumor assessment
At 10, 16, or 20 weeks of age, wild-type, Klf4+/−, ApcMin/+, and Klf4+/−/ApcMin/+ mice were sacrificed by CO2 asphyxiation. The entire small intestine and colon were dissected longitudinally and washed in PBS. The intestines were examined under a dissecting microscope for the presence of adenomas. The number and size of adenomas in both the small and large intestines were recorded. Adenomas identified in the small and large intestines were grouped by size (<1, 1–2, 2–3, and >3 mm and ≤2 and >2 mm, respectively).
Immunohistochemistry
Intestinal tissues for immunohistochemistry were fixed in 10% formalin in PBS and subsequently embedded in paraffin. Five-micrometer-thick paraffin sections were cut and applied to Superfrost Plus slides (VWR). Sections were deparaffinized in xylene, rehydrated in ethanol, and then treated with 10 mmol/L of sodium citrate buffer (pH 6.0), at 120°C for 10 min in a pressure cooker. The histologic sections were incubated with a blocking buffer (10% nonfat dry milk, 0.01% Tween 20, and 10% normal horse serum in PBS) for 1 h at room temperature. An avidin/biotin blocking kit (Vector Laboratories) was used in conjunction with the blocking buffer according to the manufacturer’s directions to reduce background and nonspecific secondary antibody binding. Sections were then stained for Klf4 (goat anti-GKLF, T-16; Santa Cruz Biotechnology), β-catenin (monoclonal anti–β-catenin; Zymed/Invitrogen), or cyclin D1 (rabbit anti–cyclin D1; Santa Cruz Biotechnology). The Klf4, β-catenin, and cyclin D1 antibodies were used at dilutions of 1:50, 1:1,000, and 1:100, respectively, in the blocking buffer for 1 h at room temperature. Detection of primary antibodies and color development were done using the Dako (k6090) kit (Dako North America, Inc.). Sections were then counterstained with hematoxylin, dehydrated, and coverslipped. Images were acquired using an Axioskop 2 plus microscope (Zeiss) equipped with an AxioCam MRc5 CCD camera (Zeiss).
Reverse transcription-PCR
Adenomas and adjacent normal-appearing intestinal tissues from ApcMin/+ and Klf4+/−/ApcMin/+ mice were retrieved under a dissecting microscope. Adenomas from six 16- to 20-week-old mice of each genotype were pooled by size (<1, 1–3, and >3 mm). Total RNA from the adenomas was then extracted using RNeasy Micro Kit (QIAGEN). Two micrograms of RNA per sample was used for cDNA synthesis using SuperScript III First-Strand Synthesis System for reverse transcription-PCR (RT-PCR) kit (Invitrogen). Klf4 cDNA amplification was carried out using primers F-5′-TGATGGGCAAGTTTGTGCTGAAGG-3′ and R-5′-ACAGTGGT-AAGGTTTCTCGCCTGT-3′ that amplified the wild-type allele. PCR reactions were carried out for 25 cycles (95°C for 30 s, 56°C for 30 s, 68°C for 45 s) in a buffer of 50 μmol/L of each deoxynucleotide triphosphate, 1.25 units of Taq polymerase, 1× Taq buffer containing 1.5 mmol/L of MgSO4. The wild-type allele produced a band of 632 bp. As a control, RT-PCR for β-actin was carried out using the primers described previously (35). Band intensities were quantified using the Scion Image software.5
Measurement of LOH of the Apc+ locus
To reduce contamination of adenomatous tissues with the surrounding normal tissues, adenomas versus normal-appearing tissues were dissected using laser capture microscopy (LCM). Intestines were rinsed thrice with PBS without being cut open, and quickly embedded in Tissue-Tek optimal cutting temperature compound (VWR), and then snap-frozen in liquid nitrogen. Eight-micrometer-thick sections were cut using a Leica CM1510-3 cryostat (Leica Microsystems) and applied to Superfrost Plus slides. Sections were counterstained for visualization using HistoGene LCM Frozen Section Staining Kit (Arcturus) following the manufacturer’s protocol. Cells from normal tissue and adenomas (<1, 1–3, and >3 mm in size) were dissected using a PixCell IIe microscope (Arcturus) and captured on CapSure Macro LCM Caps LCM 0211 (Arcturus). DNA was extracted from the captured cells using QIAmp DNA Micro Kit (QIAGEN) following the manufacturer’s protocol. PCR for the Apc locus and HindIII digestion was carried out as described previously (36). Ten microliters of each HindIII digestion reaction was electrophoresed through a 2.5% agarose gel. Bands were quantified using Scion Image software. Determination of LOH was carried out as described previously (36).
Results
The in vivo role of Klf4 in the development of intestinal adenomas
Mice null for Klf4 (Klf4−/−) are not suitable for the study of intestinal tumorigenesis because they die shortly after birth (33, 37). We therefore used an alternative approach to investigate the role of KLF4 in intestinal adenoma formation in the setting of the well-established model of intestinal tumorigenesis, the ApcMin mouse (30). We first compared the number and size of the adenomas that developed in ApcMin/+ and Klf4+/−/ApcMin/+ mice between 10 and 20 weeks of age. Neither wild-type nor Klf4+/− mice developed adenomas in this period. ApcMin/+ mice developed, on average, 18.4 ± 6.8 adenomas per mouse (n = 40) in the small intestine and less than one adenoma per mouse in the large intestine (Fig. 1A and B, respectively). The average number of adenomas in the small intestine of Klf4+/−/ApcMin/+ mice was 28.9 ± 7.8 adenomas per mouse (N = 40; Fig. 1A), 57% more than ApcMin/+ mice (P = 1.3 × 10−7 by paired two-tailed t test). In the large intestine, the average number of adenomas in Klf4+/−/ApcMin/+ mice trended higher than ApcMin/+ mice, although it did not reach statistical significance (P = 0.053; Fig. 1B). Combining the tumor burden in the small and large intestines, the average number of adenomas per mouse was 18.7 ± 6.9 and 29.7 ± 7.7 for ApcMin/+ and Klf4+/−/ApcMin/+ mice, respectively, with a highly statistically difference of 59% (P = 4 × 10−8 by paired two-tailed t test; Fig. 1C).
Figure 1.
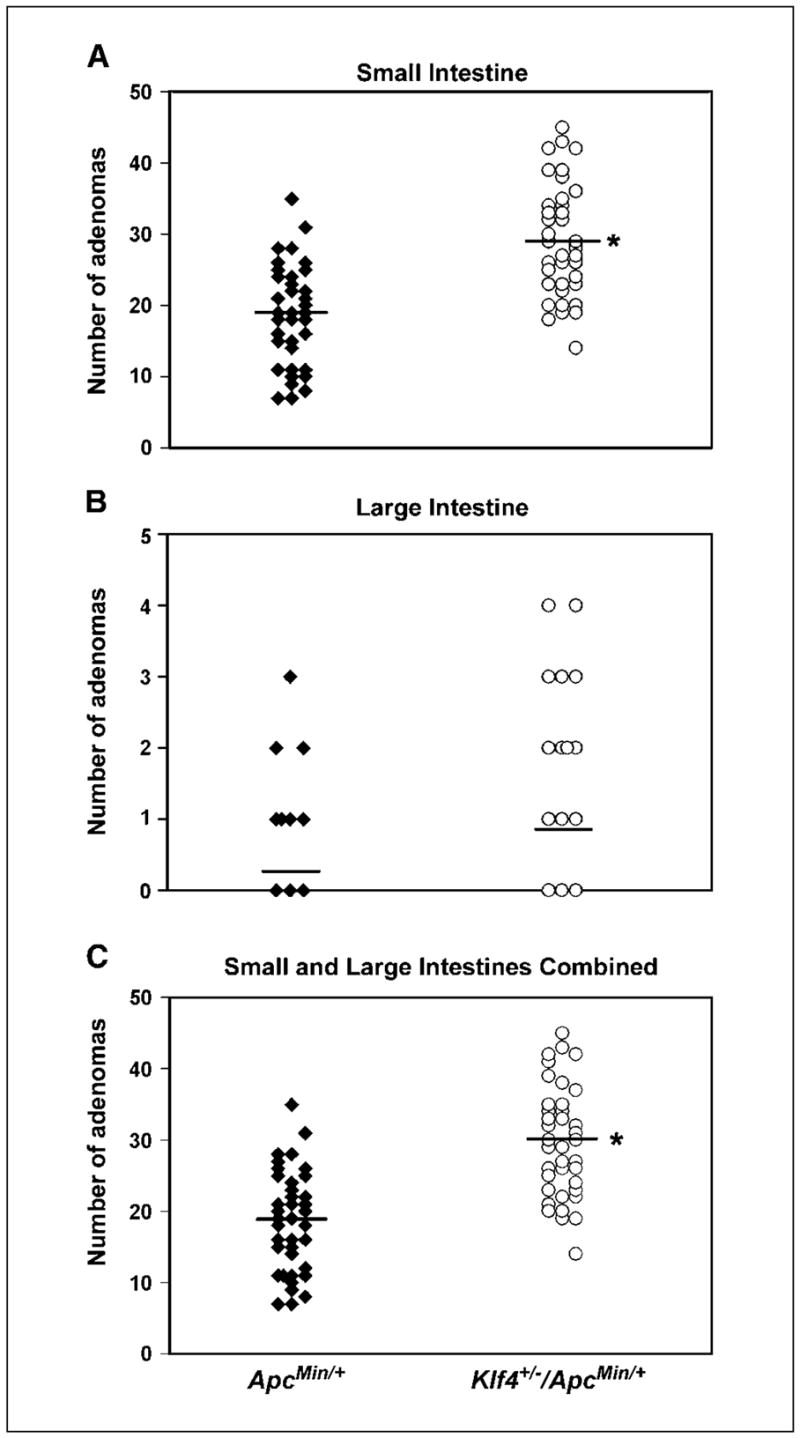
Klf4+/−/ApcMin/+ mice develop more intestinal adenomas than ApcMin/+ mice. Klf4+/− mice were bred with ApcMin/+ mice. Mice were sacrificed at 10, 16, and 20 weeks and the number of adenomas of the three age groups combined was counted and compared between the two genotypes. A, comparison of the number of adenomas per mouse in the small intestine between ApcMin/+ and Klf4+/−/ApcMin/+ mice. B, comparison of the number of adenomas per mouse in the large intestine between ApcMin/+ and Klf4+/−/ApcMin/+ mice. C, comparison of the total number of adenomas per mouse in both the small and large intestines between ApcMin/+ and Klf4+/−/ApcMin/+ mice. Points, number of adenomas per mouse; *, P < 0.0001 by paired two-tailed t test between the two genotypes.
Results in Supplementary Fig. S1 compare the number of adenomas per mouse between ApcMin/+ and Klf4+/−/ApcMin/+ mice in each of the three age groups—10, 16, and 20 weeks. Consistent with a previous report (32), ApcMin/+ mice developed adenomas early in life but tumor numbers did not increase over the life span of the animals (Supplementary Fig. S1A, C, and E). Klf4+/−/ApcMin/+ mice developed significantly more small intestinal adenomas in as early as 10 weeks (Supplementary Fig. S1A) and this increase was maintained up to 20 weeks (Supplementary Fig. S1C and E). Adenomas in the large intestine developed later in life in both groups. There was a trend for the Klf4+/−/ApcMin/+ mice to have more colonic adenomas than the ApcMin/+ mice, although it did not reach statistical significance (Supplementary Fig. S1D and F).
We also examined whether there were any differences in the size of adenomas between the small and large intestines of ApcMin/+ and Klf4+/−/ApcMin/+ mice. As shown in Supplementary Fig. S2A, the adenomas which developed in the small intestines of both ApcMin/+ and Klf4+/−/ApcMin/+ mice at 10 weeks were mostly small (<1 and 1–2 mm). By week 16, there was a shift in the size of adenomas to larger ones in both ApcMin/+ and Klf4+/−/ApcMin/+ mice, and this trend continued at 20 weeks (Supplementary Fig. S2A). Although the Klf4+/−/ApcMin/+ mice had higher numbers of adenomas in each of the size categories than the ApcMin/+ mice at any given age, there was no significant difference in the distribution of sizes between the two groups of mice. There was also no significant difference in the size distribution of adenomas formed in the large intestines of ApcMin/+ and Klf4+/−/ApcMin/+ mice (Supplementary Fig. S2B).
Immunohistochemical characterization of the normal-appearing small intestines and adenomas from ApcMin/+ and Klf4+/−/ApcMin/+ mice
To investigate the mechanisms by which the haploinsufficiency of Klf4 enhances intestinal adenoma formation in the ApcMin background, we examined the levels of Klf4, β-catenin, and cyclin D1 by immunohistochemistry in the normal-appearing intestinal tissues obtained from age-matched wild-type, Klf4+/−, ApcMin/+, and Klf4+/−/ApcMin/+ mice and in adenomas derived from mice with the latter two genotypes. As seen in Fig. 2, Klf4 was present primarily in epithelial cells lining the villi of the small intestine of wild-type mice (Fig. 2A). Consistent with previous findings (17, 24), Klf4 is absent from the proliferating crypt cell compartment in the wild-type intestine. As expected from the reduced gene dosage, Klf4+/− mice had less expression of Klf4 than wild-type mice (Fig. 2B). Also, compared with the ApcMin/+ mice (Fig. 2C), immunostaining for Klf4 was significantly decreased in the normal-appearing small intestine of Klf4+/−/ApcMin/+ mice (Fig. 2D).
Figure 2.
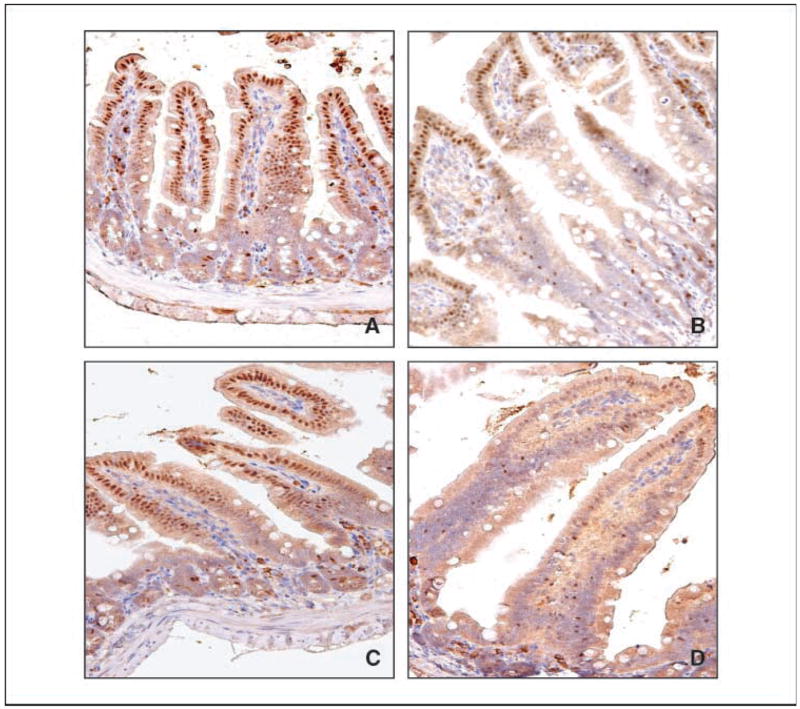
Immunohistochemical localization of Klf4 in normal-appearing small intestinal tissues. Normal-appearing small intestinal tissues obtained from age-matched wild-type (A), Klf4+/− (B), ApcMin/+ (C), and Klf4+/−/ApcMin/+ (D) mice were stained with a Klf4 antibody.
KLF4 has been shown to repress Wnt/β-catenin activity in colonic epithelial cells (24). We therefore determined the levels of β-catenin in the normal-appearing intestines of mice with the four different genotypes. Figure 3 shows that β-catenin is present in the basolateral membranes of epithelial cells lining the villi and crypts of the intestines from wild-type and Klf4+/− mice (Fig. 3A and B, respectively). The staining of β-catenin is increased in the ApcMin/+ mice and further increased in the Klf4+/−/ApcMin/+ mice (Fig. 3C and D, respectively). Notably, the staining of β-catenin seems higher in the crypts than in the villi in both ApcMin/+ and Klf4+/−/ApcMin/+ mice with a shift in the staining pattern from membranous in the villi to cytoplasmic in the crypts. The increase in β-catenin in the ApcMin/+ and Klf4+/−/ApcMin/+ mice was reflected by a corresponding increase in the staining of cyclin D1, a transcriptional target of β-catenin (ref. 10; compare Fig. 4C and D with Fig. 4A and B). Importantly, although the staining of cyclin D1 was primarily present in the crypt cells of wild-type (Fig. 4A) and Klf4+/− mice (Fig. 4B), the staining patterns in the ApcMin/+ and Klf4+/−/ApcMin/+ mice seems to have extended into the villus cells (Fig. 4C and D, respectively).
Figure 3.
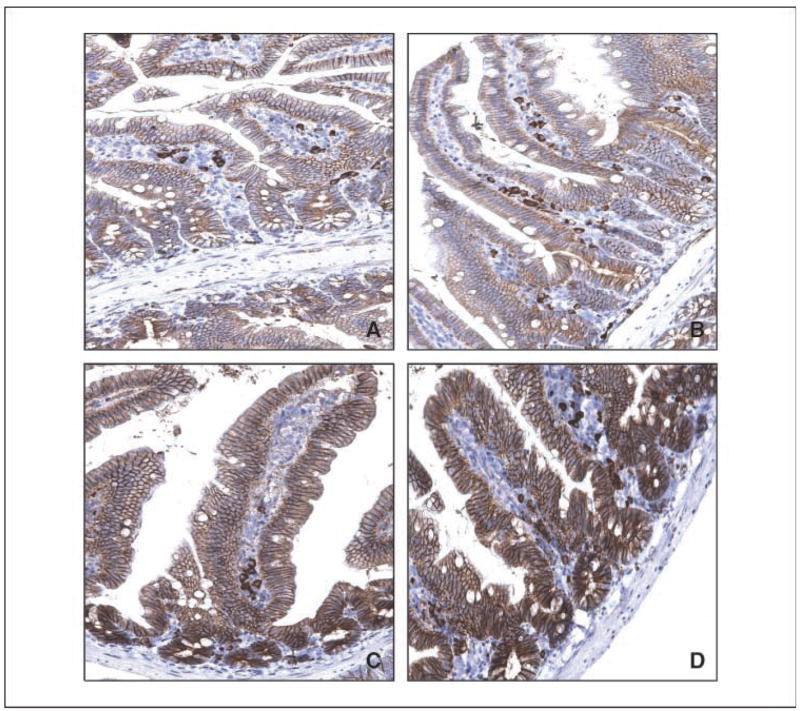
Immunostaining of β-catenin in normal-appearing small intestinal tissues. Normal-appearing small intestinal tissues obtained from age-matched wild-type (A), Klf4+/− (B), ApcMin/+ (C), and Klf4+/−/ApcMin/+ (D) mice were stained with a β-catenin antibody.
Figure 4.
Immunostaining of cyclin D1 in normal-appearing small intestinal tissues. Normal-appearing small intestinal tissues obtained from age-matched wild-type (A), Klf4+/− (B), ApcMin/+ (C), and Klf4+/−/ApcMin/+ (D) mice were stained with a cyclin D1 antibody.
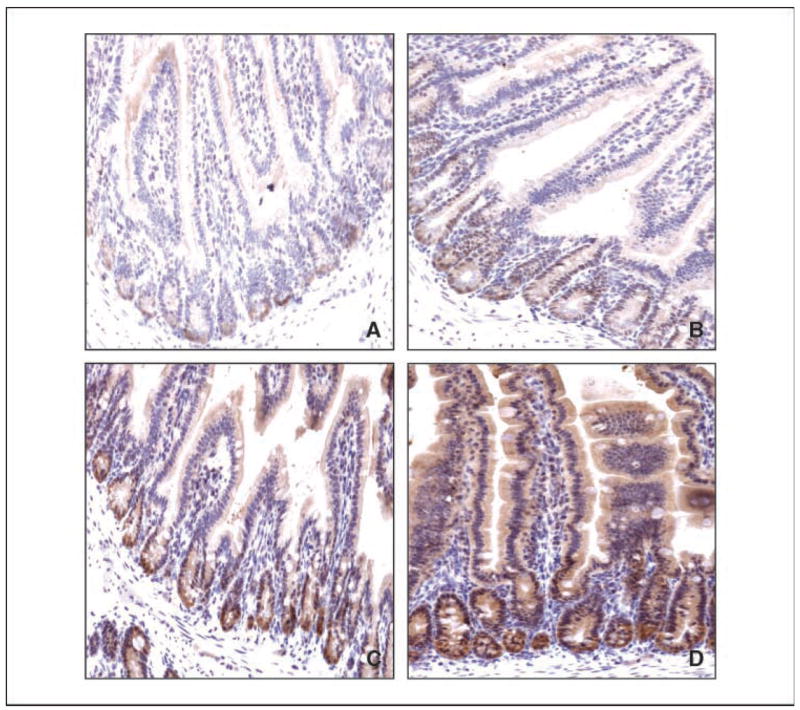
Figure 5 shows immunostaining for Klf4, β-catenin, and cyclin D1 in the adenomas derived from ApcMin/+ and Klf4+/−/ApcMin/+ mice. Klf4 levels were decreased in intestinal adenomas from both ApcMin/+ and Klf4+/−/ApcMin/+ mice (Fig. 5A) compared with their respective normal-appearing tissues (Fig. 2C and D, respectively). Reflecting this reduction, the levels of β-catenin (Fig. 5B) and cyclin D1 (Fig. 5C) were elevated in adenomas derived from both ApcMin/+ and Klf4+/−/ApcMin/+ mice. Moreover, the pattern of β-catenin staining in tumor cells from ApcMin/+ and Klf4+/−/ApcMin/+ mice now becomes cytoplasmic/nuclear (Fig. 5B).
Figure 5.

Immunohistochemical characterization of small intestinal adenomas from ApcMin/+ and Klf4+/−/ApcMin/+ mice. Adenomas from small intestines of ApcMin/+ mice and Klf4+/−/ApcMin/+ mice were stained with antibodies against Klf4 (A), β-catenin (B), and cyclin D1 (C).
The levels of Klf4 mRNA in Klf4+/−/ApcMin/+ and ApcMin/+ mice decrease as tumor sizes increase
To verify the reduction in Klf4 levels observed by immunohistochemical staining of Klf4+/−/ApcMin/+ mouse intestines, we isolated tumors of different sizes (<1, 1–3, and >3 mm) from Klf4+/−/ApcMin/+ and ApcMin/+ mouse intestines by microdissection. mRNA was purified from these tumors and semiquantitative RT-PCR was conducted using mouse Klf4-specific primers. As shown in Supplementary Fig. S3, the level of Klf4 mRNA in the normal-appearing intestinal tissues of Klf4+/−/ApcMin/+ mice was significantly lower than that of ApcMin/+ mice (Supplementary Fig. S3B). The Klf4 mRNA levels further decreased in adenomas from both Klf4+/−/ApcMin/+ and ApcMin/+ mice as the size of the adenoma increases compared with their respective normal tissues. Lastly, there was a greater reduction in Klf4 mRNA levels in adenomas 1 to 3 mm and >3 mm in size isolated from Klf4+/−/ApcMin/+ mice as compared with those from ApcMin/+ mice. Supplementary Fig. S3A shows a representative RT-PCR result for Klf4 and β-actin. Supplementary Fig. S3B is a densito-metric analysis of the amplified Klf4 bands after normalization to β-actin.
Evidence for progressive LOH in adenomas from ApcMin/+ and Klf4+/−/ApcMin/+ mice
We further investigated whether there were any differences in the extent of loss of the wild-type Apc allele (LOH) between intestinal adenomas derived from ApcMin/+ and Klf4+/3/ApcMin/+ mice. As seen in Fig. 6, there was a progressive increase in LOH of the Apc allele that correlated with the increase in tumor size in both ApcMin/+ and Klf4+/3/ApcMin/+ mice. The rates of LOH were similar in smaller tumors between the two groups. However, adenomas >3 mm in size showed a higher level of LOH in Klf4+/3/ApcMin/+ mice as compared with ApcMin/+ mice. Figure 6A shows a representative PCR result for LOH of Apc. Figure 6B is a densitometric analysis of the relative band intensity ratio of the amplified wild-type and mutated Apc bands are shown in Fig. 6A.
Figure 6.
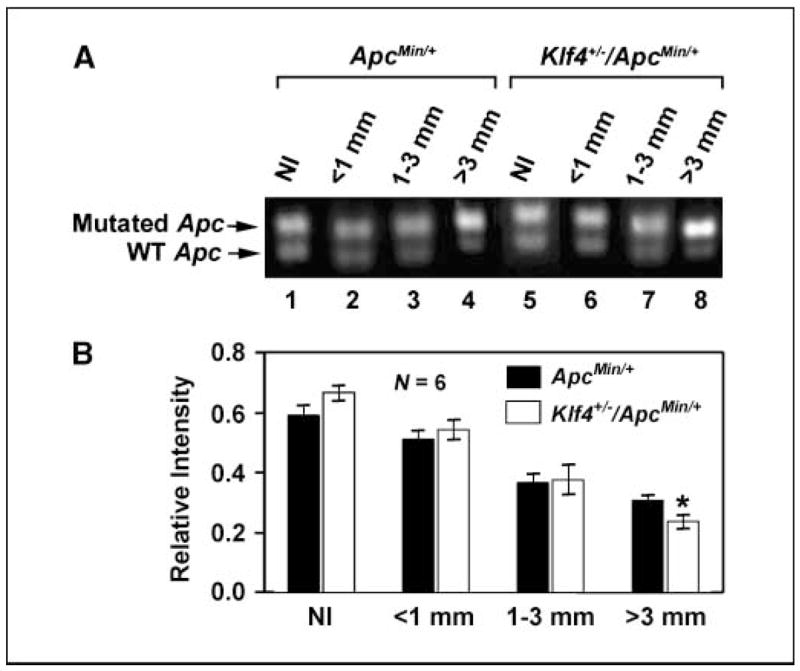
LOH of Apc increases as adenoma size increases. A, adenomas of various sizes and neighboring normal tissue were microdissected out from intestines of ApcMin/+ and Klf4+/−/ApcMin/+ mice. DNA was extracted and the extent of LOH was analyzed by semiquantitative PCR. Adenomas sizes were <1 mm (lanes 2 and 6), 1 to 3 mm (lanes 3 and 7), and >3 mm (lanes 4 and 8). Normal-appearing intestinal tissues (Nl) were also included in the analysis (lanes 1 and 5). B, densitometric analysis of the amplified Apc bands in (A).
Discussion
Previous studies indicate that KLF4 inhibits cell proliferation by activating crucial checkpoints in the cell cycle (18, 20, 21, 38–40). These findings led to the suggestion that KLF4 may function as a tumor suppressor. Indeed, expression of KLF4 was found to be significantly reduced in intestinal adenomas from ApcMin/+ mice and patients with familial adenomatous polyposis when compared with their respective matched normal-appearing tissues (41). Similarly, the levels of KLF4 mRNA were reduced in a panel of human colorectal cancers when compared with their matched control tissues (25). Moreover, there is evidence for LOH of the KLF4 locus as well as promoter hypermethylation in a subset of colorectal cancer specimens (25). Conversely, reexpression of KLF4 in a colorectal cancer cell line resulted in reduced tumorigenecity in a xenotransplantation model (42). Results from the current study provide the first in vivo evidence that haploinsufficiency of Klf4 promotes intestinal tumorigenesis in the setting of the ApcMin mutation, thus further solidifying the concept that KLF4 exerts a tumor-suppressive function in the gastrointestinal system.
The ApcMin mouse, which carries a germ line nonsense mutation in the murine Apc gene (31), has served as a remarkably useful model for the study of intestinal tumorigenesis. Similar to the ApcMin model, familial adenomatous polyposis is a human disorder in which germ line mutations of the APC gene lead to polyposis early in life (1, 43). A notable difference between the human syndrome and the mouse model is the much higher prevalence of adenomas in the small intestine than in the colon in ApcMin/+ mice (32), whereas it is the reverse in humans. Another unique nature of the ApcMin mouse model is that all adenomas were established by 100 days of age or sooner; no new tumors were formed over the remaining life span of the animals (32), a finding confirmed by the current study. Our study also shows that Klf4+/−/ApcMin/+ mice developed significantly more intestinal adenomas than ApcMin/+ mice in as early as 10 weeks of age (Supplementary Fig. S1). This increase was maintained later in life, at 16 and 20 weeks. However, the size distribution of the adenomas in the two groups of mice were comparable at all time points (Supplementary Fig. S2), suggesting that the rates of tumor growth in both ApcMin/+ and Klf4+/−/ApcMin/+ mice were similar. In the colon, although there was no statistically significant difference in the number of tumors between the two groups, the Klf4+/−/ApcMin/+ mice tended to have more tumors than the ApcMin/+ mice at 16 and 20 weeks (Supplementary Fig. S2). Analysis of intestinal tumor multiplicity in the ApcMin/+ mice suggests that tumor number does not significantly increase over time (32, 44). The fact that Klf4+/−/ApcMin/+ mice maintained a higher number of intestinal adenomas, but a similar size distribution when compared with ApcMin/+ mice throughout the course of the study, suggests that haploinsufficiency of Klf4 contributes to the initiation but not the subsequent progression of adenomas.
The results of this study show that the levels of Klf4 protein and mRNA were reduced in the normal-appearing small intestinal tissues from Klf4+/−/ApcMin/+ mice as compared with ApcMin/+ mice (Fig. 2; Supplementary Fig. S3). In addition, the level of Klf4 mRNA decreased progressively as the size of the adenomas increased in both ApcMin/+ and Klf4+/−/ApcMin/+ mice (Supplementary Fig. S3). The reduced level of Klf4 in the normal-appearing small intestinal tissues of the Klf4+/−/ApcMin/+ mice, as compared with the ApcMin/+ mice, may be a contributing factor to the early formation of intestinal adenomas in mice that are heterozygous for the Klf4 and ApcMin alleles. Reduced expression or loss of KLF4 has been reported in gastrointestinal tumors and other types of solid tumors (25–29, 45, 46). Our findings are not only consistent with these reports, but provide direct evidence to show an in vivo effect of KLF4 reduction in promoting intestinal tumorigenesis.
The human APC gene was mutated in a majority of colorectal cancer (1). Loss of the normal APC allele occurred in up to 70% of colorectal adenomas (47–49) and 80% of colorectal cancer cell lines (2). These studies also indicate that one mechanism of adenoma formation is the inactivation of both alleles of the APC tumor suppressor gene. Consistent with this, quantitative site-specific PCR assays showed extensive loss of the wild-type Apc allele in adenomas derived from ApcMin/+ mice (36). Using the same assay, we were able to show that LOH of the Apc allele occurred not only in the normal-appearing intestines of ApcMin/+ mice but also in those of Klf4+/−/ApcMin/+ mice (Fig. 6). Moreover, the LOH increased progressively as tumor size increased in both groups of mice (Fig. 6). It is of interest to note that in the largest adenomas (>3 mm), the LOH of Apc was greater in Klf4+/−/ApcMin/+ than in ApcMin/+ mice (Fig. 6). This finding raises the possibility that reduced KLF4 levels may help facilitate LOH of APC.
The results of our study support a role for KLF4 in suppressing intestinal tumorigenesis by modulating β-catenin and cyclin D1 activity. KLF4 has been shown to regulate the Wnt/β-catenin pathway by inhibiting the expression of β-catenin (23). In addition, KLF4 binds the transcriptional activation domain of β-catenin and inhibits β-catenin–mediated transcription (24). Both of these findings were confirmed by the in vivo data in the current study showing the significant increase in the levels of β-catenin in the normal-appearing small intestinal tissues in the Klf4+/−/ApcMin/+ mice. Moreover, β-catenin staining in the normal-appearing intestine of Klf4+/−/ApcMin/+ mice changes from a membranous pattern in the villi to a cytoplasmic pattern in the crypts (Fig. 3). This is further changed to a cytoplasmic/nuclear distribution in the adenomas (Fig. 4). A previous study showed that both the cytoplasmic and cytoplasmic/nuclear patterns of β-catenin staining were associated with increased proliferative index (50). The different staining patterns of β-catenin in the Klf4+/−/ApcMin/+ mice may thus represent different mechanisms of cyclin D1 induction. In cells with membranous β-catenin, Klf4 may directly repress cyclin D1 promoter; and the decrease in Klf4 releases cyclin D1 from repression. In contrast, in cells with cytoplasmic and cytoplasmic/nuclear distribution of β-catenin, cyclin D1 may be directly induced by β-catenin.
It has also been previously shown that the expression of KLF4 was dependent on APC (22). Our observations that Klf4 mRNA and protein levels were decreased in the normal-appearing intestinal tissues of Klf4+/−/ApcMin/+ mice compared with wild-type, Klf4+/−, and ApcMin/+ mice also supports the notion that Klf4 is regulated by Apc. Moreover, the cellular localization of KLF4 and APC in the intestinal epithelium was similar to each other (2, 17). Taken together, the findings of the previous studies and the current study suggest a synergistic mechanism by which KLF4 and APC may function to regulate intestinal epithelial cell proliferation.
The increase in adenoma formation in Klf4+/−/ApcMin/+ mice may thus be viewed in the context of crosstalk between KLF4 and the Wnt/β-catenin pathway. The decrease in the wild-type Apc level by LOH in the Klf4+/−/ApcMin/+ mice leads to a decrease in Klf4 to a level below that of ApcMin/+ mice. This decrease is accentuated by having only one wild-type Klf4 allele in the Klf4+/−/ApcMin/+ mice compared with ApcMin/+ mice. As KLF4 has been shown to inhibit β-catenin–mediated gene expression (23, 24), the simultaneous reduction in Klf4 and Apc could exacerbate the dysregulation of β-catenin in the intestinal epithelium, leading to an increased rate of proliferation, a greater chance for early LOH, and earlier initiation of tumorigenesis. We also speculate that mutations in the APC gene, along with haploinsufficiency of the KLF4 gene in the intestine, would result in the constitutive activation of other β-catenin target genes that promote cell proliferation, and eventually, transformation.
In conclusion, we show for the first time that reduced Klf4 expression in vivo plays a role in the onset of intestinal adenoma formation in ApcMin/+ mice. Our findings suggest that genetic events which inactivate KLF4 may promote tumorigenesis in human colorectal cancers, particularly in those with mutations in the APC gene. Examination of human colorectal cancer tissues for evidence of KLF4 mutations may further help to substantiate the role of KLF4 in colorectal carcinogenesis.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: NIH grants DK52230, DK64399, and CA84197. V.W. Yang is the recipient of a Georgia Cancer Coalition Distinguished Cancer Clinician Scientist Award.
Footnotes
References
- 1.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 2.Smith KJ, Johnson KA, Bryan TM, et al. The APC gene product in normal and tumor cells. Proc Natl Acad Sci U S A. 1993;90:2846–50. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baeg GH, Matsumine A, Kuroda T, et al. The tumour suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMBO J. 1995;14:5618–25. doi: 10.1002/j.1460-2075.1995.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinen CD, Goss KH, Cornelius JR, et al. The APC tumor suppressor controls entry into S-phase through its ability to regulate the cyclin D/RB pathway. Gastroenterology. 2002;123:751–63. doi: 10.1053/gast.2002.35382. [DOI] [PubMed] [Google Scholar]
- 5.Olmeda D, Castel S, Vilaro S, Cano A. β-Catenin regulation during the cell cycle: implications in G2/M and apoptosis. Mol Biol Cell. 2003;14:2844–60. doi: 10.1091/mbc.E03-01-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trzepacz C, Lowy AM, Kordich JJ, Groden J. Phos-phorylation of the tumor suppressor adenomatous polyposis coli (APC) by the cyclin-dependent kinase p34. J Biol Chem. 1997;272:21681–4. doi: 10.1074/jbc.272.35.21681. [DOI] [PubMed] [Google Scholar]
- 7.Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 8.Morin PJ, Sparks AB, Korinek V, et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 9.Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–2. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 10.Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 11.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 12.Bieker JJ. Krüppel-like factors: three fingers in many pies. J Biol Chem. 2001;276:34355–8. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 13.Black AR, Black JD, Azizkhan-Clifford J. Sp1 and Krüppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–60. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 14.Dang DT, Pevsner J, Yang VW. The biology of the mammalian Krüppel-like family of transcription factors. Int J Biochem Cell Biol. 2000;32:1103–21. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaczynski J, Cook T, Urrutia R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics. 2005;85:551–6. doi: 10.1016/j.ygeno.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Krüppel-like factor expressed during growth arrest. J Biol Chem. 1996;271:20009–17. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Krüppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–6. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Shields JM, Sogawa K, Fujii-Kuriyama Y, Yang VW. The gut-enriched Krüppel-like factor suppresses the activity of the CYP1A1 promoter in an Sp1-dependent fashion. J Biol Chem. 1998;273:17917–25. doi: 10.1074/jbc.273.28.17917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon HS, Chen X, Yang VW. Krüppel-like factor 4 mediates p53-dependent G1/S cell cycle arrest in response to DNA damage. J Biol Chem. 2003;278:2101–5. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoon HS, Yang VW. Requirement of Krüppel-like factor 4 in preventing entry into mitosis following DNA damage. J Biol Chem. 2004;279:5035–41. doi: 10.1074/jbc.M307631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang DT, Mahatan CS, Dang LH, Agboola IA, Yang VW. Expression of the gut-enriched Krüppel-like factor (Krüppel-like factor 4) gene in the human colon cancer cell line RKO is dependent on CDX2. Oncogene. 2001;20:4884–90. doi: 10.1038/sj.onc.1204645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone CD, Chen ZY, Tseng CC. Gut-enriched Krüppel-like factor regulates colonic cell growth through APC/β-catenin pathway. FEBS Lett. 2002;530:147–52. doi: 10.1016/s0014-5793(02)03449-x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Chen X, Kato Y, et al. Novel cross talk of Krüppel-like factor 4 and β-catenin regulates normal intestinal homeostasis and tumor repression. Mol Cell Biol. 2006;26:2055–64. doi: 10.1128/MCB.26.6.2055-2064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Krüppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402. doi: 10.1038/sj.onc.1207067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi BJ, Cho YG, Song JW, et al. Altered expression of the KLF4 in colorectal cancers. Pathol Res Pract. 2006;202:585–9. doi: 10.1016/j.prp.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Wei D, Gong W, Kanai M, et al. Drastic down-regulation of Krüppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005;65:2746–54. doi: 10.1158/0008-5472.CAN-04-3619. [DOI] [PubMed] [Google Scholar]
- 28.Wei D, Kanai M, Huang S, Xie K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis. 2006;27:23–31. doi: 10.1093/carcin/bgi243. [DOI] [PubMed] [Google Scholar]
- 29.Wang N, Liu ZH, Ding F, Wang XQ, Zhou CN, Wu M. Down-regulation of gut-enriched Krüppel-like factor expression in esophageal cancer. World J Gastroenterol. 2002;8:966–70. doi: 10.3748/wjg.v8.i6.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 31.Su LK, Kinzler KW, Vogelstein B, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 32.Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–26. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katz JP, Perreault N, Goldstein BG, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002;129:2619–28. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietrich WF, Lander ES, Smith JS, et al. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–9. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 35.Dang DT, Bachman KE, Mahatan CS, Dang LH, Giardiello FM, Yang VW. Decreased expression of the gut-enriched Krüppel-like factor gene in intestinal adenomas of multiple intestinal neoplasia mice and in colonic adenomas of familial adenomatous polyposis patients. FEBS Lett. 2000;476:203–7. doi: 10.1016/s0014-5793(00)01727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–52. [PubMed] [Google Scholar]
- 37.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–60. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 38.Chen X, Johns DC, Geiman DE, et al. Krüppel-like factor 4 (gut-enriched Krüppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. 2001;276:30423–8. doi: 10.1074/jbc.M101194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Whitney EM, Gao SY, Yang VW. Transcriptional profiling of Krüppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J Mol Biol. 2003;326:665–77. doi: 10.1016/S0022-2836(02)01449-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitney EM, Ghaleb AM, Chen X, Yang VW. Transcriptional profiling of the cell cycle checkpoint gene Krüppel-like factor 4 reveals a global inhibitory function in macromolecular biosynthesis. Gene Expr. 2006;13:85–96. doi: 10.3727/000000006783991908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ton-That H, Kaestner KH, Shields JM, Mahatanankoon CS, Yang VW. Expression of the gut-enriched Krüppel-like factor gene during development and intestinal tumorigen-esis. FEBS Lett. 1997;419:239–43. doi: 10.1016/s0014-5793(97)01465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang DT, Chen X, Feng J, Torbenson M, Dang LH, Yang VW. Overexpression of Krüppel-like factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenecity. Oncogene. 2003;22:3424–30. doi: 10.1038/sj.onc.1206413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lal G, Gallinger S. Familial adenomatous polyposis. Semin Surg Oncol. 2000;18:314–23. doi: 10.1002/(sici)1098-2388(200006)18:4<314::aid-ssu6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 44.Shoemaker AR, Gould KA, Luongo C, Moser AR, Dove WF. Studies of neoplasia in the Min mouse. Biochim Biophys Acta. 1997;1332:F25–48. doi: 10.1016/s0304-419x(96)00041-8. [DOI] [PubMed] [Google Scholar]
- 45.Shie JL, Chen ZY, O’Brien MJ, Pestell RG, Lee ME, Tseng CC. Role of gut-enriched Krüppel-like factor in colonic cell growth and differentiation. Am J Physiol Gastrointest Liver Physiol. 2000;279:G806–14. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- 46.Ohnishi S, Ohnami S, Laub F, et al. Downregulation and growth inhibitory effect of epithelial-type Krüppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun. 2003;308:251–6. doi: 10.1016/s0006-291x(03)01356-1. [DOI] [PubMed] [Google Scholar]
- 47.Miyoshi Y, Nagase H, Ando H, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–33. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 48.Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 49.Ichii S, Horii A, Nakatsuru S, Furuyama J, Utsunomiya J, Nakamura Y. Inactivation of both APC alleles in an early stage of colon adenomas in a patient with familial adenomatous polyposis (FAP) Hum Mol Genet. 1992;1:387–90. doi: 10.1093/hmg/1.6.387. [DOI] [PubMed] [Google Scholar]
- 50.Kotsinas A, Evangelou K, Zacharatos P, Kittas C, Gorgoulis VG. Proliferation, but not apoptosis, is associated with distinct β-catenin expression patterns in non-small-cell lung carcinomas: relationship with adenomatous polyposis coli and G(1)-to S-phase cell-cycle regulators. Am J Pathol. 2002;161:1619–34. doi: 10.1016/s0002-9440(10)64440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).