

Abstract
Bax/Bak activation and cardiolipin peroxidation are essential for cytochrome c release during apoptosis, yet, the link between them remains elusive. We report that sequence of events after exposure of mouse embryonic fibroblast (MEF) cells to actinomycin D followed the order: Bax translocation -> superoxide production -> cardiolipin peroxidation. Genetic ablation of Bax/Bak inhibited actinomycin D induced superoxide production and cardiolipin peroxidation. Rotenone caused robust superoxide generation but did not trigger cardiolipin peroxidation in Bax/Bak double knockout MEF cells. This suggests that, in addition to participating in ROS generation, Bax/Bak play another specific role in cardiolipin oxidation. In isolated mitochondria, recombinant Bax enhanced succinate induced cardiolipin oxidation and cytochrome c release. Mitochondrial peroxidase activity, likely involved in cardiolipin peroxidation, was enhanced upon incubation with recombinant Bax. Thus cardiolipin peroxidation may be causatively and time-dependently related to Bax/Bak effects on ROS generation and peroxidase activation of cytochrome c.
Keywords: Bax/Bak, cardiolipin oxidation, cytochrome c, mitochondria, reactive oxygen species, superoxide, peroxidase
Introduction
Release of cytochrome (cyt) c from mitochondria into the cytosol is a defining event that marks the point of no return in apoptotic signaling pathway. The precise mechanisms of cyt c release remain elusive. Activation of Bax/Bak to form a requisite gateway on the outer mitochondrial membrane (OMM) has been pointed as a direct trigger [1,2]. In addition to the critical role in OMM permeabilization, Bax/Bak have been associated with earlier events of the mitochondrial stage of apoptosis. It has been reported that Bax acts upstream of increased reactive oxygen species (ROS) production in nerve growth factor-deprived neurons and that Bax induced ROS burst is both necessary and sufficient for cyt c redistribution in these cells [3]. Priault et al. [4] suggested that Bax induced cell death is related to generation of ROS and membrane lipid peroxidation.
Accumulating evidence indicates that cardiolipin (CL), a mitochondria specific phospholipid, is involved in the apoptotic pathways that initiate release of cyt c from the mitochondrial intermembrane space [5-7]. Disassociation of cyt c from the inner mitochondria membrane (IMM) [8,9], by peroxidation of CL is considered the first step for the departure of cyt c from mitochondria [10]. A likely involvement of CL peroxidation products in mitochondrial permeabilization is supported by the restored ability of oxidized CL to release Smac/Diablo from mitochondria isolated from cyt c deficient cells [11].
While the link between Bax/Bak activation and CL oxidation in the regulation of cyt c release during apoptosis remains unspecified [4,6,12], two processes - generation of ROS and activation of cyt c into a CL-specific peroxidase [11] - are essential requisites for the oxidation of CL. Here, we report that Bax/Bak controls CL oxidation by affecting both of these processes.
Materials and Methods
Cell culture
Wild type (WT) and Bax/Bak double knockout (DKO) mouse embryonic fibroblast (MEF) cells are courtesy of Dr. SJ Korsmeyer's lab (Dana-Farber Cancer Institute, Boston, MA) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat inactivated fetal bovine serum, 0.1 mmol/L nonessential amino acids, 0.1 μmol/L 2-mercaptoethanol, and antibiotics at 37°C in an atmosphere of 95% air and 5% CO2.
Superoxide generation
Oxidation-dependent fluorogenic dye, dihydroethidium (DHE) was used to evaluate intracellular production of superoxide radicals. Briefly, cells were incubated with 5 μM DHE for 30 min at the indicated time point. Cells were collected by trypsinization and resuspended in PBS. The fluorescence of ethidium was measured using a FACScan flow cytometer (Becton-Dickinson, Rutherford, NJ) supplied with the CellQuest software. Mean fluorescence intensity from 10,000 cells was acquired using a 585/42 nm bandpass filter.
HPTLC separation of phospholipids and CL oxidation assay
Lipids were extracted from cells and resolved by 2D HPTLC as previously described [13]. Spots of phospholipids were scraped from the HPTLC plates and phospholipids were extracted from silica. Lipid phosphorus was determined by a micro-method [13]. CL hydroperoxides were determined by fluorescence HPLC of products formed in Microperoxidase (MP)-11-catalyzed reaction with a fluorogenic substrate, Amplex Red. Oxidized phospholipids were hydrolyzed by pancreatic phospholipase A2 (2 U/ml) in 25 mM phosphate buffer containing 1 mM CaCl2, 0.5 mM EDTA and 0.5 mM SDS (pH 8.0 at RT for 30 min). After that Amplex Red and MP-11 were added and samples were incubated for 40 min at 4°C. Shimadzu LC-100AT vp HPLC system (Shimadzu, Columbia, MD) equipped with fluorescence detector (RF-10Axl, Excitation/Emission=560/590 nm) and autosampler (SIL-10AD vp) were used for the analysis of products separated by HPLC (Eclipse XDB-C18 column, 5 μm, 150×4.6 mm, Agilent Technology, Santa Clara , CA). Mobile phase was composed of NaH2PO4 (25 mM, pH 7.0)/methanol (60:40 v/v).
Immunoblotting
Harvested cells were resuspended in lysis buffer containing 250 mM sucrose, 20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 0.05% digitonin for 4 min on ice, and then centrifuged at 10,000×g for 5 min. The resulting supernatants and pellets were subjected to 12% SDS-PAGE and then transferred to a nitrocellulose membrane, which was probed with antibodies against Bax (clone 6A7, Pharmingen, San Diego, CA) or cyt c (clone 7H8.2C12, Pharmingen) followed by horseradish peroxidase-coupled detection. Duplicated membranes were probed with actin (anti-actin antibody, Calbiochem, San Diego, CA) or COX-IV (cyt c oxidase subunit IV, anti COX-IV antibody, Abcam, Cambridge, MA) as loading controls, respectively. Semi-quantitation of the bands was carried out by densitometry using Labworks Image Acquisition and Analysis Software (UVP, Upland, CA). The levels of Bax were expressed as the mean densitometry ratio of Bax over actin or COX-IV and then normalized to control.
Phosphatidylserine (PS) externalization
Externalization of PS was analyzed by flow cytometry using annexin-V kit (Biovision, Mountain View, CA). Briefly, harvested cells were stained with annexin-V-FITC and PI for 5 min in dark prior to flow cytometry analysis. Ten thousand events were collected on a FACScan flow cytometer supplied with CellQuest software.
Mitochondria preparation
Mitochondrial fractions were prepared by differential centrifugation. Briefly, MEF DKO cells were suspended in mitochondria isolation buffer (MIB, 210 mM mannitol, 70 mM sucrose, 10 mM HEPES, pH 7.4, 1 mM EDTA) and lysed by Dounce homogenization. Unbroken cells, nuclei and debris were removed by 10 min of centrifugation at 700×g at 4°C. A mitochondria rich fraction was obtained by 10 min centrifugation at 5,000×g, and washed twice with MIB.
Recombinant Bax induced release of pro-apoptotic factors from isolated mitochondria
Isolated mitochondria (1 mg/mL) were resuspended in the incubation buffer A (210 mM sucrose, 70 mM KCl, 10 mM HEPES, pH7.4, 3 mM sodium phosphate, 0.5 mM EGTA) in the presence or absence of 5 mM succinate, and then incubated with mouse recombinant Bax (200, 400 nM, ProteinX Lab, San Diego, CA) for 20 min at room temperature. Samples were then centrifuged at 10,000×g for 5 min. The resulting supernatants and pellets were analyzed for cyt c, Smac/Diablo (clone 7, Pharmingen) or COX-IV (loading control to confirm equal amount of mitochondria was used) content by Western Blot. The levels of released proteins were expressed as the mean densitometry ratio of cyt c or Smac/Diablo over COX-IV and then normalized to control sample without Bax incubation.
Mitochondrial peroxidase activity
Peroxidase activity in mitochondria was evaluated using 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA). Mitochondria (0.5 mg/mL) were resuspended in incubation buffer A with 100 μM DTPA and 10 μM DCFH-DA. Recombinant and Bax (200 nM) was then added to mitochondria for 15-min incubation. The reaction was initiated by adding 20 μM H2O2 to the samples. Fluorescence spectra were recorded with a Shimadzu RF-5301PC fluorescence spectrophotometer (Shimadzu, Kyoto, Japan) after 30 min incubation (Excitation=488 nm).
Statistical analysis
All data are expressed as means ± S.D. of at least three independent experiments. Statistical comparisons between groups were performed by Student's t-test.
Results and Discussion
Translocation of Bax precedes superoxide generation and cardiolipin oxidation in actinomycin D (ActD) induced apoptosis in MEF cells
WT and Bax/Bak DKO MEF cells were incubated with 50 ng/mL ActD for indicated periods of time (4, 5, 6, 7, 8, 9, 10, 16 and 18 h). Lowered levels of cytosolic Bax were detectable as early as 5 h after exposure to ActD and the decrease became significant after 7 h incubation. Accordingly, time dependent increase of Bax accumulation in the mitochondrial fraction was observed (Fig. 1A, B). A significant increase of superoxide generation occurred after 8 h incubation in WT MEF cells. In line with previous studies [14,15], no increased superoxide generation was observed in Bax/Bak DKO MEF cells under the same conditions (Fig. 1C). Significant accumulation of CL hydroperoxides (31.8 ± 6.3 vs 11.3 ± 5.6 pmol/nmol CL in control, p<0.05) was only observed after 16 h incubation with ActD in WT MEF cells, but not in Bax/Bak DKO MEF cells even after 18 h incubation (13.4 ± 4.2 vs 7.3 ± 3.6 pmol/nmol CL in control) (Fig. 1D). Consequently, accumulation of cyt c in the cytosol and PS externalization occurred in ActD treated MEF cells but were not observed in Bax/Bak DKO MEF cells (Fig. 2A, E). These results indicate that oxidation of CL was a downstream event following Bax/Bak translocation in ActD treated MEF cells.
Fig. 1.
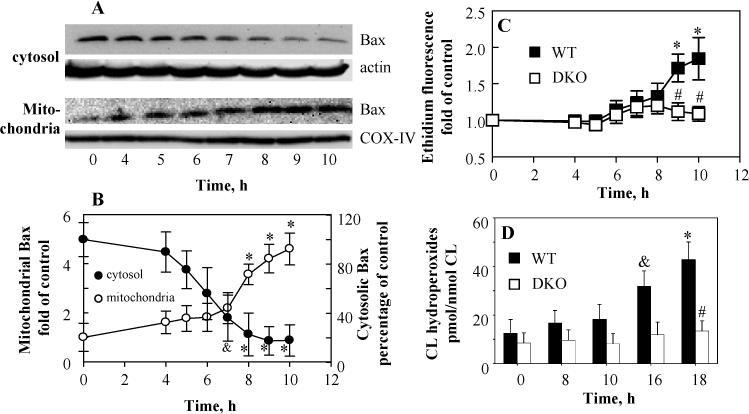
Time course of Bax translocation, superoxide generation and CL hydroperoxides accumulation in actinomycin D (ActD) treated MEF cells. MEF cells were treated with 50 ng/mL ActD for indicated period of time (0, 4, 5, 6, 7, 8, 9, 10, 16, and 18 h). (A)(B) Determination of subcellular relocation of Bax in WT MEF cells by Western blot. The levels of Bax were expressed as the mean densitometry ratio of Bax over actin or COX-IV and then normalized to control. (C) Time dependent generation of superoxide in ActD treated MEF cells evaluated by dihydroethidium assay. (D) Time dependent accumulation of CL hydroperoxides in ActD treated MEF cells. CL hydroperoxides were determined using an Amplex Red based fluorescent HPLC assay. Data presented are means ± S.D. (n=3), *(&) p<0.01 (0.05) vs control cells, # p<0.01 vs WT MEF cells under the same condition.
Fig. 2.
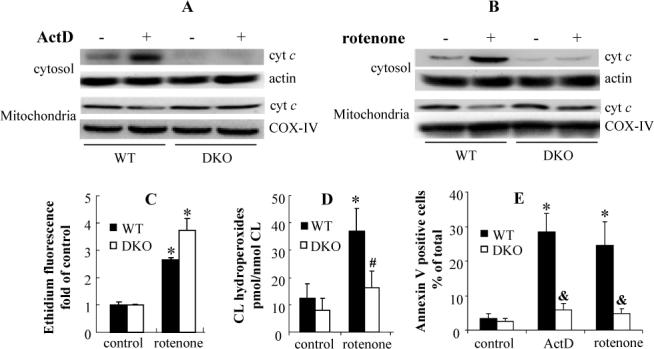
(A)(B) Redistribution of cyt c in ActD and rotenone treated MEF cells. WT and Bax/Bak DKO MEF cells were treated with 50 ng/mL ActD or 1 μM rotenone for 18 h. Harvested cells were resuspended in digitonin containing lysis buffer. The cyt c amounts in the cytosolic and mitochondrial fraction were analyzed by Western Blot. Actin and COX-IV were used as loading control respectively. (C) Determination of rotenone (1 μM, 4 h) induced superoxide generation in WT and DKO MEF cells using dihydroethidium assay. (D) Assessment of rotenone (1 μM, 18 h) induced CL oxidation in WT and Bax/Bak DKO MEF cells. CL hydroperoxides were determined using an Amplex Red based fluorescent HPLC assay. (E) ActD (50 ng/mL, 18 h) and rotenone (1 μM, 18 h) induced PS externalization. Data presented are means ± S.D.(n=3). * p<0.01 vs control cells. # (&) p<0.05 (0.01) vs WT MEF cells under the same condition.
Rotenone induced superoxide generation is not sufficient for CL oxidation and cyt c release
To determine whether Bax/Bak involvement in CL oxidation was exclusively due to their effects on ROS production, WT and Bax/Bak DKO MEF cells were treated with rotenone, a mitochondria respiratory complex I inhibitor, to induce superoxide generation independently of Bax/Bak. Indeed, comparable amounts of superoxide were detected in WT and DKO MEF cells (2.6 and 3.7 fold over control, respectively) following rotenone treatment (1 μM, 4 h) (Fig. 2C). No accumulation of CL hydroperoxides and release of cyt c were detected in Bax/Bak DKO MEF cells after 18-h incubation with rotenone (Fig. 2B, D). In contrast, CL hydroperoxides mounted up to 36.9 ± 7.6 pmol/nmol CL in rotenone treated WT MEF cells (p<0.01 vs 12.6 ± 5.3 pmol/nmol CL in control, Fig. 2D). This was accompanied by the release of cyt c into the cytosolic fraction and exposure of PS on cell surface (Fig. 2B, E). Thus Bax/Bak participation in CL oxidation is not limited to their role in activation of ROS generation.
Recombinant Bax enhances succinate induced CL oxidation and cyt c release in isolated mitochondria
In mitochondria isolated from Bax/Bak DKO MEF cells, succinate, a complex II substrate known to generate superoxide due to the reverse electron flow, induced an increase of CL oxidation (24.6 ±7.8 vs 9.8 ± 6.5 pmol/nmol CL in non-energized mitochondria). This effect was markedly enhanced (53.8 ± 9.7 pmol/nmol CL) in the presence of recombinant Bax (Fig. 3A). These data further indicate that Bax/Bak play additional role(s) in CL oxidation distinct from their effects on ROS generation. Consistent with a previous study [6], release of cyt c was correlated with oxidation of CL; neither succinate alone nor recombinant Bax alone was sufficient to induce massive cyt c release (Fig. 3B,C).
Fig. 3.
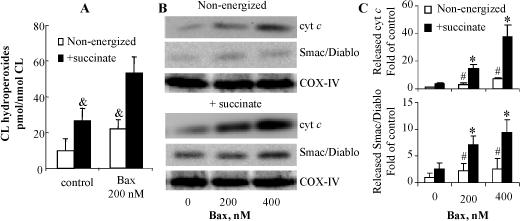
(A) Effect of succinate and recombinant Bax on CL oxidation in mitochondria isolated from Bax/Bak DKO MEF cells. Mitochondria (1 mg/mL) were incubated with 200 nM Bax in the presence or absence of 5 mM succinate for 20 min at room temperature. The contents of CL hydroperoxides were determined using Amplex Red assay. (B) Recombinant Bax induced release of cyt c and Smac/Diablo from mitochondria isolated from Bax/Bak DKO MEF cells. Mitochondria (1 mg/ml) were incubated with recombinant Bax (200 and 400 nM) for 20 min at room temperature in the presence or absence or succinate. The released cyt c and Smac/Diablo were examined by Western Blot. (C) The levels of released proteins were expressed as the mean densitometry ratio of cyt c or Smac/Diablo over COX-IV and then normalized to control sample without Bax treatment. Data presented are mean ± S.D. (n=3). & p<0.05 vs energized mitochondria treated with recombinant Bax. * p<0.01 vs mitochondria without Bax treatment. # p<0.01 vs succinate energized mitochondria under the same condition.
Recombinant Bax increases mitochondrial peroxidase activity
It is generally accepted that the major pool of cyt c is free or loosely (electrostatically) attached to the outer leaflet of IMM. This cyt c maintains hexa-coordinated low-spin configuration of heme iron and does not exert peroxidase activity [11]. Strong electrostatic and hydrophobic interactions along with hydrogen bonding between CL and cyt c induce protein unfolding which confers the peroxidase activity on partially denatured cyt c [11]. These CL-dependent structural transitions include the disturbance of heme iron hexa-coordination, opening of heme crevice, and the adjustment of the hydrophobic face of amphipathic helices toward the hydrocarbon core of the lipid bilayer [16-18]. It has been demonstrated that Bax induces a rapid transbilayer movement of lipids [19]. Moreover, we and others reported that CL moves to the outer leaflet of IMM in the early phase of apoptosis [11,20], Thus, it is tempting to speculate that Bax facilitates the formation of cyt c/CL complexes with peroxidase activity. Therefore, we examined the peroxidase activity in recombinant Bax pretreated mitochondria. As shown in Fig. 4, oxidation of DCFH was markedly enhanced in the Bax treated mitochondria (p<0.05 vs mitochondria without Bax incubation). Notably, other anionic phospholipids, such as PS, phosphatidylinositol (PI), phosphatidic acid (PA), and phosphatidylglycerol (PG), are also capable of inducing the conformational changes of cyt c, thus increasing its peroxidase activity. In fact, in a recent study [21], we performed comparative quantitative analysis of the ability of different anionic phospholipids to unfold cyt c and induce its peroxidase activity. CL was found to be the most effective “destabilizer” of the cyt c's structure as compared to other anionic phospholipids such as PA, PS, and PIs. It should be also noted that only marginal amounts (<1.0%) of PS, PA and PG are present in mitochondria [22,23]. The content of CL is about 10 times higher than that of PI in the inner mitochondrial membrane [22]. In addition, the estimated binding constant of CL with cyt c is markedly higher than that with PS (1.7 ± 1.0×109 M−1 for tetraoleoyl-CL/cyt c vs 3.8 ± 1.8×106 M−1 for dioleoyl-PS/cyt c) [24,25]. Therefore, it is reasonable to assume that CL is the major contributor to the mitochondrial activation of cyt c into a peroxidase during apoptosis.
Fig. 4.
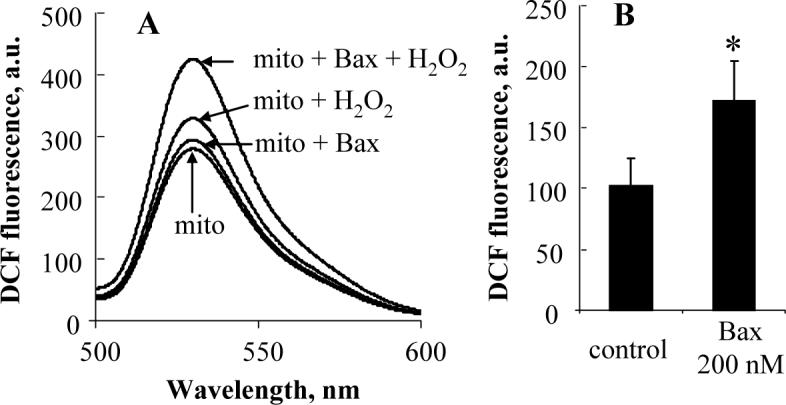
Effect of recombinant Bax on mitochondrial peroxidase activity. The peroxidase activity was evaluated using DCFH-DA assay. Fluorescence spectra were recorded with a Shimadzu RF-5301PC fluorescence spectrophotometer (Shimadzu, Kyoto, Japan) after 30 min incubation (after excitation at 488 nm). (A) Spectra are representative of three independent experiments. (B) Data are mean ± S.D.( n=3). *p<0.05 vs mitochondria without Bax treatment.
The specific molecular mechanisms through which Bax may be involved in regulation of cardiolipin peroxidation have not been established yet, although previous studies have elucidated that Bax acts upstream of accumulation of ROS [3,4,26] that required for lipid peroxidation. Recently, we reported that mitochondria targeted electron scavengers - hemi-gramicidin S conjugated nitroxides - prevented both superoxide accumulation and CL peroxidation and apoptosis initiated by ActD or irradiation without affecting Bax translocation [27,28]. Combined with the results of this work showing that Bax/Bak translocation precedes superoxide production and CL peroxidation and that genetic ablation of Bax/Bak inhibited ActD and rotenone induced CL oxidation and apoptosis, we speculate that CL peroxidation not only correlates but is likely causatively and time-dependently related to mitochondrial relocation of Bax/Bak. This is mainly due to Bax/Bak effect on ROS generation and peroxidase activation of cyt c. This molecular scenario portrays the link between proteins of Bcl-2 family and CL peroxidation in the regulation of release of cyt c during apoptosis. The detailed mechanisms through which oxidized CL induces permeabilization of mitochondria still remain elusive, although several models have been proposed [5,6,11,29]. The fact that the recombinant Bax induced a release of Smac/Diablo from non-energized mitochondria similarly to cyt c (Fig. 3B,C) indicates that the role of CL oxidation is far broader than simply creation of a pool of cyt c available for the release. The effects of oxidized CL on Bax/Bak and other mitochondrial components such as adenine nucleotide translocator and voltage-dependent anion channel ultimately leading to the OMM permeabilization during apoptosis await further exploration.
Acknowledgements
This work is supported by NIH NIAID U19 AI068021 and The Human Frontier Science Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Antonsson B. Mitochondria and the Bcl-2 family proteins in apoptosis signaling pathways. Mol. Cell Biochem. 2004;256−257:141–155. doi: 10.1023/b:mcbi.0000009865.70898.36. [DOI] [PubMed] [Google Scholar]
- 3.Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J. Neurosci. 2002;22:6480–6490. doi: 10.1523/JNEUROSCI.22-15-06480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Priault M, Bessoule JJ, Grelaud-Coq A, Camougrand N, Manon S. Bax-induced cell death in yeast depends on mitochondrial lipid oxidation. Eur. J. Biochem. 2002;269:5440–5450. doi: 10.1046/j.1432-1033.2002.03234.x. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–885. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- 6.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 7.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett. 2006;580:6311–6316. doi: 10.1016/j.febslet.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 8.Shidoji Y, Hayashi K, Komura S, Ohishi N, Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation, Biochem. Biophys. Res. Commun. 1999;264:343–347. doi: 10.1006/bbrc.1999.1410. [DOI] [PubMed] [Google Scholar]
- 9.Ostrander DB, Sparagna GC, Amoscato AA, McMillin JB, Dowhan W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J. Biol. Chem. 2001;276:38061–38067. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- 10.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 12.Perier C, Tieu K, Guégan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc. Natl. Acad. Sci. USA. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyurina YY, Kawai K, Tyurin VA, Liu SX, Kagan VE, Fabisiak JP. The plasma membrane is the site of selective phosphatidylserine oxidation during apoptosis: role of cytochrome c. Antioxid. Redox. Signal. 2004;6:209–225. doi: 10.1089/152308604322899288. [DOI] [PubMed] [Google Scholar]
- 14.Kirkland RA, Franklin JL. Prooxidant effects of NGF withdrawal and MEK inhibition in sympathetic neurons. Antioxid. Redox. Signal. 2003;5:635–639. doi: 10.1089/152308603770310301. [DOI] [PubMed] [Google Scholar]
- 15.Boya P, Morales MC, Gonzalez-Polo RA, Andreau K, Gourdier I, Perfettini JL, Larochette N, Deniaud A, Baran-Marszak F, Fagard R, Feuillard J, Asumendi A, Raphael M, Pau B, Brenner C, Kroemer G. The chemopreventive agent N-(4-hydroxyphenyl) retinamide induces apoptosis through a mitochondrial pathway regulated by proteins from the Bcl-2 family. Oncogene. 2003;22:6220–6230. doi: 10.1038/sj.onc.1206827. [DOI] [PubMed] [Google Scholar]
- 16.Pinheiro TJ, Elöve GA, Watts A, Roder H. Structural and kinetic description of cytochrome c unfolding induced by the interaction with lipid vesicles. Biochemistry. 1997;36:13122–13132. doi: 10.1021/bi971235z. [DOI] [PubMed] [Google Scholar]
- 17.Cortese JD, Voglino AL, Hackenbrock CR. Multiple conformations of physiological membrane-bound cytochrome c. Biochemistry. 1998;37:6402–6409. doi: 10.1021/bi9730543. [DOI] [PubMed] [Google Scholar]
- 18.Sanghera N, Pinheiro TJ. Unfolding and refolding of cytochrome c driven by the interaction with lipid micelles. Protein Sci. 2000;9:1194–1202. doi: 10.1110/ps.9.6.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A, Basañez G. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J. Biol. Chem. 2004;279:30081–30091. doi: 10.1074/jbc.M313420200. [DOI] [PubMed] [Google Scholar]
- 20.Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ. 2002;13:449–455. [PubMed] [Google Scholar]
- 21.Kapralov AA, Kurnikov IV, Vlasova II, Belikova NA, Tyurin VA, Basova LV, Zhao Q, Tyurina YY, Jiang J, Bayir H, Vladimirov YA, Kagan VE. The Hierarchy of Structural Transitions Induced in Cytochrome c by Anionic Phospholipids Determines Its Peroxidase Activation and Selective Peroxidation during Apoptosis in Cells. Biochemistry. 2007;46:14232–14244. doi: 10.1021/bi701237b. [DOI] [PubMed] [Google Scholar]
- 22.Andreoli TE, Fanestil DD, Hoffman JF, Schultz SG. Membrane Physiology. second ed. Springer; New York: 1987. [Google Scholar]
- 23.Choi SY, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007;14:597–606. doi: 10.1038/sj.cdd.4402020. [DOI] [PubMed] [Google Scholar]
- 24.Jiang J, Kini V, Belikova N, Serinkan BF, Borisenko GG, Tyurina YY, Tyurin VA, Kagan VE. Cytochrome c release is required for phosphatidylserine peroxidation during Fas-triggered apoptosis in lung epithelial A549 cells. Lipids. 2004;39:1133–1142. doi: 10.1007/s11745-004-1340-1. [DOI] [PubMed] [Google Scholar]
- 25.Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, Kagan VE. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin containing membranes. Biochemistry. 2006;45:4998–5009. doi: 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manon S. Utilization of yeast to investigate the role of lipid oxidation in cell death. Antioxid. Redox. Signal. 2004;6:259–267. doi: 10.1089/152308604322899323. [DOI] [PubMed] [Google Scholar]
- 27.Jiang J, Kurnikov I, Belikova NA, Xiao J, Zhao Q, Amoscato AA, Braslau R, Studer A, Fink MP, Greenberger JS, Wipf P, Kagan VE. Structural requirements for optimized delivery, inhibition of oxidative stress, and antiapoptotic activity of targeted nitroxides. J. Pharmacol. Exp. Ther. 2007;320:1050–1060. doi: 10.1124/jpet.106.114769. [DOI] [PubMed] [Google Scholar]
- 28.Jiang J, Belikova NA, Hoye AT, Zhao Q, Epperly MW, Greenberger JS, Wipf P, Kagan VE. A Mitochondria-Targeted Nitroxide/Hemi-Gramicidin S Conjugate Protects Mouse Embryonic Cells Against γ-Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2008 doi: 10.1016/j.ijrobp.2007.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson JD, Zhivotovsky B, Gogvadze V, Orrenius S. Outer mitochondrial membrane permeabilization: an open-and-shut case. Cell Death Differ. 2003;10:485–487. doi: 10.1038/sj.cdd.4401218. [DOI] [PubMed] [Google Scholar]